
Nasopharyngeal Microbiome Community Composition and Structure Is Associated with Severity of COVID-19 Disease and Breathing Treatment

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Clinical Attributes
2.3. Metagenomic Sequencing and Classification
2.4. Alpha Diversity
2.5. Beta Diversity
2.6. Community Composition
2.7. Differential Abundance
3. Results
3.1. Classification
3.2. Alpha Diversity
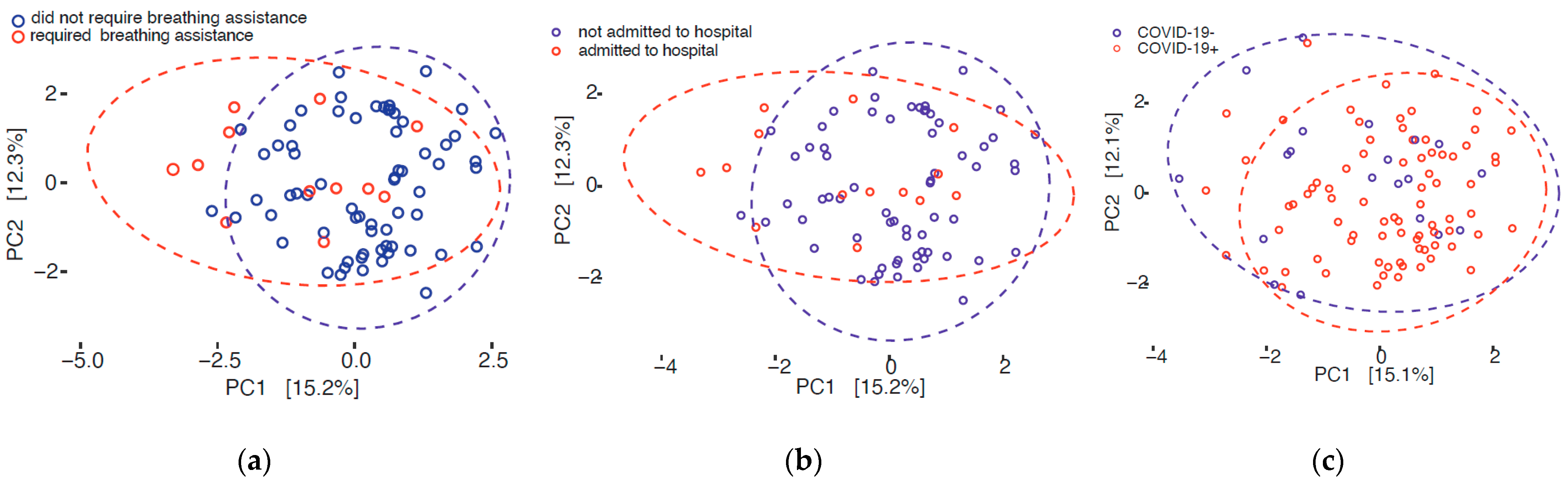
3.3. Beta Diversity
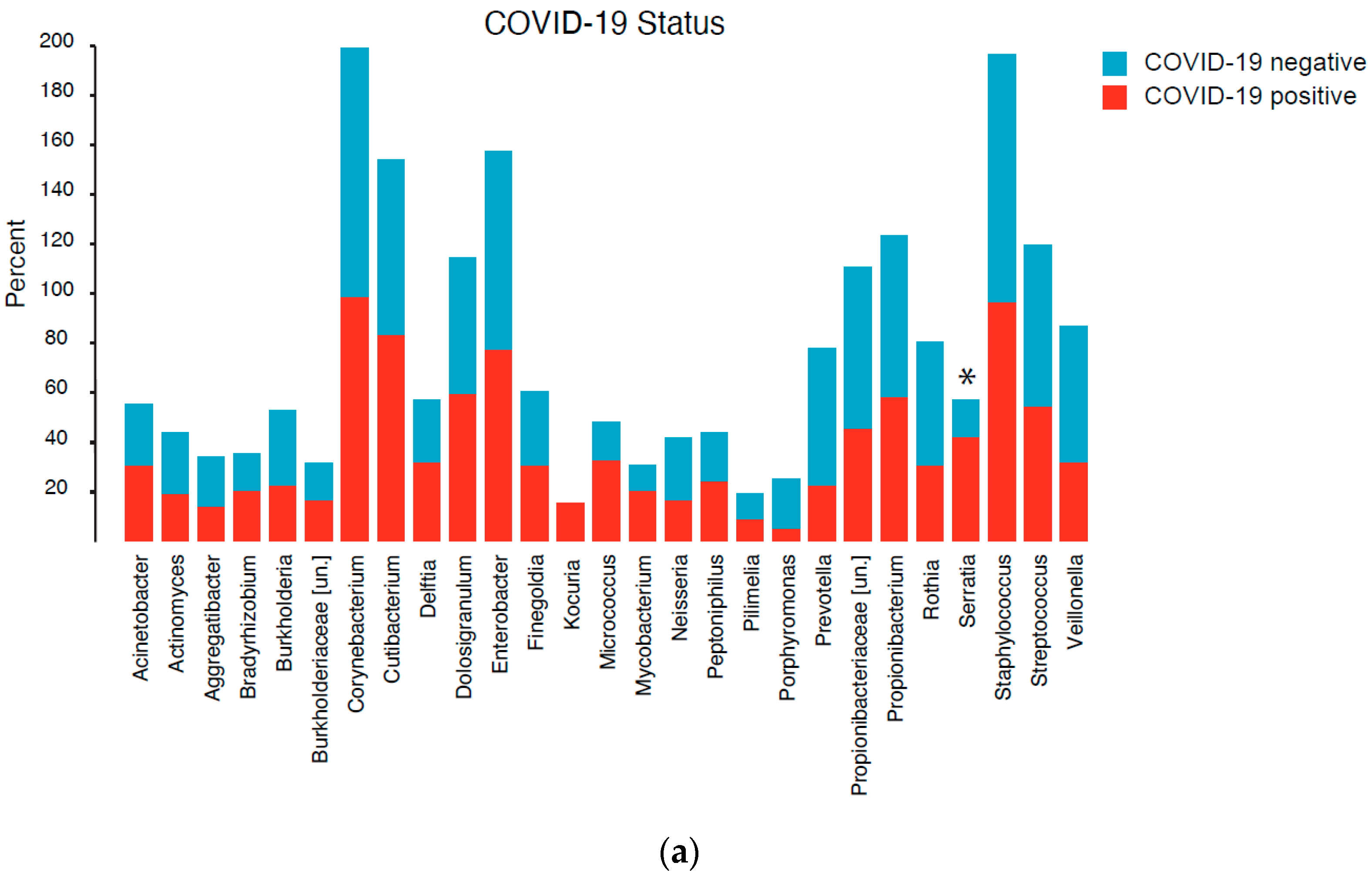
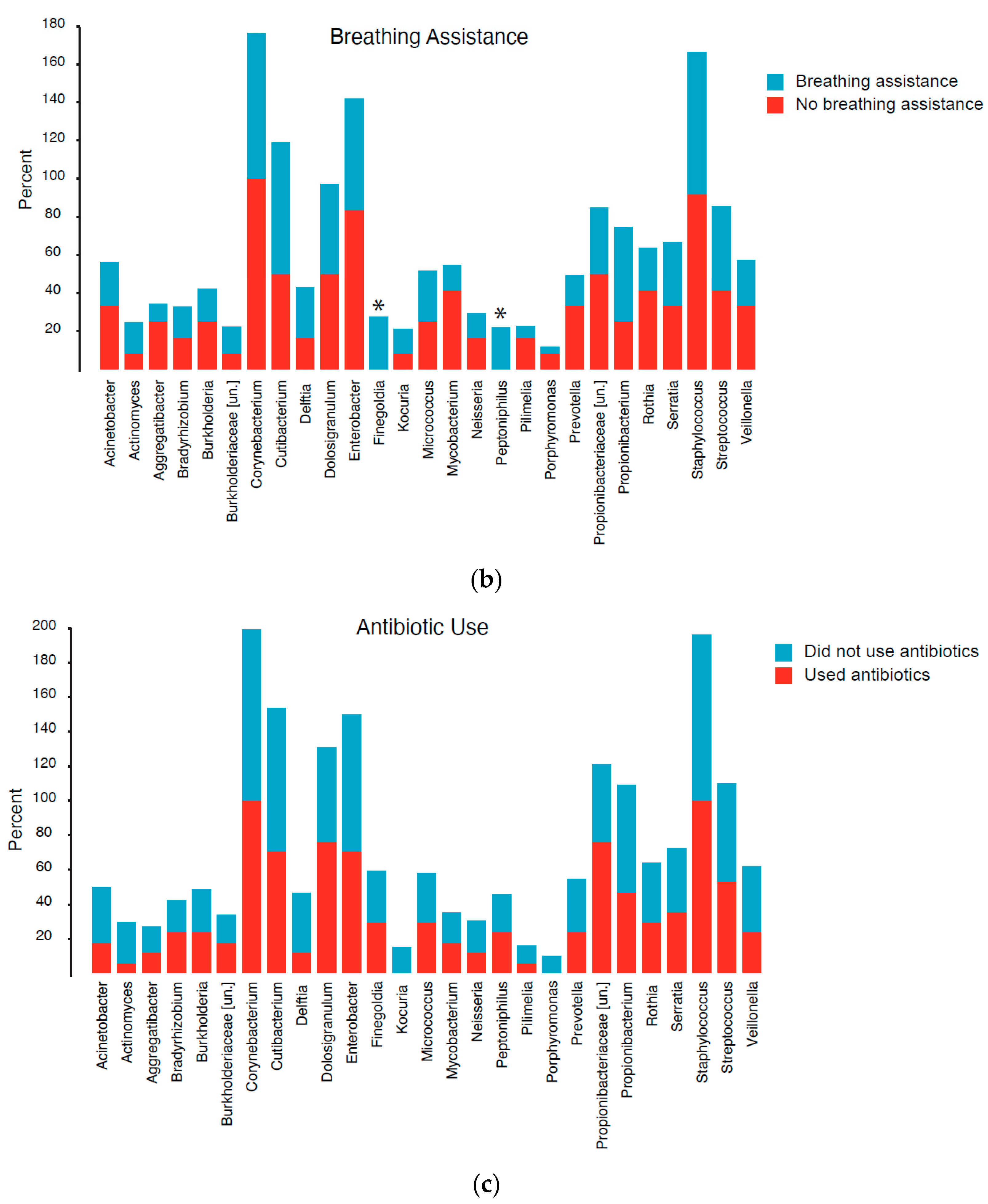
3.4. Community Composition
3.5. Differential Abundance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Maio, F.; Posteraro, B.; Ponziani, F.R.; Cattani, P.; Gasbarrini, A.; Sanguinetti, M. Nasopharyngeal Microbiota Profiling of SARS-CoV-2 Infected Patients. Biol. Proced. Online 2020, 22, 18. [Google Scholar] [CrossRef] [PubMed]
- Engen, P.A.; Naqib, A.; Jennings, C.; Green, S.J.; Landay, A.; Keshavarzian, A.; Voigt, R.M. Nasopharyngeal Microbiota in SARS-CoV-2 Positive and Negative Patients. Biol. Proced. Online 2021, 23, 10. [Google Scholar] [CrossRef]
- Mostafa, H.H.; Fissel, J.A.; Fanelli, B.; Bergman, Y.; Gniazdowski, V.; Dadlani, M.; Carroll, K.C.; Colwell, R.R.; Simner, P.J. Metagenomic Next-Generation Sequencing of Nasopharyngeal Specimens Collected from Confirmed and Suspect COVID-19 Patients. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, C.; Gentile, I.; Setaro, M.; Di Domenico, C.; Pinchera, B.; Buonomo, A.R.; Zappulo, E.; Scotto, R.; Scaglione, G.L.; Castaldo, G.; et al. Nasopharyngeal Microbiome Signature in COVID-19 Positive Patients: Can We Definitively Get a Role to Fusobacterium periodonticum? Front. Cell. Infect. Microbiol. 2021, 11, 625581. