
Curcumin Analogues as a Potential Drug against Antibiotic Resistant Protein, β-Lactamases and L, D-Transpeptidases Involved in Toxin Secretion in Salmonella typhi: A Computational Approach

 , , , , , ,
, , , , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drug-Likeness Features Interpretation
2.1.1. Protein Preparation for Docking
Sequence Retrieval
Physiochemical Property Identification
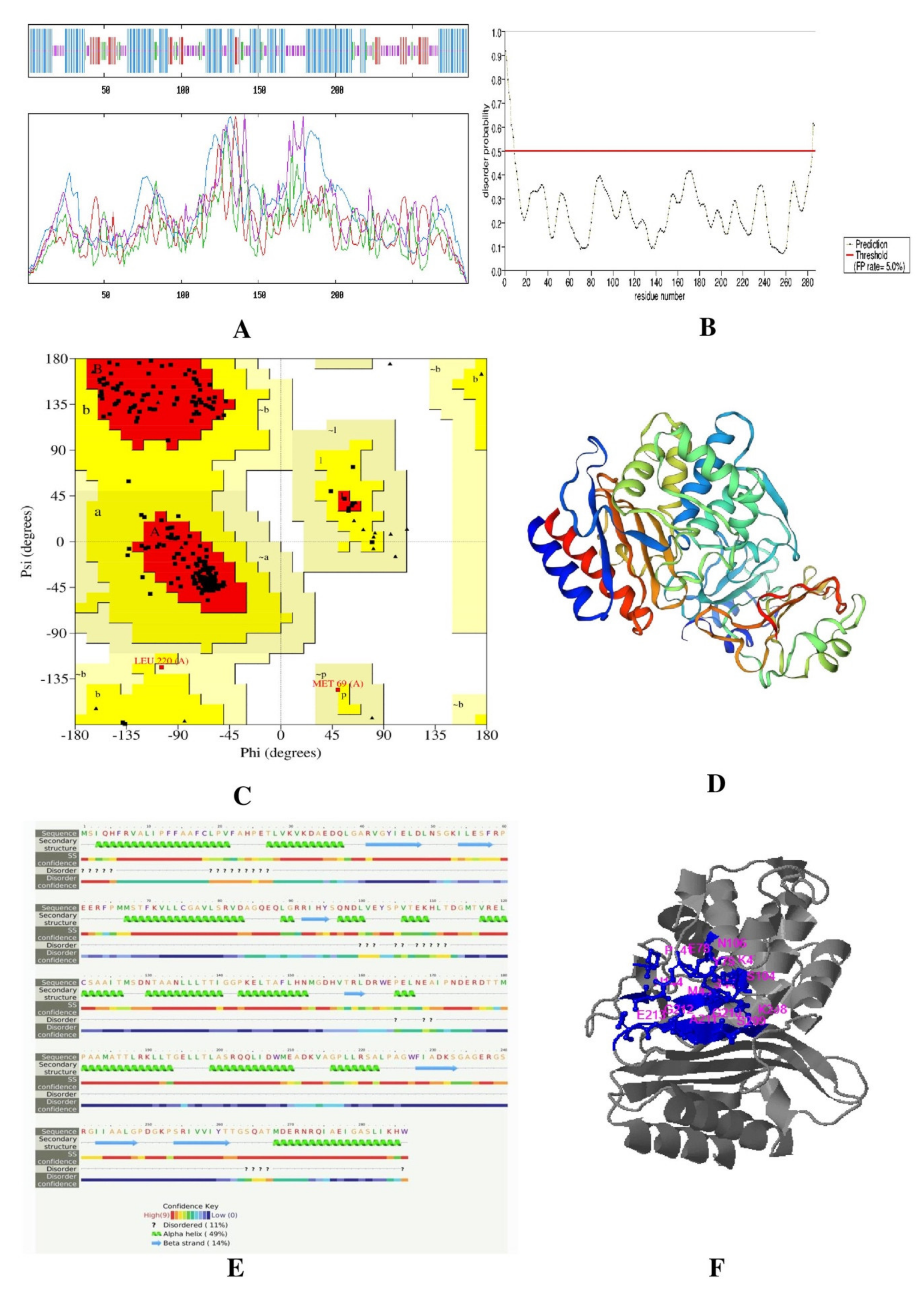
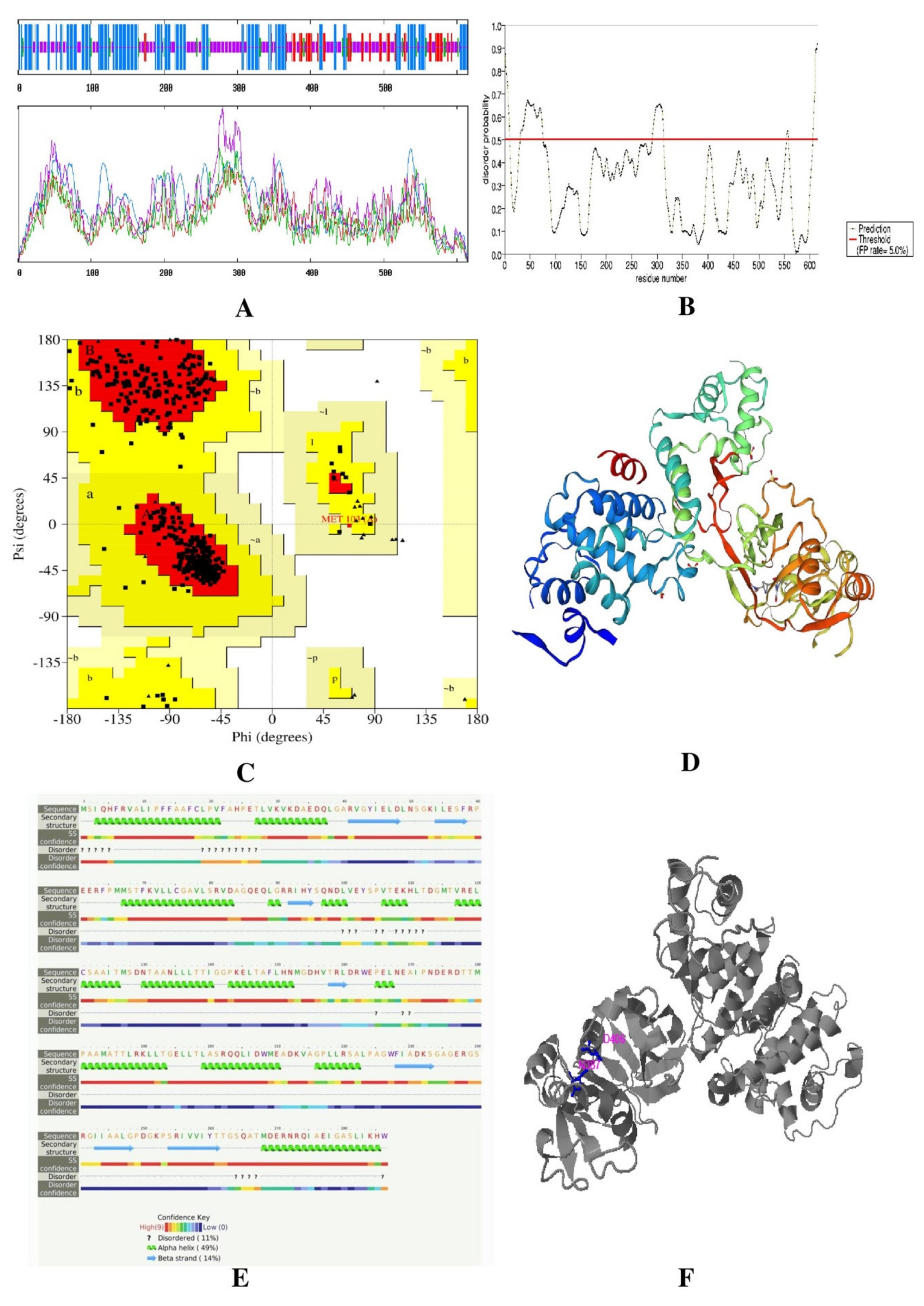
Secondary and Tertiary Structure Prediction
Disordered Regions Prediction
Validation of Tertiary Protein Model
Active Site Prediction
2.1.2. Ligand and Protein Preparation for Docking
2.2. ADME/T Prediction
Validation of Tertiary Protein Model
2.3. Pharmacological and Biological Activity Prediction
2.4. Pred. P450 Site of Metabolism Iction
3. Results
3.1. Drug-Likeness Features Interpretation
3.2. Protein Preparation for Docking
3.2.1. Sequence Salvation
3.2.2. Physiochemical Property Identification
3.2.3. Secondary and Tertiary Structure Prediction
3.2.4. Validation of Tertiary Protein Model
3.2.5. Active Site Prediction
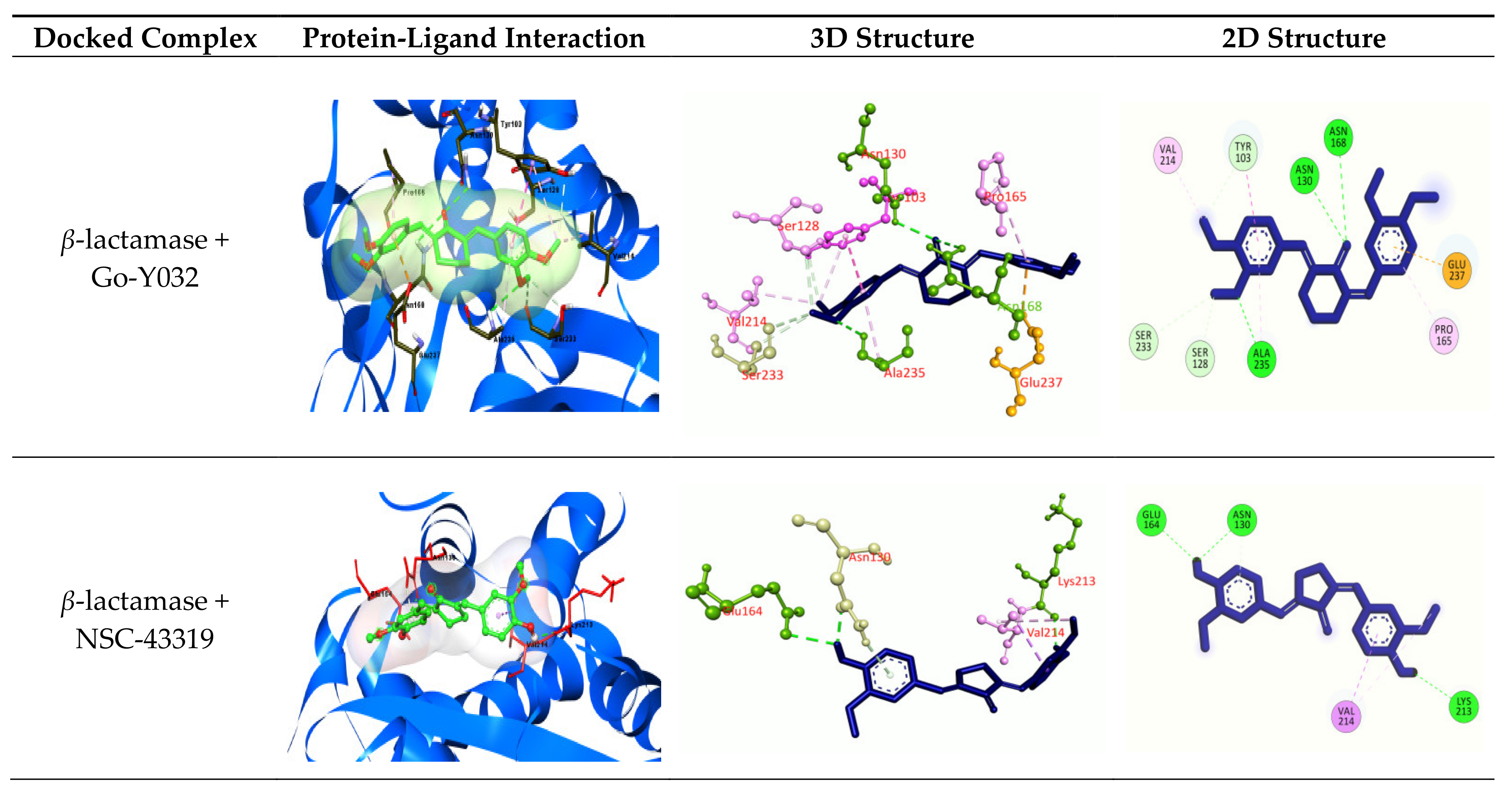
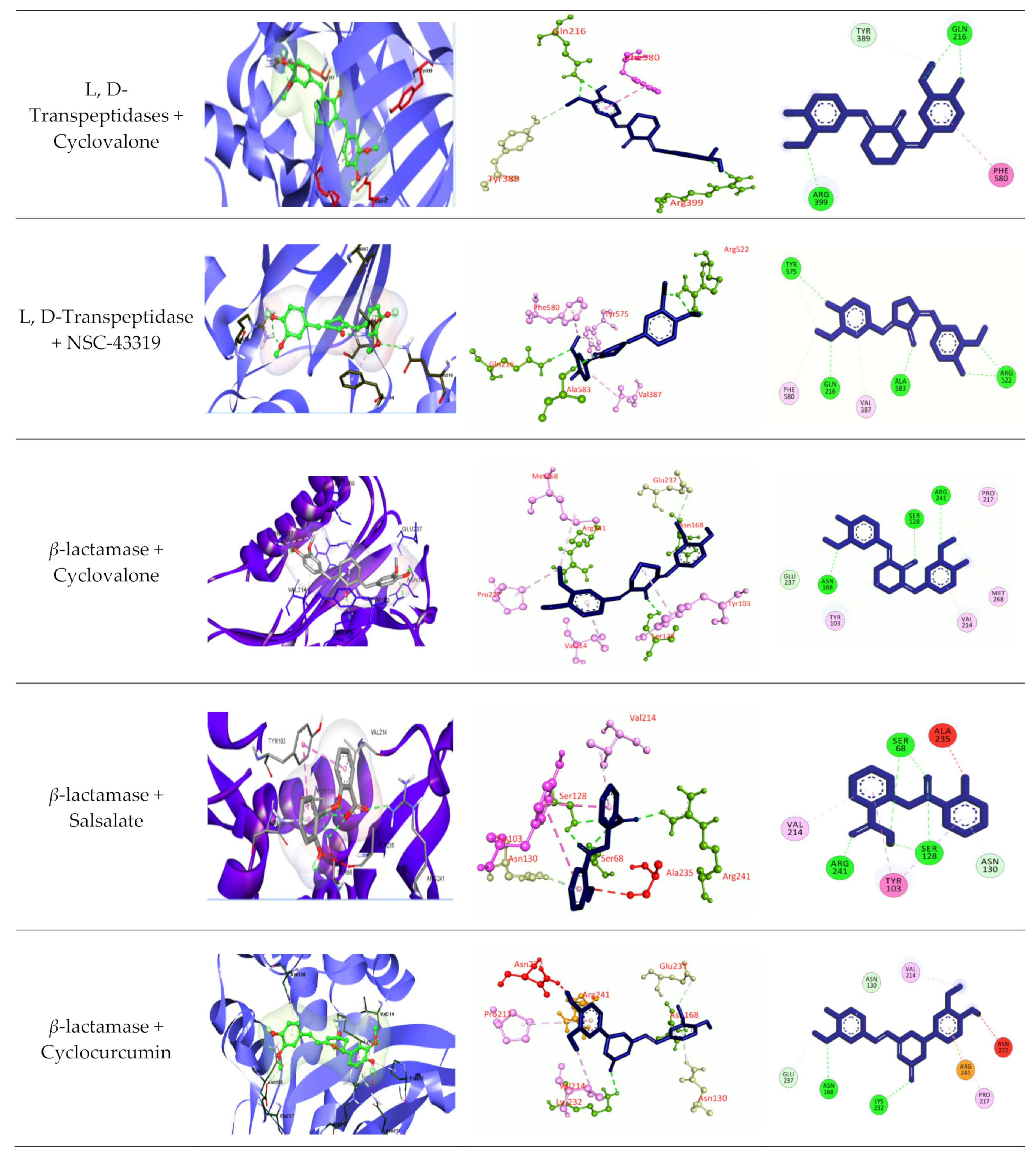
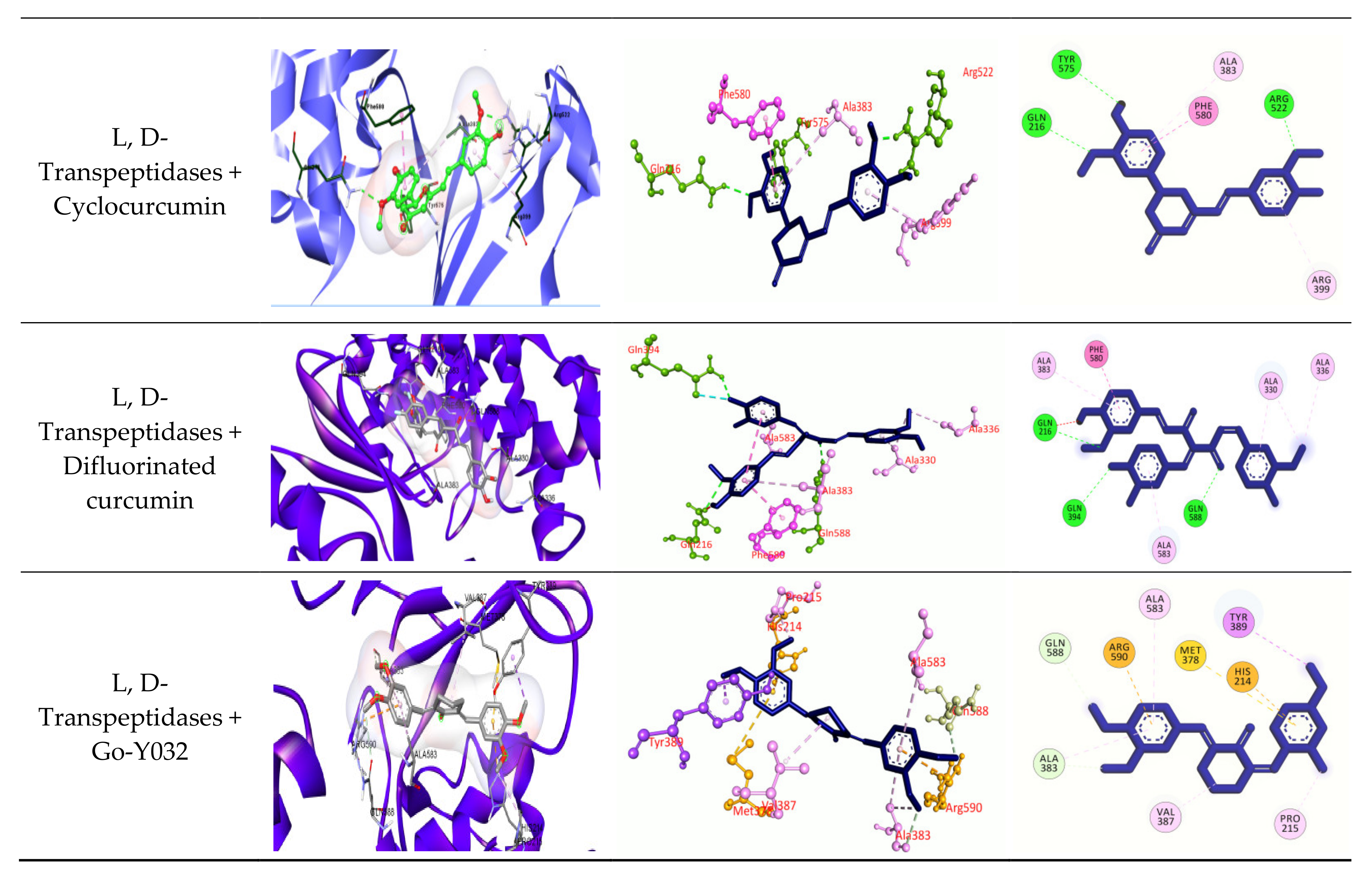
3.2.6. Molecular Docking
3.2.7. ADME/T Prognosis
3.3. Prediction of Pharmacological and Biological Activity
3.4. Prediction of P450-Mediated Sites of Metabolism (SOMs)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mogasale, V.; Maskery, B.; Ochiai, R.L.; Lee, J.S.; Mogasale, V.V.; Ramani, E.; Kim, Y.E.; Park, J.K.; Wierzba, T.F. Burden of typhoid fever in low-income and middle-income countries: A systematic, literature-based update with risk-factor adjustment. Lancet Glob. Health 2014, 2, e570–e580. [Google Scholar] [CrossRef] [Green Version]
- Crump, J.A.; Mintz, E.D. Global trends in typhoid and paratyphoid fever. Clin. Infect. Dis. 2010, 50, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Naheed, A.; Ram, P.K.; Brooks, W.A.; Hossain, M.A.; Parsons, M.B.; Talukder, K.A.; Mintz, E.; Luby, S.; Breiman, R.F. Burden of typhoid and paratyphoid fever in a densely populated urban community, Dhaka, Bangladesh. Int. J. Infect. Dis. 2010, 14, e93–e99. [Google Scholar] [CrossRef] [Green Version]
- Theiss-Nyland, K.; Qadri, F.; Colin-Jones, R.; Zaman, K.; Khanam, F.; Liu, X.; Voysey, M.; Khan, A.; Hasan, N.; Ashher, F. Assessing the impact of a Vi-polysaccharide conjugate vaccine in preventing typhoid infection among Bangladeshi children: A protocol for a phase IIIb trial. Clin. Infect. Dis. 2019, 68, S74–S82. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Reiner, R.C.; Blacker, B.F.; Goldberg, E.M.; Khalil, I.A.; Troeger, C.E.; Andrews, J.R.; Bhutta, Z.A.; Crump, J.A.; Im, J. The global burden of typhoid and paratyphoid fevers: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect. Dis. 2019, 19, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Kariuki, S.; Revathi, G.; Kiiru, J.; Mengo, D.M.; Mwituria, J.; Muyodi, J.; Munyalo, A.; Teo, Y.Y.; Holt, K.E.; Kingsley, R.A. Typhoid in Kenya is associated with a dominant multidrug-resistant Salmonella enterica serovar Typhi haplotype that is also widespread in Southeast Asia. J. Clin. Microbiol. 2010, 48, 2171–2176. [Google Scholar] [CrossRef] [Green Version]
