
Living in a Puddle of Mud: Isolation and Characterization of Two Novel Caulobacteraceae Strains Brevundimonas pondensis sp. nov. and Brevundimonas goettingensis sp. nov.

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of the Bacteria and DNA Extraction
2.2. Amplicon Based 16S rRNA Gene Sequencing of Enrichment Cultures
2.3. Amplicon Sequence Analysis
2.4. Genome Sequencing, Assembly and Annotation
2.5. Preparation and Sequencing of Prophages and Visualization Using TraV
2.6. Phylogenetic Classification of Brevundimonas sp. nov. LVF1T and LVF2T
2.7. Comparative Genomics
2.8. Cell Morphology and Gram Staining Procedure
2.9. Transmission Electron Microscopy
2.10. Determination of Temperature Optimum and Salt Tolerance
2.11. Determination of Growth Kinetics
2.12. Anaerobic Growth
2.13. Metabolic Activity and Antibiotic Resistances
3. Results
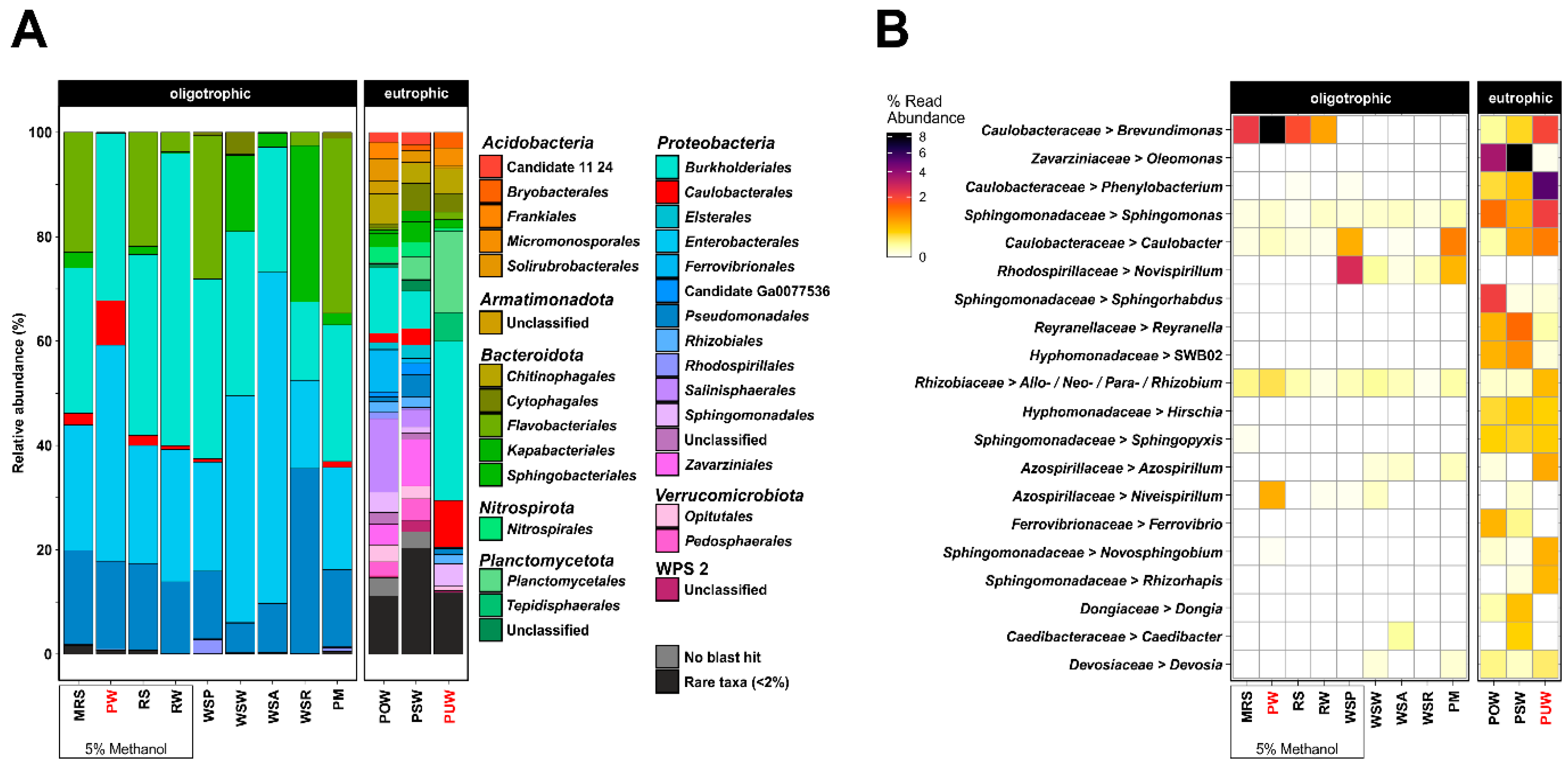
3.1. Enrichment of Caulobacteraceae from the Environment
3.2. Caulobacteraceae Isolation from Enriched Environmental Samples
3.3. Phylogeny of LVF1T and LVF2T Based on Their Full Genome Sequence
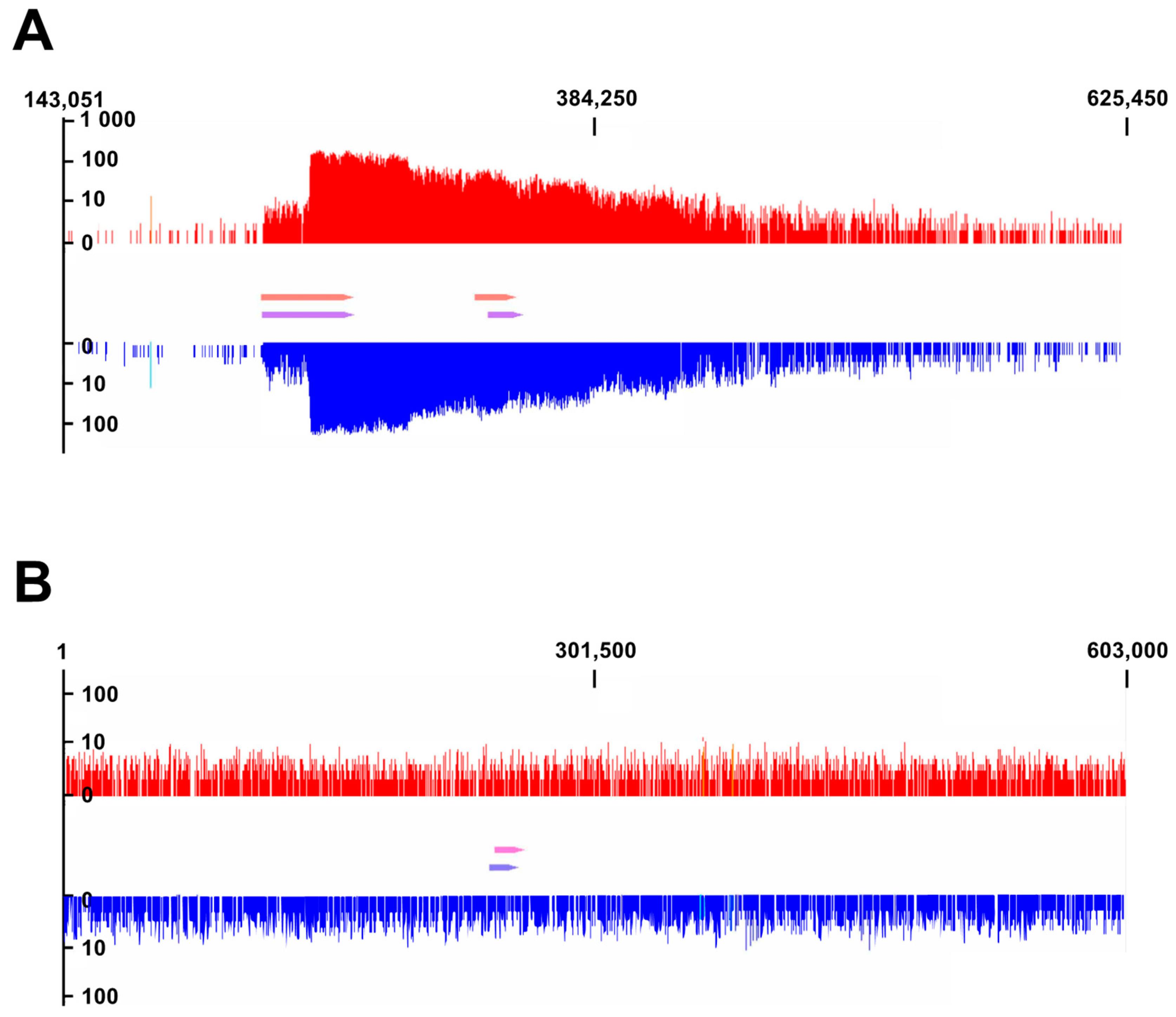
3.4. Identification of Prophage Regions
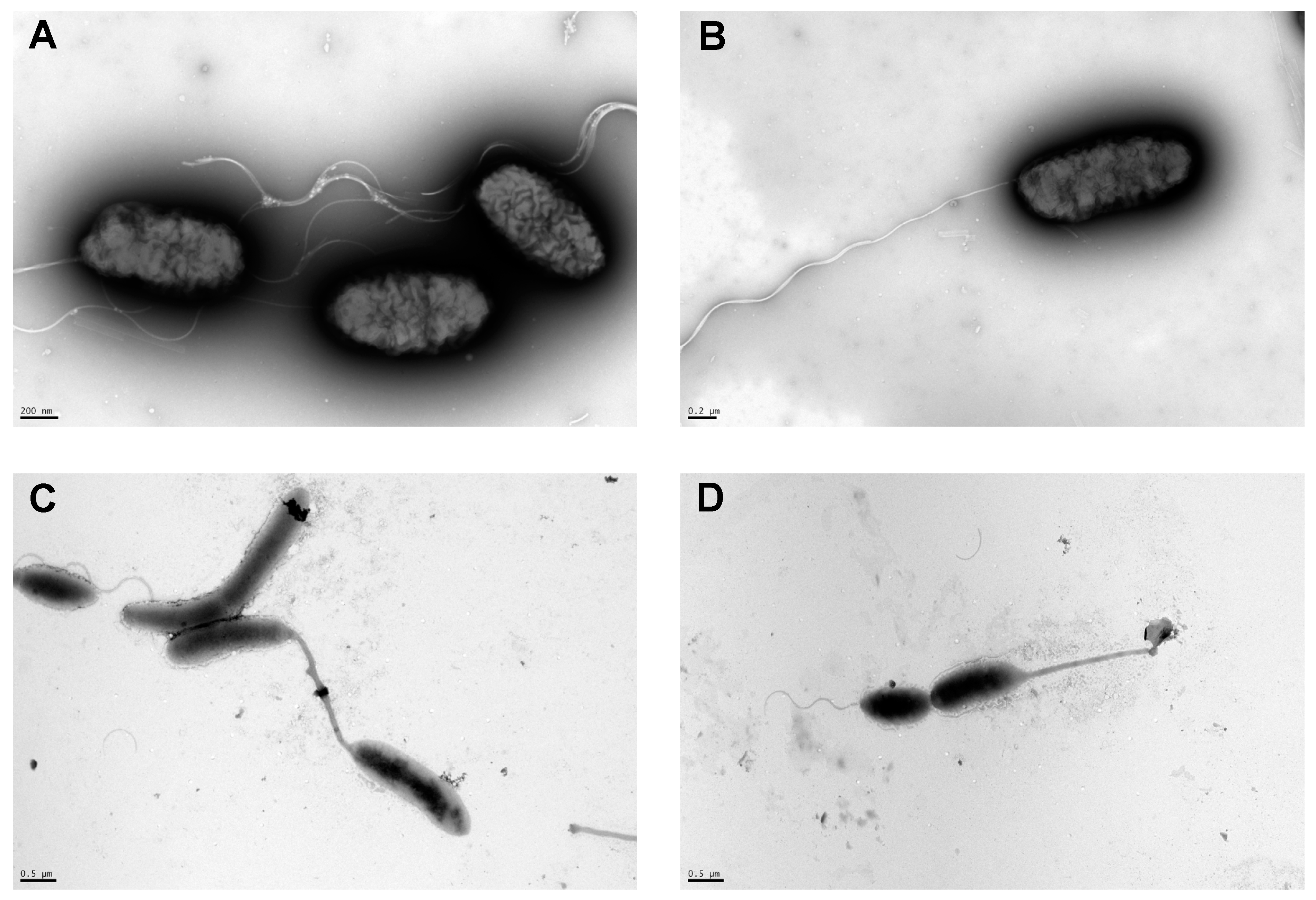
3.5. Morphological Analysis of LVF1T and LVF2T
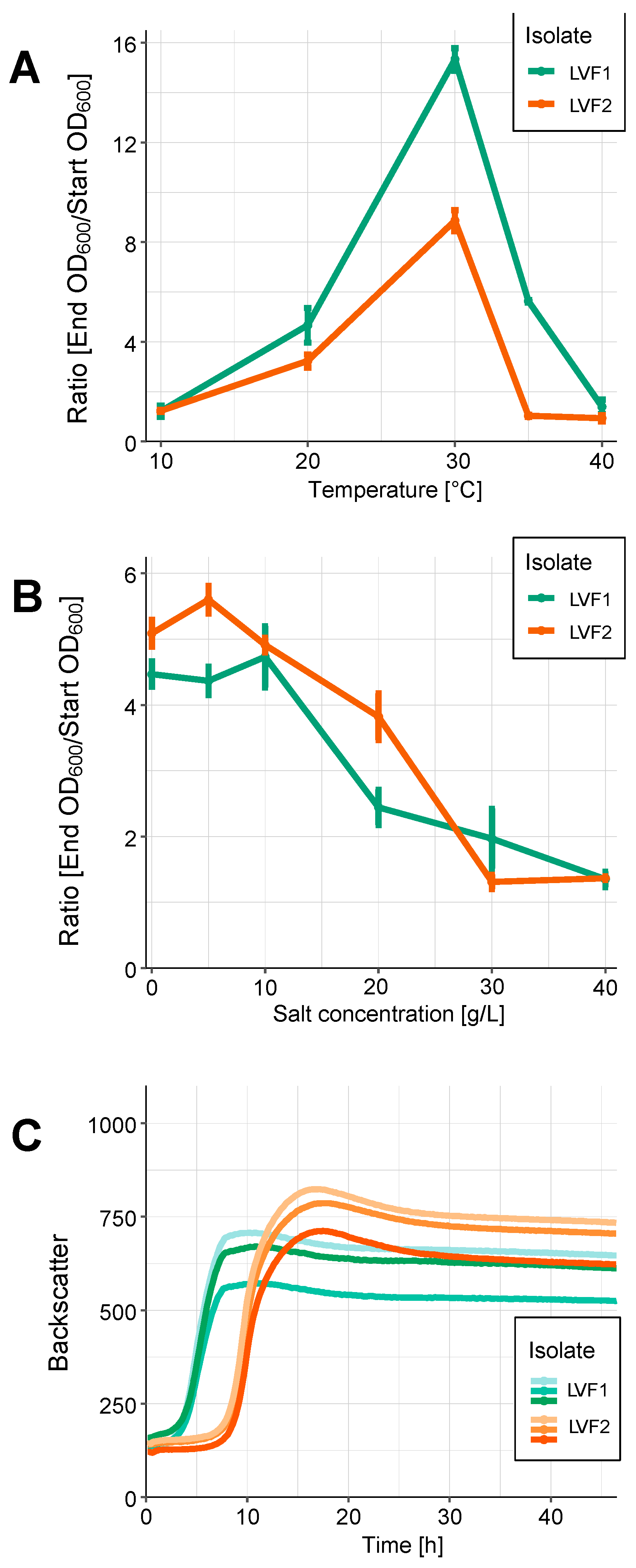
3.6. Physiological Characterization
4. Discussion
4.1. Description of Brevundimonas Pondensis sp. nov.
4.2. Description of Brevundimonas Goettingensis sp. nov.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henrici, A.T.; Johnson, D.E. Studies of Freshwater Bacteria: II. Stalked Bacteria, a New Order of Schizomycetes. J. Bacteriol. 1935, 30, 61–93. [Google Scholar] [CrossRef] [Green Version]