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Sahni, S.; Siddiqui, S.; Mishra, N.; Sharma, P.; Sharma, S.; Tyagi, A.; Chattopadhyay, P.; Vivekanand, A.; Devi, P.; et al. Respiratory Co-Infections: Modulators of SARS-CoV-2 Patients’ Clinical Sub-Phenotype. Front. Microbiol. 2021, 12, 653399. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea, J.A.; Bamford, C.G. SARS-CoV-2, bacterial co-infections, and AMR: The deadly trio in COVID-19? EMBO Mol. Med. 2020, 12, e12560. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.K.; Yi, H.; Kearns, D.B.; Mainou, B.A. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLoS Pathog. 2017, 13, e1006768. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 2015, 347, 266–269. [Google Scholar] [CrossRef] [Green Version]
- Mastromarino, P.; Cacciotti, F.; Masci, A.; Mosca, L. Antiviral activity of Lactobacillus brevis towards herpes simplex virus type 2: Role of cell wall associated components. Anaerobe 2011, 17, 334–336. [Google Scholar] [CrossRef]
- Bandoro, C.; Runstadler, J.A. Bacterial Lipopolysaccharide Destabilizes Influenza Viruses. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.W.; Liu, P.F.; Liu, Y.T.; Kuo, S.; Zhang, X.Q.; Schooley, R.T.; Rohde, H.; Gallo, R.L.; Huang, C.M. Nasal commensal Staphylococcus epidermidis counteracts influenza virus. Sci. Rep. 2016, 6, 27870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Ma, W.T.; Pang, M.; Fan, Q.L.; Hua, J.L. The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef]
- Rosas-Salazar, C.; Kimura, K.S.; Shilts, M.H.; Strickland, B.A.; Freeman, M.H.; Wessinger, B.C.; Gupta, V.; Brown, H.M.; Rajagopala, S.V.; Turner, J.H.; et al. SARS-CoV-2 infection and viral load are associated with the upper respiratory tract microbiome. J. Allergy Clin. Immunol. 2021, 147, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, I.; Chung, M.; Angel, L.; Koralov, S.; Wu, B.; Yeung, S.; Krolikowski, K.; Li, Y.; Duerr, R.; Schluger, R.; et al. Microbial signatures in the lower airways of mechanically ventilated Covid19 patients associated with poor clinical outcome. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Aardema, H.; Lisotto, P.; Kurilshikov, A.; Diepeveen, J.R.J.; Friedrich, A.W.; Sinha, B.; de Smet, A.; Harmsen, H.J.M. Marked Changes in Gut Microbiota in Cardio-Surgical Intensive Care Patients: A Longitudinal Cohort Study. Front. Cell. Infect. Microbiol. 2019, 9, 467. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic Nanopore Sequencing of Influenza Virus Direct from Clinical Respiratory Samples. J. Clin. Microbiol. 2019, 58. [Google Scholar] [CrossRef] [Green Version]
- Minich, J.; Ali, F.; Marotz, C.; Belda-Ferre, P.; Chiang, L.; Shaffer, J.P.; Carpenter, C.S.; McDonald, D.; Gilbert, J.; Allard, S.M.; et al. Feasibility of using alternative swabs and storage solutions for paired SARS-CoV-2 detection and microbiome analysis in the hospital environment. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, M.; Xue, J.; Huang, J.; Zhuang, R.; Zhou, X.; Zhang, H.; Fu, Q.; Hao, Y. Body Mass Index Differences in the Gut Microbiota Are Gender Specific. Front. Microbiol. 2018, 9, 1250. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zheng, W.; Cai, Q.; Shrubsole, M.J.; Pei, Z.; Brucker, R.; Steinwandel, M.; Bordenstein, S.R.; Li, Z.; Blot, W.J.; et al. Racial Differences in the Oral Microbiome: Data from Low-Income Populations of African Ancestry and European Ancestry. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, M.B.; Galenkamp, H.; Prins, M.; Derks, E.M.; Peters, R.J.G.; Zwinderman, A.H.; Stronks, K. Cohort profile: The Healthy Life in an Urban Setting (HELIUS) study in Amsterdam, The Netherlands. BMJ Open 2017, 7, e017873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarsy, R.; Pean de Ponfilly, G.R.; Benmansour, H.A.; Jacquier, H.; Cambau, E.E.; Megarbane, B. Serratia marcescens outbreak in the intensive care unit during the COVID-19 pandemic: A paradoxical risk? Méd. Mal. Infect. 