- Geiger, T.; Lara-Tejero, M.; Xiong, Y.; Galán, J.E. Mechanisms of substrate recognition by a typhoid toxin secretion-associated muramidase. eLife 2020, 9, e53473. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; van der Bij, A.K.; Freeman, J.L.; Poirel, L.; Nordmann, P.; Costello, M.; Tchesnokova, V.L.; Pitout, J.D. Characteristics of Escherichia coli sequence type 131 isolates that produce extended-spectrum β-lactamases: Global distribution of the H 30-Rx sublineage. Antimicrob. Agents Chemother. 2014, 58, 3762–3767. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Palumbo, P.E.; Edelson, P.J. Salmonella strains resistant to multiple antibiotics: Therapeutic implications. Pediatric Infect. Dis. 1984, 3, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Samajpati, S.; Ray, U.; Roy, I.; Dutta, S. Antimicrobial resistance and molecular subtypes of Salmonella enterica serovar Typhi isolates from Kolkata, India over a 15 years period 1998–2012. Int. J. Med. Microbiol. 2017, 307, 28–36. [Google Scholar] [CrossRef]
- Melchiorre, M.G.; Chiatti, C.; Lamura, G.; Torres-Gonzales, F.; Stankunas, M.; Lindert, J.; Ioannidi-Kapolou, E.; Barros, H.; Macassa, G.; Soares, J.F. Social support, socio-economic status, health and abuse among older people in seven European countries. PLoS ONE 2013, 8, e54856. [Google Scholar]
- Iyer, R.N.; Jangam, R.R.; Jacinth, A.; Venkatalakshmi, A.; Nahdi, F.B. Prevalence and trends in the antimicrobial susceptibility pattern of Salmonella enterica serovars Typhi and Paratyphi A among children in a pediatric tertiary care hospital in South India over a period of ten years: A retrospective study. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2399–2404. [Google Scholar] [CrossRef] [PubMed]
- Tanmoy, A.M.; Westeel, E.; De Bruyne, K.; Goris, J.; Rajoharison, A.; Sajib, M.S.; van Belkum, A.; Saha, S.K.; Komurian-Pradel, F.; Endtz, H.P. Salmonella enterica serovar Typhi in Bangladesh: Exploration of genomic diversity and antimicrobial resistance. MBio 2018, 9, e02112-18. [Google Scholar] [CrossRef] [Green Version]
- Parvin, M.S.; Hasan, M.M.; Ali, M.Y.; Chowdhury, E.H.; Rahman, M.T.; Islam, M.T. Prevalence and Multidrug Resistance Pattern of Salmonella Carrying Extended-Spectrum β-Lactamase in Frozen Chicken Meat in Bangladesh. J. Food Prot. 2020, 83, 2107–2121. [Google Scholar] [CrossRef]