- Abraham, W.-R.; Strompl, C.; Meyer, H.; Lindholst, S.; Moore, E.R.B.; Christ, R.; Vancanneyt, M.; Tindall, B.J.; Bennasar, A.; Smit, J.; et al. Phylogeny and Polyphasic Taxonomy of Caulobacter Species. Proposal of Maricaulis gen. nov. with Maricaulis maris (Poindexter) comb. nov. as the Type Species, and Emended Description of the Genera Brevundimonas and Caulobacter. Int. J. Syst. Bacteriol. 1999, 49, 1053–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, W.-R.; Rohde, M.; Bennasar, A. The family Caulobacteraceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 179–205. ISBN 978-3-642-30197-1. [Google Scholar]
- Staley, J.T. Prosthecomicrobium and Ancalomicrobium: New Prosthecate Freshwater Bacteria. J. Bacteriol. 1968, 95, 1921–1942. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Lee, H.-G.; Kim, H.-S.; Ahn, C.-Y.; Oh, H.-M. Caulobacter daechungensis sp. nov., a Stalked Bacterium Isolated from a Eutrophic Reservoir. Int. J. Syst. Evol. Microbiol. 2014, 63, 2559–2564. [Google Scholar] [CrossRef] [Green Version]
- Stove, J.L.; Stanier, R.Y. Cellular Differentiation in Stalked Bacteria. Nature 1962, 196, 1189–1192. [Google Scholar] [CrossRef]
- Poindexter, J.S. Biological Properties and Classification of the Caulobacter Group. Bacteriol. Rev. 1964, 28, 231–295. [Google Scholar] [CrossRef]
- Wilhelm, R.C. Following the Terrestrial Tracks of Caulobacter—Redefining the Ecology of a Reputed Aquatic Oligotroph. ISME J. 2018, 12, 3025–3037. [Google Scholar] [CrossRef]
- Segers, P.; Vancanneyt, M.; Pot, B.; Torck, U.; Hoste, B.; Dewettinck, D.; Falsen, E.; Kersters, K.; De Vos, P. Classification of Pseudomonas diminuta Leifson and Hugh 1954 and Pseudomonas vesicularis Büsing, Döll, and Freytag 1953 in Brevundimonas gen. nov. as Brevundimonas diminuta comb. nov. and Brevundimonas vesicularis comb. nov., Respectively. Int. J. Syst. Evol. Microbiol. 1994, 44, 499–510. [Google Scholar] [CrossRef]