2020, 50, 750–751. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Choi, S.H.; Ryu, D.W. Descending necrotizing Mediastinitis caused by Kocuria rosea: A case report. BMC Infect. Dis. 2013, 13, 475. [Google Scholar] [CrossRef] [Green Version]
- De Pascale, G.; De Maio, F.; Carelli, S.; De Angelis, G.; Cacaci, M.; Montini, L.; Bello, G.; Cutuli, S.L.; Pintaudi, G.; Tanzarella, E.S.; et al. Staphylococcus aureus ventilator-associated pneumonia in patients with COVID-19: Clinical features and potential inference with lung dysbiosis. Crit. Care 2021, 25, 197. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.; Wu, J.; Li, Y.; Zhou, X.; Li, X.; Chen, H.; Guo, M.; Chen, S.; Sun, F.; et al. Risks and features of secondary infections in severe and critical ill COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 1958–1964. [Google Scholar] [CrossRef]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Elvers, K.T.; Wilson, V.J.; Hammond, A.; Duncan, L.; Huntley, A.L.; Hay, A.D.; van der Werf, E.T. Antibiotic-induced changes in the human gut microbiota for the most commonly prescribed antibiotics in primary care in the UK: A systematic review. BMJ Open 2020, 10, e035677. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Positive | SARS-CoV-2 Negative | |||
|---|---|---|---|---|
| n | % | n | % | |
| Total | 79 | 100% | 20 | 100 |
| Inpatient | 14 | 18% | 1 | 5% |
| Any Breathing Assist Device | 12 | 15% | 0 | 0% |
| Disposition (dead) | 5 | 6% | 2 | 10% |
| Severity 1 | ||||
| low | 2 | 3% | N/A | |
| mild | 43 | 54% | ||
| moderate | 20 | 25% | ||
| high | 2 | 3% | ||
| severe | 12 | 15% | ||
| Proton Pump Inhibitor | 14 | 18% | 5 | 25% |
| Smoker (yes) | 8 | 10% | 3 | 15% |
| Inhaler | 21 | 27% | 9 | 45% |
| Antibiotic | 15 | 19% | 2 | 10% |