- Alam, S.B.; Mahmud, M.; Akter, R.; Hasan, M.; Sobur, A.; Nazir, K.; Noreddin, A.; Rahman, T.; El Zowalaty, M.E.; Rahman, M. Molecular detection of multidrug resistant Salmonella species isolated from broiler farm in Bangladesh. Pathogens 2020, 9, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, A.; Gopalakrishnan, R.; Senthur, N.P.; Ramasubramanian, V.; Ghafur, K.A.; Thirunarayan, M. Antimicrobial susceptibility of Salmonella enterica serovars in a tertiary care hospital in southern India. Indian J. Med. Res. 2013, 137, 800. [Google Scholar] [PubMed]
- Zhang, J.; Jin, H.; Hu, J.; Yuan, Z.; Shi, W.; Ran, L.; Zhao, S.; Yang, X.; Meng, J.; Xu, X. Serovars and antimicrobial resistance of non-typhoidal Salmonella from human patients in Shanghai, China, 2006–2010. Epidemiol. Infect. 2014, 142, 826–832. [Google Scholar] [CrossRef]
- Kuki, Á.; Nagy, L.; Zsuga, M.; Kéki, S. Fast identification of phthalic acid esters in poly (vinyl chloride) samples by direct analysis in real time (DART) tandem mass spectrometry. Int. J. Mass Spectrom. 2011, 303, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Eggleston, K.; Rotimi, V.; Zeckhauser, R. Antibiotic resistance as a global threat: Evidence from China, Kuwait and the United States. Glob. Health 2006, 2, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Lunguya, O.; Lejon, V.; Phoba, M.-F.; Bertrand, S.; Vanhoof, R.; Glupczynski, Y.; Verhaegen, J.; Muyembe-Tamfum, J.-J.; Jacobs, J. Antimicrobial resistance in invasive non-typhoid Salmonella from the Democratic Republic of the Congo: Emergence of decreased fluoroquinolone susceptibility and extended-spectrum beta lactamases. PLoS Negl. Trop. Dis. 2013, 7, e2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvey, M.R.; Boyd, D.A.; Finley, R.; Fakharuddin, K.; Langner, S.; Allen, V.; Ang, L.; Bekal, S.; El Bailey, S.; Haldane, D. Ciprofloxacin-resistant Salmonella enterica serovar Kentucky in Canada. Emerg. Infect. Dis. 2013, 19, 999. [Google Scholar] [CrossRef] [PubMed]
- Threlfall, E.; Ward, L. Ciprofloxacin-resistant Salmonella typhi and treatment failure. Lancet 1999, 353, 1590–1591. [Google Scholar] [CrossRef]
- Rushdy, A.A.; Mabrouk, M.I.; Abu-Sef, F.A.-H.; Kheiralla, Z.H.; Abdel-All, S.M.; Saleh, N.M. Contribution of different mechanisms to the resistance to fluoroquinolones in clinical isolates of Salmonella enterica. Braz. J. Infect. Dis. 2013, 17, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Chandrasiri, P.; Elwitigala, J.; Nanayakkara, G. A multi centre laboratory study of Gram negative bacterial blood stream infections in Sri Lanka. Ceylon Med. J. 2013, 58, 56–61. [Google Scholar] [CrossRef]