- Yoon, J.H.; Kang, S.J.; Oh, H.W.; Lee, J.S.; Oh, T.K. Brevundimonas kwangchunensis sp. nov., Isolated from an Alkaline Soil in Korea. Int. J. Syst. Evol. Microbiol. 2006, 56, 613–617. [Google Scholar] [CrossRef]
- Ryu, S.H.; Park, M.; Lee, J.R.; Yun, P.-Y.; Jeon, C.O. Brevundimonas aveniformis sp. nov., a Stalked Species Isolated from Activated Sludge. Int. J. Syst. Evol. Microbiol. 2007, 57, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Estrela, A.B.; Abraham, W.R. Brevundimonas vancanneytii sp. nov., Isolated from Blood of a Patient with Endocarditis. Int. J. Syst. Evol. Microbiol. 2010, 60, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kim, M.S.; Roh, S.W.; Bae, J.W. Brevundimonas basaltis sp. nov., Isolated from Black Sand. Int. J. Syst. Evol. Microbiol. 2010, 60, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.T.T.; Manangkil, O.E.; Mori, N.; Yoshida, S.; Nakamura, C. Post-Germination Seedling Vigor under Submergence and Submergence-Induced SUB1A Gene Expression in Indica and Japonica Rice (Oryza sativa L.). Aust. J. Crop Sci. 2010, 4, 264–272. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Ding, K.; Xin, Y.; Pang, H. Brevundimonas viscosa sp. nov., Isolated from Saline Soil. Int. J. Syst. Evol. Microbiol. 2012, 62, 2475–2479. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, T.; Koyama, S.; Mori, K.; Shimane, Y.; Usui, K.; Tokuda, M.; Tame, A.; Uematsu, K.; Maruyama, T.; Hatada, Y. Brevundimonas denitrificans sp. nov., a Denitrifying Bacterium Isolated from Deep Subseafloor Sediment. Int. J. Syst. Evol. Microbiol. 2014, 64, 3709–3716. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.-R.; Estrela, A.B.; Nikitin, D.I.; Smit, J.; Vancanneyt, M. Brevundimonas halotolerans sp. nov., Brevundimonas poindexterae sp. nov. and Brevundimonas staleyi sp. nov., Prosthecate Bacteria from Aquatic Habitats. Int. J. Syst. Evol. Microbiol. 2010, 60, 1837–1843. [Google Scholar] [CrossRef]