| Average BMI (±SD) | 32.6 | ±8.2 | 28.8 | ±5.9 |
| Average Age (±SD) | 51.6 | ±17.0 | 50.3 | ±14.0 |
| Race | ||||
| Black | 44 | 56% | 11 | 55% |
| White | 35 | 44% | 9 | 45% |
| Sex | ||||
| Male | 27 | 34% | 7 | 35% |
| Female | 52 | 66% | 13 | 65% |
| Characteristic | Patient Subgroup | Adonis p-Value | Dispersion p-Value |
|---|---|---|---|
| Breathing assistance | COVID-19+ | 0.003 | 0.104 |
| Disposition | COVID-19+ | 0.010 | 0.637 |
| Inpatient | COVID-19+ | 0.004 | 0.283 |
| Severity | COVID-19+ | 0.109 | |
| Antibiotic | All | 0.213 | |
| COVID-19 status | All | 0.012 | 0.834 |
| Inhaler | All | 0.072 | |
| Proton pump inhibitor | All | 0.953 | |
| Smoker | All | 0.070 | |
| Race | All | 0.001 | 0.001 |
| Sex | All | 0.138 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feehan, A.K.; Rose, R.; Nolan, D.J.; Spitz, A.M.; Graubics, K.; Colwell, R.R.; Garcia-Diaz, J.; Lamers, S.L. Nasopharyngeal Microbiome Community Composition and Structure Is Associated with Severity of COVID-19 Disease and Breathing Treatment. Appl. Microbiol. 2021, 1, 177-188. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1020014
Feehan AK, Rose R, Nolan DJ, Spitz AM, Graubics K, Colwell RR, Garcia-Diaz J, Lamers SL. Nasopharyngeal Microbiome Community Composition and Structure Is Associated with Severity of COVID-19 Disease and Breathing Treatment. Applied Microbiology. 2021; 1(2):177-188. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1020014
Chicago/Turabian StyleFeehan, Amy K., Rebecca Rose, David J. Nolan, Austin M. Spitz, Karlis Graubics, Rita R. Colwell, Julia Garcia-Diaz, and Susanna L. Lamers. 2021. "Nasopharyngeal Microbiome Community Composition and Structure Is Associated with Severity of COVID-19 Disease and Breathing Treatment" Applied Microbiology 1, no. 2: 177-188. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1020014