- Chen, S.; Cui, S.; McDermott, P.F.; Zhao, S.; White, D.G.; Paulsen, I.; Meng, J. Contribution of target gene mutations and efflux to decreased susceptibility of Salmonella enterica serovar Typhimurium to fluoroquinolones and other antimicrobials. Antimicrob. Agents Chemother. 2007, 51, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Gaind, R.; Paglietti, B.; Murgia, M.; Dawar, R.; Uzzau, S.; Cappuccinelli, P.; Deb, M.; Aggarwal, P.; Rubino, S. Molecular characterization of ciprofloxacin-resistant Salmonella enterica serovar Typhi and Paratyphi A causing enteric fever in India. J. Antimicrob. Chemother. 2006, 58, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Hashimoto, A.; Tamura, K.; Kawamura, Y.; Ezaki, T.; Sagara, H.; Watanabe, H. DNA sequence analysis of DNA gyrase and DNA topoisomerase IV quinolone resistance-determining regions of Salmonella enterica serovar Typhi and serovar Paratyphi A. Antimicrob. Agents Chemother. 2002, 46, 3249–3252. [Google Scholar] [CrossRef] [Green Version]
- Menezes, G.A.; Harish, B.N.; Khan, M.A.; Goessens, W.; Hays, J. Antimicrobial resistance trends in blood culture positive Salmonella Paratyphi A isolates from Pondicherry, India. Indian J. Med. Microbiol. 2016, 34, 222–227. [Google Scholar] [CrossRef]
- Wilke, M.S.; Lovering, A.L.; Strynadka, N.C. β-Lactam antibiotic resistance: A current structural perspective. Curr. Opin. Microbiol. 2005, 8, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.M.; Campbell, A.M.; Park, C.R.; Halonen, J.; Zoladz, P.R. The temporal dynamics model of emotional memory processing: A synthesis on the neurobiological basis of stress-induced amnesia, flashbulb and traumatic memories, and the Yerkes-Dodson law. Neural Plast. 2007, 2007, 060803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, A.; Lee, S.; Yang, Y.-A.; Song, J. Focus: Infectious diseases: The role of typhoid toxin in salmonella typhi virulence. Yale J. Biol. Med. 2017, 90, 283. [Google Scholar]
- Geiger, T.; Pazos, M.; Lara-Tejero, M.; Vollmer, W.; Galán, J.E. Peptidoglycan editing by a specific LD-transpeptidase controls the muramidase-dependent secretion of typhoid toxin. Nat. Microbiol. 2018, 3, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.D.; Vollmer, W.; Foster, S.J. Different walls for rods and balls: The diversity of peptidoglycan. Mol. Microbiol. 2014, 91, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, A.J.; Biboy, J.; van’t Veer, I.; Breukink, E.; Vollmer, W. Activities and regulation of peptidoglycan synthases. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20150031. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [Green Version]
- Knapp, K.M.; English, B.K. Carbapenems. In Seminars in Pediatric Infectious Diseases; WB Saunders: Philadelphia, PA, USA, 2001; pp. 175–185. [Google Scholar]
- Khanna, N.R.; Gerriets, V. Beta Lactamase Inhibitors. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sienkiewicz, N.; Członka, S.; Kairyte, A.; Vaitkus, S. Curcumin as a natural compound in the synthesis of rigid polyurethane foams with enhanced mechanical, antibacterial and anti-ageing properties. Polym. Test. 2019, 79, 106046. [Google Scholar] [CrossRef]