- Tóth, E.; Szuróczki, S.; Kéki, Z.; Kosztik, J.; Makk, J.; Bóka, K.; Spröer, C.; Márialigeti, K.; Schumann, P. Brevundimonas balnearis sp. nov., Isolated from the Well Water of a Thermal Bath. Int. J. Syst. Evol. Microbiol. 2017, 67, 1033–1038. [Google Scholar] [CrossRef]
- Gorbatyuk, B.; Marczynski, G.T. Regulated Degradation of Chromosome Replication Proteins DnaA and CtrA in Caulobacter crescentus. Mol. Microbiol. 2005, 55, 1233–1245. [Google Scholar] [CrossRef]
- Parte, A.C.; Carbasse, J.S.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic Names with Standing in Nomenclature (LPSN) Moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Beilstein, F.; Dreiseikelmann, B. Bacteriophages of Freshwater Brevundimonas vesicularis Isolates. Res. Microbiol. 2006, 157, 213–219. [Google Scholar] [CrossRef]
- West, D.; Lagenaur, C.; Agabian, N. Isolation and Characterization of Caulobacter crecentus Bacteriophage Phi Cd1. J. Virol. 1976, 17, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, H.-W. Bacteriophage electron microscopy. In Advances in Virus Research; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 82, pp. 1–32. ISBN 978-0-12-394621-8. [Google Scholar]
- Schmidt, J.M.; Stainer, R.Y. Isolation and Characterization of Bacteriophages Active against Stalked Bacteria. J. Gen. Microbiol. 1965, 39, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Kazaks, A.; Voronkova, T.; Rumnieks, J.; Dishlers, A.; Tars, K. Genome Structure of Caulobacter Phage PhiCb5. J. Virol. 2011, 85, 4628–4631. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, I.; Hollensteiner, J.; Schneider, D.; Poehlein, A.; Hertel, R.; Daniel, R. First Complete Genome Sequences of Janthinobacterium lividum EIF1 and EIF2 and Their Comparative Genome Analysis. Genome Biol. Evol. 2020, 12, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, I.; Hollensteiner, J.; Scherf, J.; Weyergraf, J.; Klassen, A.; Poehlein, A.; Hertel, R.; Daniel, R. Complete Genome Sequence of Stenotrophomonas indicatrix DAIF1. Microbiol. Resour. Announc. 2021, 10, 15–17. [Google Scholar] [CrossRef]