- Alizadeh, N.; Malakzadeh, S. Antioxidant, antibacterial and anti-cancer activities of β-and γ-CDs/curcumin loaded in chitosan nanoparticles. Int. J. Biol. Macromol. 2020, 147, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, A.; Damani, A. A Comparative Study of the Performance of Selected Mutual Fund Growth Schemes from the Private Sector and Public Sector Schemes in India. Anvesha 2014, 7, 1–9. [Google Scholar]
- Rahayu, S.I.; Nurdiana, N.; Santoso, S. The effect of curcumin and cotrimoxazole in Salmonella typhimurium infection in vivo. Int. Sch. Res. Not. 2013, 2013, 601076. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, N.A.; Habil, N.Y. Evaluation of antimicrobial activity of curcumin against two oral bacteria. Autom. Control Intell. Syst. 2015, 3, 18. [Google Scholar] [CrossRef]
- Sarkar, A.; De, R.; Mukhopadhyay, A.K. Curcumin as a potential therapeutic candidate for Helicobacter pylori associated diseases. World J. Gastroenterol. 2016, 22, 2736. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, A.; Ożarowski, M.; Karpiński, T.M. Curcumin, a natural antimicrobial agent with strain-specific activity. Pharmaceuticals 2020, 13, 153. [Google Scholar] [CrossRef]
- Teow, S.-Y.; Liew, K.; Ali, S.A.; Khoo, A.S.-B.; Peh, S.-C. Antibacterial action of curcumin against Staphylococcus aureus: A brief review. J. Trop. Med. 2016, 2016, 2853045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Huang, C.; Huang, H.; Zhao, Y.; Khan, M.R.U.; Zhao, H.; Huang, L. Antibacterial mechanism of curcumin: A review. Chem. Biodivers. 2020, 17, e2000171. [Google Scholar] [CrossRef] [PubMed]
- Zorofchian Moghadamtousi, S.; Abdul Kadir, H.; Hassandarvish, P.; Tajik, H.; Abubakar, S.; Zandi, K. A review on antibacterial, antiviral, and antifungal activity of curcumin. BioMed Res. Int. 2014, 2014, 186864. [Google Scholar] [CrossRef]
- Marathe, S.A.; Balakrishnan, A.; Negi, V.D.; Sakorey, D.; Chandra, N.; Chakravortty, D. Curcumin reduces the motility of Salmonella enterica serovar Typhimurium by binding to the flagella, thereby leading to flagellar fragility and shedding. J. Bacteriol. 2016, 198, 1798–1811. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ullah, A.; Prottoy, N.I.; Araf, Y.; Hossain, S.; Sarkar, B.; Saha, A. Molecular docking and pharmacological property analysis of phytochemicals from Clitoria ternatea as potent inhibitors of cell cycle checkpoint proteins in the cyclin/CDK pathway in cancer cells. Comput. Mol. Biosci. 2019, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated platform of small molecules and biological activities. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2008; Volume 4, pp. 217–241. [Google Scholar]
- Molinspiration Cheminformatics, Nova ulica, SK-900 26, Slovensky Grob, Slovak Republic. 2018. Available online: https://www.molinspiration.com/ (accessed on 20 October 2020).
- Walker, J.M. The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvement in protein secondary structure prediction by c prediction from alignments and joint prediction. CABIOS 1995, 11, 681–684. [Google Scholar] [PubMed]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo-and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 1–15. [Google Scholar]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, M.; Kaptein, R.; Thornton, J. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar]
- Laskowski, R.; MacArthur, M.; Moss, D.; Thornton, J. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Liu, D.; Liu, F.; Wu, J.; Zou, J.; Xiao, X.; Zhao, F.; Zhu, B. HTQC: A fast quality control toolkit for Illumina sequencing data. BMC Bioinform. 2013, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2012, 41, D1096–D1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, S.; Sarkar, B.; Prottoy, M.N.I.; Araf, Y.; Taniya, M.A.; Ullah, M.A. Thrombolytic activity, drug likeness property and ADME/T analysis of isolated phytochemicals from ginger (Zingiber officinale) using in silico approaches. Mod. Res. Inflamm. 2019, 8, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Adedoyin, A. ADME–Tox in drug discovery: Integration of experimental and computational technologies. Drug Discov. Today 2003, 8, 852–861. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.; Lagunin, A.; Gloriozova, T.; Rudik, A.; Druzhilovskii, D.; Pogodin, P.; Poroikov, V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Tarcsay, Á.; Keserű, G.M. In silico site of metabolism prediction of cytochrome P450-mediated biotransformations. Expert Opin. Drug Metab. Toxicol. 2011, 7, 299–312. [Google Scholar] [CrossRef]
- Gschwend, D.A.; Good, A.C.; Kuntz, I.D. Molecular docking towards drug discovery. J. Mol. Recognit. Interdiscip. J. 1996, 9, 175–186. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef] [Green Version]
- Li, A.P. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov. Today 2001, 6, 357–366. [Google Scholar] [CrossRef]
- Paul Gleeson, M.; Hersey, A.; Hannongbua, S. In-silico ADME models: A general assessment of their utility in drug discovery applications. Curr. Top. Med. Chem. 2011, 11, 358–381. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, C.; Vermeulen, N.P.; Feenstra, K.A. Cytochrome P450 in silico: An integrative modeling approach. J. Med. Chem. 2005, 48, 2725–2755. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Waterman, M.R.; Kelly, S.L.; Guengerich, F.P. Cytochromes P450 and drug discovery. Curr. Opin. Biotechnol. 2007, 18, 504–512. [Google Scholar] [CrossRef]
- Anzenbacher, P.; Anzenbacherova, E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. CMLS 2001, 58, 737–747. [Google Scholar] [CrossRef]
- Xu, C.; Cheng, F.; Chen, L.; Du, Z.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico prediction of chemical Ames mutagenicity. J. Chem. Inf. Model. 2012, 52, 2840–2847. [Google Scholar] [CrossRef]
- Ames, B.N.; Gurney, E.G.; Miller, J.A.; Bartsch, H. Carcinogens as frameshift mutagens: Metabolites and derivatives of 2-acetylaminofluorene and other aromatic amine carcinogens. Proc. Natl. Acad. Sci. USA 1972, 69, 3128–3132. [Google Scholar] [CrossRef] [Green Version]
- Priest, B.; Bell, I.M.; Garcia, M. Role of hERG potassium channel assays in drug development. Channels 2008, 2, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Hacker, K.; Maas, R.; Kornhuber, J.; Fromm, M.F.; Zolk, O. Substrate-dependent inhibition of the human organic cation transporter OCT2: A comparison of metformin with experimental substrates. PLoS ONE 2015, 10, e0136451. [Google Scholar] [CrossRef] [Green Version]
- Stepanchikova, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. Prediction of biological activity spectra for substances: Evaluation on the diverse sets of drug-like structures. Curr. Med. Chem. 2003, 10, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Values | |
|---|---|---|
| β-lactamases | L, D-Transpeptidases | |
| Number of Amino Acid | 286 | 615 |
| Molecular Weight (gm) | 31,515.20 | 67,812.49 |
| Theoretical PI | 5.69 | 8.63 |
| Total number of negatively residues (Asp + Glu) | 36 | 55 |
| Total number of negatively residues (Arg + Lys) | 30 | 59 |
| Instability index | 40.74 | 43.79 |
| Aliphatic index | 93.53 | 86.98 |
| GRAVY | −0.109 | −0.274 |
| Parameters | Values | |
|---|---|---|
| β-lactamases | L, D-Transpeptidases | |
| Alpha helix | 49.30% | 39.35% |
| 310 helix | 0.00% | 0.00% |
| Pi helix | 0.00% | 0.00% |
| Beta bridge | 0.00% | 0.00% |
| Extended strand | 12.94% | 11.54% |
| Beta turn | 8.39% | 5.53% |
| Bend region | 0.00% | 0.00% |
| Random coil | 29.37% | 43.58% |
| Ambiguous status | 0.00% | 0.00% |
| Other status | 0.00% | 0.00% |
| Parameters | β-lactamase | L, D-Transpeptidases |
|---|---|---|
| Biounit Oligo State | Monomer | Monomer |
| QSQE | 0.00 | 0.00 |
| Method | X-ray, 1.55 A° | X-ray, 2.76 A° |