- Hollensteiner, J.; Friedrich, I.; Hollstein, L.; Lamping, J.; Wolf, K.; Liesegang, H.; Poehlein, A.; Hertel, R.; Daniel, R. Complete Genome Sequence of Kinneretia sp. Strain DAIF2, Isolated from a Freshwater Pond. Microbiol. Resour. Announc. 2021, 10, 5–7. [Google Scholar] [CrossRef]
- Poindexter, J.S. Dimorphic prosthecate bacteria: The genera Caulobacter, Asticcacaulis, Hyphomicrobium, Pedomicrobium, Hyphomonas and Thiodendron. In The Prokaryotes; Springer: New York, NY, USA, 2006; Volume 5, pp. 72–90. ISBN 978-0-387-30745-9. [Google Scholar]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Fredriksson, N.J.; Hermansson, M.; Wilén, B.-M. The Choice of PCR Primers Has Great Impact on Assessments of Bacterial Community Diversity and Dynamics in a Wastewater Treatment Plant. PLoS ONE 2013, 8, e76431. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A Fast and Accurate Illumina Paired-End ReAd MergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G.I. Denoising the Denoisers: An Independent Evaluation of Microbiome Sequence Error-Correction Approaches. PeerJ 2018, 2018, e5364. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Wickham, H. Ggplot2—Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; Volume 77, p. 3. ISBN 978-0-387-98140-6. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and Accurate de novo Genome Assembly from Long Uncorrected Reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- BLAST Command Line Applications User Manual [Internet]. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK279690/ (accessed on 20 November 2020).