| Sequence Similarity | 0.61 | 0.62 |
| Coverage | 1.00 | 0.95 |
| Range | 24–286 | 37–615 |
| Residues | 263 | 505 |
| Parameters | Factors | β-Lactamase | L, D-Transpeptidases |
|---|---|---|---|
| ERRAT | Overall Quality Factor | 97.2549 | 84.1141 |
| Verified 3D | Pass | 98.48% of the residues have averaged 3D-1D score ≥ 0.2 | 92.28% of the residues have averaged 3D-1D score ≥ 0.2 |
| Ramachandran plot | Residues in most favored region | 93.4% | 91.8% |
| Number of end-residues (excl. Gly and Pro) | 2 | 8 | |
| Number of Glycine residues | 21 | 31 | |
| Number of Proline residues | 12 | 39 |
| Serial No | Ligand | PubChem CID | Affinity (kcal/mol) |
|---|---|---|---|
| 1 | (8)-Shogaol | CID_6442560 | −5.5 |
| 2 | Desmethoxycurcumin | CID_5469424 | −6.5 |
| 3 | Tetrahydro curcumin | CID_56965746 | −6.1 |
| 4 | 6-Dehydrogingerdione | CID_22321203 | −6.6 |
| 5 | Difluorinated curcumin | CID_54597187 | −7.3 |
| 6 | EAC | CID_8868 | −4.7 |
| 8 | Chalcone | CID_637760 | −6.9 |
| 9 | Cyclovalone | CID_1550234 | −7.6 |
| 10 | Curcumin PE | CID_5281767 | −7.0 |
| 11 | Salsalate | CID_5161 | −7.7 |
| 12 | Go-Y016 | CID_1550385 | −6.2 |
| 13 | Petasiphenol | CID_6438779 | −7.7 |
| 14 | Benzyl ferulate | CID_7766335 | −6.3 |
| 15 | Calebin A | CID_637429 | −6.6 |
| 16 | ACMC-1AEIO | CID_2889 | −6.9 |
| 17 | MFCD00012210 | CID_14121 | −5.7 |
| 18 | Khi-201 | CID_99844 | −6.7 |
| 19 | Go-Y032 | CID_1714173 | −7.8 |
| 20 | 3,3′-dimethoxystilbene-4,4′-diol | CID_5280698 | −6.4 |
| 21 | AO-002 | CID_5318278 | −5.6 |
| 22 | phenylethyl-trans-isoferulate | CID_5468215 | −6.6 |
| 23 | NSC-43319 | CID_5470829 | −7.8 |
| 24 | 3,4-dimethoxy-4′-hydroxychalcone | CID_5930244 | −6.1 |
| 25 | PHSK | CID_6123890 | −6.6 |
| 26 | Go-Y022 | CID_6474893 | −6.5 |
| 27 | BRD-89483 | CID_6477637 | −7.2 |
| 28 | 3,3′-dimethoxy-cis-stilbene-4,4′-diol | CID_9548762 | −7.6 |
| 29 | CHEMBL482607 | CID_10904292 | −6.4 |
| 30 | ZINC100190381 | CID_11895692 | −6.7 |
| 31 | SCHEMBL18672270 | CID_16087306 | −5.8 |
| 32 | EI-135 | CID_16760039 | −6.2 |
| 33 | 3,4′-Dimethoxystilbene-4-ol | CID_23652110 | −6.1 |
| 34 | BDBM149243 | CID_44538441 | −6.8 |
| 35 | Go-Y078 | CID_46231908 | −6.3 |
| 36 | CHEMBL3940632 | CID_68556085 | −6.6 |
| 37 | CHEMBL494826 | CID_71717791 | −6.5 |
| 38 | SCHEMBL1374497 | CID_86590085 | −6.6 |
| 39 | CHEMBL3290186 | CID_90644814 | −6.9 |
| 40 | BDBM145855 | CID_91809442 | −6.6 |
| 41 | BDBM145853 | CID_91809620 | −6.6 |
| 42 | 1,5-Bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadien-3-one | CID_131752986 | −6.0 |
| 43 | Coniferyl ferulate | CID_6441913 | −6.4 |
| 44 | Curcumin sulfate | CID_66645351 | −7.1 |
| 45 | Dihydrocurcumin | CID_10429233 | −6.6 |
| 46 | Dimethoxycurcumin | CID_9952605 | −6.3 |
| 47 | Dimethylcurcumin | CID_6477182 | −6.3 |
| 48 | Ethyl curcumin | CID_11474949 | −6.6 |
| 49 | Griffithane D | CID_56597215 | −6.4 |
| 50 | Monodemethylcurcumin | CID_5469426 | −6.7 |
| 51 | Phenylethyl 3-methylcaffeate | CID_5284444 | −6.3 |
| 52 | p-Hydroxyphenethyl trans-ferulate | CID_637308 | −6.8 |
| 53 | Piperkadsin A | CID_11717379 | −6.2 |
| 54 | Shogaol | CID_5281794 | −5.2 |
| 55 | Tetrahydrocurcumin | CID_124072 | −6.6 |
| 56 | Tetrahydrodemethoxydiferuloylmethane | CID_9906039 | −6.1 |
| 57 | Tetramethylcurcumin | CID_11487078 | −6.9 |
| 58 | Wallichinine | CID_5315280 | −6.3 |
| 59 | 6-paradol | CID_94378 | −5.7 |
| 60 | Bisdemethoxycurcumin | CID_5315472 | −6.7 |
| 61 | Curcumin | CID_969516 | −6.6 |
| 62 | Cyclocurcumin | CID_69879809 | −7.9 |
| 63 | Dehydrozingerone | CID_5354238 | −5.6 |
| 64 | Dibenzoylmethane | CID_8433 | −7.0 |
| 65 | 6-Gingerol | CID_442793 | −5.1 |
| 66 | Isoeugenol | CID_853433 | −5.6 |
| 67 | Yakuchinone A | CID_133145 | −6.3 |
| 68 | Yakuchinone B | CID_6440365 | −6.6 |
| 69 | Clavulanic Acid | CID_5280980 | −6.0 |
| 70 | Tazobactum | CID_23663400 | −7.5 |
| Serial No | Ligand | PubChem CID | Affinity (kcal/mol) |
|---|---|---|---|
| 1 | (8)-Shogaol | CID_6442560 | −5.3 |
| 2 | Desmethoxycurcumin | CID_5469424 | −6.8 |
| 3 | Tetrahydro curcumin | CID_56965746 | −5.7 |
| 4 | 6-Dehydrogingerdione | CID_22321203 | −5.7 |
| 5 | Difluorinated curcumin | CID_54597187 | −8.2 |
| 6 | EAC | CID_8868 | −4.2 |
| 8 | Chalcone | CID_637760 | −6.2 |
| 9 | Cyclovalone | CID_1550234 | −7.6 |
| 10 | Curcumin PE | CID_5281767 | −7.2 |
| 11 | Salsalate | CID_5161 | −6.7 |
| 12 | Go-Y016 | CID_1550385 | −5.8 |
| 13 | Petasiphenol | CID_6438779 | −6.5 |
| 14 | Benzyl ferulate | CID_7766335 | −6.8 |
| 15 | Calebin A | CID_637429 | −6.1 |
| 16 | ACMC-1AEIO | CID_2889 | −6.7 |
| 17 | MFCD00012210 | CID_14121 | −5.9 |
| 18 | Khi-201 | CID_99844 | -6.3 |
| 19 | Go-Y032 | CID_1714173 | −7.5 |
| 20 | 3,3′-dimethoxystilbene-4,4′-diol | CID_5280698 | −6.7 |
| 21 | AO-002 | CID_5318278 | −5.7 |
| 22 | phenylethyl-trans-isoferulate | CID_5468215 | −6.2 |
| 23 | NSC-43319 | CID_5470829 | −7.6 |
| 24 | 3,4-dimethoxy-4′-hydroxychalcone | CID_5930244 | −6.6 |
| 25 | PHSK | CID_6123890 | −6.4 |
| 26 | Go-Y022 | CID_6474893 | −6.7 |
| 27 | BRD-89483 | CID_6477637 | −6.9 |
| 28 | 3,3′-dimethoxy-cis-stilbene-4,4′-diol | CID_9548762 | −6.4 |
| 29 | CHEMBL482607 | CID_10904292 | −7.1 |
| 30 | ZINC100190381 | CID_11895692 | −6.1 |
| 32 | EI-135 | CID_16760039 | −6.6 |
| 33 | 3,4′-Dimethoxystilbene-4-ol | CID_23652110 | −6.5 |
| 34 | BDBM149243 | CID_44538441 | −7.0 |
| 35 | Go-Y078 | CID_46231908 | −6.3 |
| 36 | CHEMBL3940632 | CID_68556085 | −6.6 |
| 37 | CHEMBL494826 | CID_71717791 | −6.4 |
| 38 | SCHEMBL1374497 | CID_86590085 | −6.5 |
| 39 | CHEMBL3290186 | CID_90644814 | −6.9 |
| 40 | BDBM145855 | CID_91809442 | −6.5 |
| 41 | BDBM145853 | CID_91809620 | −6.3 |
| 42 | 1,5-Bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadien-3-one | CID_131752986 | −6.3 |
| 43 | Coniferyl ferulate | CID_6441913 | −6.3 |
| 44 | Curcumin sulfate | CID_66645351 | −7.2 |
| 45 | Dihydrocurcumin | CID_10429233 | −5.8 |
| 46 | Dimethoxycurcumin | CID_9952605 | −6.1 |
| 47 | Dimethylcurcumin | CID_6477182 | −7.0 |
| 48 | Ethyl curcumin | CID_11474949 | −6.1 |
| 49 | Griffithane D | CID_56597215 | −6.6 |
| 50 | Monodemethylcurcumin | CID_5469426 | −7.8 |
| 51 | Phenylethyl 3-methylcaffeate | CID_5284444 | −6.0 |
| 52 | p-Hydroxyphenethyl trans-ferulate | CID_637308 | −6.3 |
| 53 | Piperkadsin A | CID_11717379 | −6.6 |
| 54 | Shogaol | CID_5281794 | −5.6 |
| 55 | Tetrahydrocurcumin | CID_124072 | −5.7 |
| 56 | Tetrahydrodemethoxydiferuloylmethane | CID_9906039 | −5.5 |
| 57 | Tetramethylcurcumin | CID_11487078 | −7.2 |
| 58 | Wallichinine | CID_5315280 | −6.2 |
| 59 | 6-paradoL | CID_94378 | −6.0 |
| 60 | Bisdemethoxycurcumin | CID_5315472 | −7.7 |
| 61 | Curcumin | CID_969516 | −7.0 |
| 62 | Cyclocurcumin | CID_69879809 | −8.3 |
| 63 | Dehydrozingerone | CID_5354238 | −6.0 |
| 64 | Dibenzoylmethane | CID_8433 | −6.6 |
| 65 | 6-Gingerol | CID_442793 | −5.9 |
| 66 | Isoeugenol | CID_853433 | −5.7 |
| 67 | Yakuchinone A | CID_133145 | −5.7 |
| 68 | Yakuchinone B | CID_6440365 | −6.8 |
| 69 | Carbapenem | CID_5280980 | −4.3 |
| 70 | Cephalosporin | CID_ 25058126 | −6.8 |
| Compound Name | IUPAC Name | Chemical Formula | 2D Structure |
|---|---|---|---|
| Go-Y032 | (2E,6E)-2,6-bis[(3,4-dimethoxyphenyl)methylidene]cyclohexan-1-one | C24H26O5 |  |
| NSC-43319 | (2E,5E)-2,5-bis[(4-hydroxy-3-methoxyphenyl)methylidene]cyclopentan-1-one | C21H20O5 |  |
| Cyclovalone | (2E,6E)-2,6-bis[(4-hydroxy-3-methoxyphenyl)methylidene]cyclohexan-1-one | C22H22O5 |  |
| Difluorinated Curcumin | (1E,6E)-4-[(3,4-difluorophenyl)methylidene]-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione | C28H22F2O6 |  |
| Salsalate | 2-(2-hydroxybenzoyl)oxybenzoic acid | C14H10O5 |  |
| Cyclocurcumin | 2-(4-hydroxy-3-methoxyphenyl)-6-[(E)-2-(4-hydroxy-3-methoxyphenyl)ethenyl]-2,3-dihydropyran-4-one | C21H20O6 |  |
| Enzyme | Go-Y032 | NSC-43319 | Cyclovalone | Cyclocurcumin | Difluorinated Curcumin | Salsalate |
|---|---|---|---|---|---|---|
| CYP3A4 |  |  |  |  |  |  |
| CYP2D6 |  |  |  |  |  |  |
| CYP2C9 |  |  |  |  |  |  |
| Enzyme | Ligands | AutoDock | SwissDock |
|---|---|---|---|
| Result (kcal/mol) | Result (kcal/mol) | ||
| β-lactamase | Go-Y032 | −7.8 | −8.15 |
| NSC-43319 | −7.8 | −8.04 | |
| Cyclovalone | −7.6 | −7.51 | |
| Salsalate | −7.7 | −7.56 | |
| Cyclocurcumin | −7.9 | −7.66 | |
| L, D-Transpeptidases | Cyclovalone | −7.6 | −7.90 |
| NSC-43319 | −7.6 | −8.01 | |
| Cyclocurcumin | −8.3 | −7.55 | |
| Difluorinated curcumin | −8.2 | −7.80 | |
| Go-Y032 | −7.5 | −8.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akter, T.; Chakma, M.; Tanzina, A.Y.; Rumi, M.H.; Shimu, M.S.S.; Saleh, M.A.; Mahmud, S.; Sami, S.A.; Emran, T.B. Curcumin Analogues as a Potential Drug against Antibiotic Resistant Protein, β-Lactamases and L, D-Transpeptidases Involved in Toxin Secretion in Salmonella typhi: A Computational Approach. BioMedInformatics 2022, 2, 77-100. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedinformatics2010005
Akter T, Chakma M, Tanzina AY, Rumi MH, Shimu MSS, Saleh MA, Mahmud S, Sami SA, Emran TB. Curcumin Analogues as a Potential Drug against Antibiotic Resistant Protein, β-Lactamases and L, D-Transpeptidases Involved in Toxin Secretion in Salmonella typhi: A Computational Approach. BioMedInformatics. 2022; 2(1):77-100. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedinformatics2010005
Chicago/Turabian StyleAkter, Tanzina, Mahim Chakma, Afsana Yeasmin Tanzina, Meheadi Hasan Rumi, Mst. Sharmin Sultana Shimu, Md. Abu Saleh, Shafi Mahmud, Saad Ahmed Sami, and Talha Bin Emran. 2022. "Curcumin Analogues as a Potential Drug against Antibiotic Resistant Protein, β-Lactamases and L, D-Transpeptidases Involved in Toxin Secretion in Salmonella typhi: A Computational Approach" BioMedInformatics 2, no. 1: 77-100. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedinformatics2010005