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, K.; Gosling, J.; Holmes, D. The Java Programming Language, 4th ed.; Addison-Wesley Professional: Lebanon, IN, USA, 2005; ISBN 978-0-321-34980-4. [Google Scholar]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A Web Tool to Identify Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, N.; Sekiguchi, M.; Iijima, T.; Takagi, Y. Induction of Phage Formation in the Lysogenic Escherichia coli K-12 by Mitomycin C. Nature 1959, 184, 1079–1080. [Google Scholar] [CrossRef]
- Dietrich, S.; Wiegand, S.; Liesegang, H. TraV: A Genome Context Sensitive Transcriptome Browser. PLoS ONE 2014, 9, e93677. [Google Scholar] [CrossRef]
- Hertel, R.; Volland, S.; Liesegang, H. Conjugative Reporter System for the Use in Bacillus licheniformis and Closely Related Bacilli. Lett. Appl. Microbiol. 2015, 60, 162–167. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. Selection of Representative Genomes for 24,706 Bacterial and Archaeal Species Clusters Provide a Complete Genome-Based Taxonomy. BioRxiv 2019, 771964. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. AntiSMASH: Rapid Identification, Annotation and Analysis of Secondary Metabolite Biosynthesis Gene Clusters in Bacterial and Fungal Genome Sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the Secondary Metabolite Genome Mining Pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, M.K.; Forsberg, K.J.; Dantas, G. Improved Annotation of Antibiotic Resistance Determinants Reveals Microbial Resistomes Cluster by Ecology. ISME J. 2015, 9, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Claus, D. A Standardized Gram Staining Procedure. World J. Microbiol. Biotechnol. 1992, 8, 451–452. [Google Scholar] [CrossRef] [Green Version]
- Bruder, S.; Reifenrath, M.; Thomik, T.; Boles, E.; Herzog, K. Parallelied Online Biomass Monitoring in Shake Flasks Enables Efficient Strain and Carbon Source Dependent Growth Characterisation of Saccharomyces cerevisiae. Microb. Cell Fact. 2016, 15, 127. [Google Scholar] [CrossRef] [Green Version]
- Macy, J.M.; Snellen, J.E.; Hungate, R.E. Use of Syringe Methods for Anaerobiosis. Am. J. Clin. Nutr. 1972, 25, 1318–1323. [Google Scholar] [CrossRef]
- Clarke, P.H.; Cowan, S.T. Biochemical Methods for Bacteriology. J. Gen. Microbiol. 1952, 6, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the Genomic Gold Standard for the Prokaryotic Species Definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Reimer, L.C.; Vetcininova, A.; Carbasse, J.S.; Söhngen, C.; Gleim, D.; Ebeling, C.; Overmann, J. BacDive in 2019: Bacterial Phenotypic Data for High-Throughput Biodiversity Analysis. Nucleic Acids Res. 2019, 47, D631–D636. [Google Scholar] [CrossRef] [Green Version]
- Milke, J.; Gałczyńska, M.; Wróbel, J. The Importance of Biological and Ecological Properties of Phragmites australis (Cav.) Trin. Ex Steud., in Phytoremendiation of Aquatic Ecosystems—The Review. Water 2020, 12, 1770. [Google Scholar] [CrossRef]
- Fazi, S.; Amalfitano, S.; Piccini, C.; Zoppini, A.; Puddu, A.; Pernthaler, J. Colonization of Overlaying Water by Bacteria from Dry River Sediments. Environ. Microbiol. 2008, 10, 2760–2772. [Google Scholar] [CrossRef] [Green Version]
- Eberspächer, J.; Lingens, F. The genus Phenylobacterium. In The Prokaryotes; Springer: New York, NY, USA, 2006; pp. 250–256. ISBN 978-0-387-30745-9. [Google Scholar]
- Vancanneyt, M.; Segers, P.; Abraham, W.; Vos, P.D. Brevundimonas . In Bergey’s Manual of Systematics of Archaea and Bacteria; Trujillo, M.E., Dedysh, S., DeVos, P., Hedlund, B., Kämpfer, P., Rainey, F.A., Whitman, W.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1–14. ISBN 978-1-118-96060-8. [Google Scholar]
- Willms, I.M.; Yuan, J.; Penone, C.; Goldmann, K.; Vogt, J.; Wubet, T.; Schöning, I.; Schrumpf, M.; Buscot, F.; Nacke, H. Distribution of Medically Relevant Antibiotic Resistance Genes and Mobile Genetic Elements in Soils of Temperate Forests and Grasslands Varying in Land Use. Genes 2020, 11, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Du, C.; de Barsy, F.; Liem, M.; Liakopoulos, A.; van Wezel, G.P.; Choi, Y.H.; Claessen, D.; Rozen, D.E. Antibiotic Production in Streptomyces Is Organized by a Division of Labor through Terminal Genomic Differentiation. Sci. Adv. 2020, 6, eaay5781. [Google Scholar] [CrossRef] [Green Version]
- Tomasch, J.; Wang, H.; Hall, A.T.K.; Patzelt, D.; Preusse, M.; Petersen, J.; Brinkmann, H.; Bunk, B.; Bhuju, S.; Jarek, M.; et al. Packaging of Dinoroseobacter shibae DNA into Gene Transfer Agent Particles Is Not Random. Genome Biol. Evol. 2018, 10, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, J.R.; Wong, H.C.; Ting, Y.E.; Van Arsdell, J.N.; Chang, S. Control of Lysogeny and Immunity of Bacillus subtilis Temperate Bacteriophage SP Beta by Its d Gene. J. Bacteriol. 1986, 167, 952–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rettenmier, C.W.; Gingell, B.; Hemphill, H.E. The Role of Temperate Bacteriophage SP Beta in Prophage-Mediated Interference in Bacillus subtilis. Can. J. Microbiol. 1979, 25, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Obana, N.; Yee, L.M.; Asai, K.; Nomura, N.; Nakamura, K. SP10 Infectivity Is Aborted after Bacteriophage SP10 Infection Induces NonA Transcription on the Prophage SPβ Region of the Bacillus subtilis Genome. J. Bacteriol. 2014, 196, 693–706. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Coordinates | Date |
|---|---|---|
| MRS | 51°33′58″ N 9°56′18″ E 230 m | 6 September 2018 |
| PW | 51°33′57″ N 9°57′20″ E 230 m | 6 September 2018 |
| RS | 51°33′58″ N 9°56′18″ E 230 m | 6 September 2018 |
| RW | 51°33′58″ N 9°56′18″ E 230 m | 6 September 2018 |
| WSP | 51°33′59″ N 9°56′22″ E 230 m | 11 September 2018 |
| WSW | 51°33′59″ N 9°56′23″ E 230 m | 11 September 2018 |
| WSA | 51°33′58″ N 9°56′22″ E 230 m | 11 September 2018 |
| WSR | 51°33′58″ N 9°56′22″ E 230 m | 11 September 2018 |
| PM | 51°33′58″ N 9°56′22″ E 230 m | 11 September 2018 |
| POW | 51°33′29″ N 9°56′41″ E 173 m | 24 September 2018 |
| PSW | 51°33′29″ N 9°56′41″ E 173 m | 24 September 2018 |
| PUW | 51°33′27″ N 9°56′40″ E 173 m | 24 September 2018 |
| Features | Brevundimonas pondensis sp. nov. LVF1T | Brevundimonas goettingensis sp. nov. LVF2T |
|---|---|---|
| Genome size (bp) | 3,550,773 | 3,984,955 |
| GC content (%) | 67.04 | 67.79 |
| Coverage | 252.9-fold | 245.4-fold |
| CDS | 3445 | 3857 |
| rRNA genes | 58 | 57 |
| tRNA genes | 48 | 48 |
| ncRNA | 4 | 3 |
| CRISPR | 0 | 0 |
| Prophage(s) | 2 | 1 |
| Characteristics | B. pondensis LVF1T | B. goettingensis LVF2T | B. diminuta NCTC 9239T | B. lenta DSM 23960T | B. naejangsensis DSM 23858T | B. subvibrioides ATCC 15264T |
|---|---|---|---|---|---|---|
| Source of isolation | Oligotrophic pond water | Puddle water | Water | Soil | Soil | Pond water |
| Colony pigmentation | Gray-white (PYE/R2A) | Yellow (PYE/R2A) | None (NA) | Grayish-yellow (NA) | Grayish-yellow (TSA) | Dark orange (PYE) |
| Stalk formation | − | + | n/a | n/a | − | + |
| Anaerobic growth | + | − | − | − | + | − |
| Temperature (°C) | ||||||
| Range | 10–40 | 10–40 | n/a | 4–34 | 4–50 | n/a |
| Optimum | 30 | 30 | 28 | 25 | 30 | 30 |
| NaCl (g/L) | ||||||
| Range | 0–40 | 0–40 | n/a | 0–10 | 0–40 | 0–20 |
| Optimum | 0–10 | 0–5 | n/a | 0 | 5 | 20 |
| Enzymatic activity | ||||||
| Alkaline phosphatase | + | + | + | + | + | n/a |
| Esterase | + | + | + | + | + | n/a |
| Esterase lipase | + | + | + | + | + | n/a |
| Lipase | − | − | − | − | − | n/a |
| Leucine arylamidase | + | + | + | + | + | v |
| Valine arylamidase | + | − | − | − | − | − |
| Cysteine arylamidase | − | − | − | − | − | n/a |
| Trypsin | + | + | + | + | + | n/a |
| α-Chymotrypsin | + | − | + | − | + | n/a |
| Acid phosphatase | + | + | + | + | + | n/a |
| Naphthol-AS-BI-phosphohydrolase | + | + | + | + | + | n/a |
| α-Galactosidase | − | − | − | − | − | n/a |
| β-Galactosidase | − | − | − | − | − | n/a |
| β-Glucuronidase | − | − | − | − | − | n/a |
| α-Glucosidase | − | − | − | n/a | − | n/a |
| β-Glucosidase | − | + | − | n/a | − | n/a |
| N-Acetyl-β-glucosaminidase | − | − | − | − | − | n/a |
| α-Mannosidase | − | − | − | − | − | n/a |
| α-Fucosidase | − | − | − | − | − | n/a |
| Utilization of | ||||||
| Potassium nitrate | − | − | − | n/a | − | − |
| L-Tryptophane | − | − | − | n/a | − | n/a |
| D-Glucose (fermentation) | − | − | − | n/a | − | n/a |
| L-Arginine | − | − | − | n/a | − | − |
| Urea | − | − | − | n/a | − | n/a |
| Esculin/ferric citrate | + | + | − | n/a | − | n/a |
| Gelatin | − | − | − | n/a | − | n/a |
| 4-Nitrophenyl-β-D-galactopyranoside | − | − | − | n/a | − | n/a |
| D-Glucose (assimilation) | − | − | − | − | − | + |
| L-Arabinose | − | − | − | − | − | v |
| D-Mannose | − | − | − | − | − | − |
| D-Mannitol | − | − | − | − | − | n/a |
| N-Acetyl-D-glucosamine | − | − | − | − | − | n/a |
| D-Maltose | + | + | − | − | − | + |
| Potassium gluconate | − | − | − | − | − | n/a |
| Capric acid | + | + | − | n/a | − | n/a |
| Adipic acid | − | − | − | n/a | − | n/a |
| Malic acid | − | − | − | n/a | + | n/a |
| Trisodium citrate | − | − | − | n/a | − | n/a |
| Phenylacetic acid | − | − | − | n/a | − | n/a |
| Oxidase | + | + | + | n/a | + | + |
| Catalase | + | + | + | n/a | + | + |
| Resistance to | ||||||
| Ampicillin | − | − | + | + | + | n/a |
| Chloramphenicol | − | − | − | − | − | n/a |
| Doxycycline | − | − | − | n/a | n/a | n/a |
| Erythromycin | + | + | − | n/a | n/a | n/a |
| Kanamycin | − | − | − | − | − | n/a |
| Meropenem | + | + | n/a | n/a | n/a | n/a |
| Oxytetracycline | − | − | n/a | n/a | n/a | n/a |
| Rifampicin | − | − | n/a | n/a | n/a | n/a |
| Streptomycin | + | + | n/a | n/a | − | − |
| Tetracycline | + | + | − | − | − | n/a |
| Vancomycin | + | + | − | n/a | n/a | n/a |
| G + C % | 67.04 | 67.79 | 67 | 68.7 | 67 | 67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrich, I.; Klassen, A.; Neubauer, H.; Schneider, D.; Hertel, R.; Daniel, R. Living in a Puddle of Mud: Isolation and Characterization of Two Novel Caulobacteraceae Strains Brevundimonas pondensis sp. nov. and Brevundimonas goettingensis sp. nov. Appl. Microbiol. 2021, 1, 38-59. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1010005
Friedrich I, Klassen A, Neubauer H, Schneider D, Hertel R, Daniel R. Living in a Puddle of Mud: Isolation and Characterization of Two Novel Caulobacteraceae Strains Brevundimonas pondensis sp. nov. and Brevundimonas goettingensis sp. nov. Applied Microbiology. 2021; 1(1):38-59. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1010005
Chicago/Turabian StyleFriedrich, Ines, Anna Klassen, Hannes Neubauer, Dominik Schneider, Robert Hertel, and Rolf Daniel. 2021. "Living in a Puddle of Mud: Isolation and Characterization of Two Novel Caulobacteraceae Strains Brevundimonas pondensis sp. nov. and Brevundimonas goettingensis sp. nov." Applied Microbiology 1, no. 1: 38-59. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1010005