
The Gut Microbiota of Naturally Occurring and Laboratory Aquaculture Lytechinus variegatus Revealed Differences in the Community Composition, Taxonomic Co-Occurrence, and Predicted Functional Attributes


 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Description and High-Throughput Sequencing
2.2. Taxonomic Distribution
2.3. Co-Occurrence Analysis of Microbial Taxa
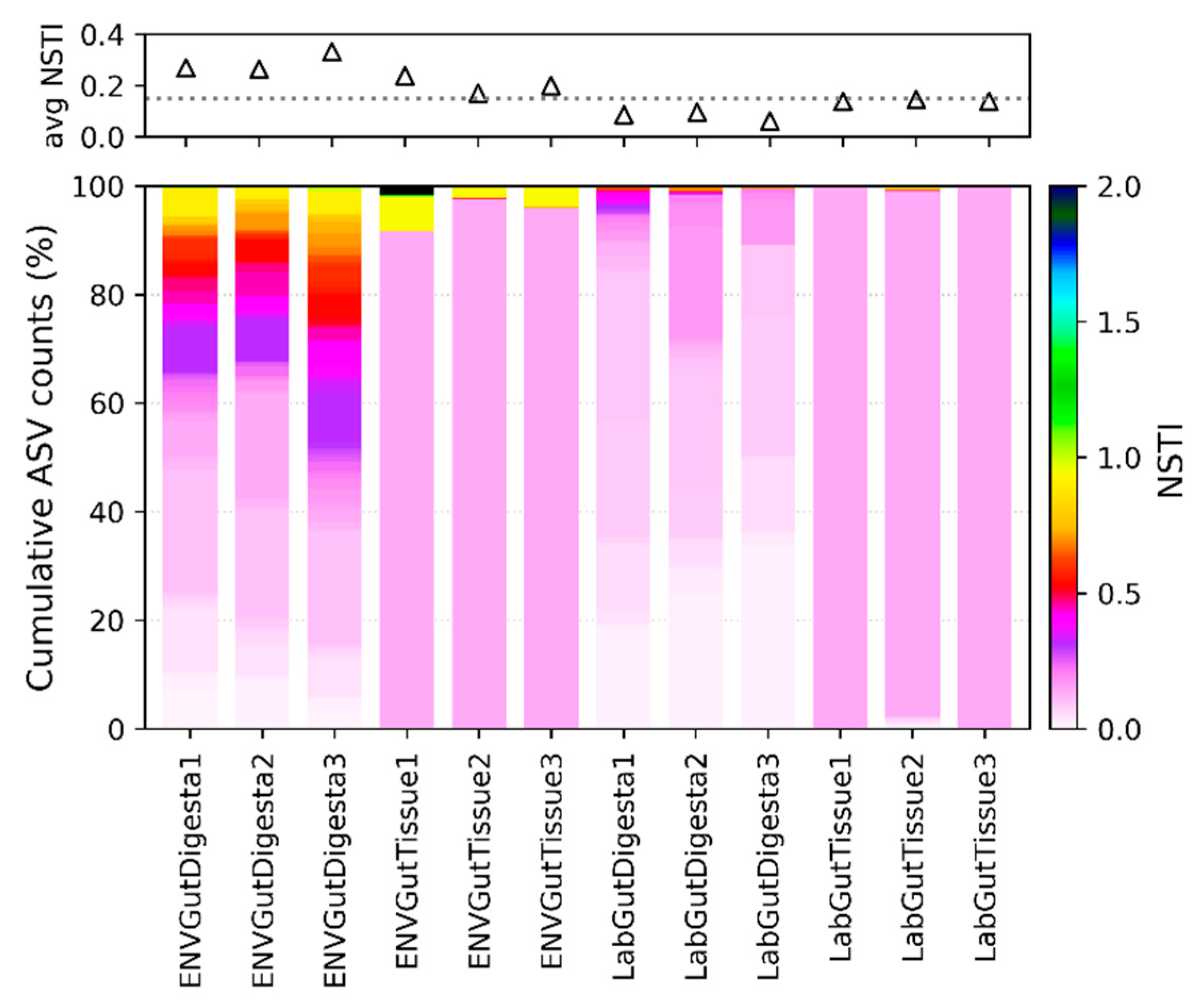
2.4. Predicted Functional Analysis
3. Results
3.1. Read Quality and Sample Statistics
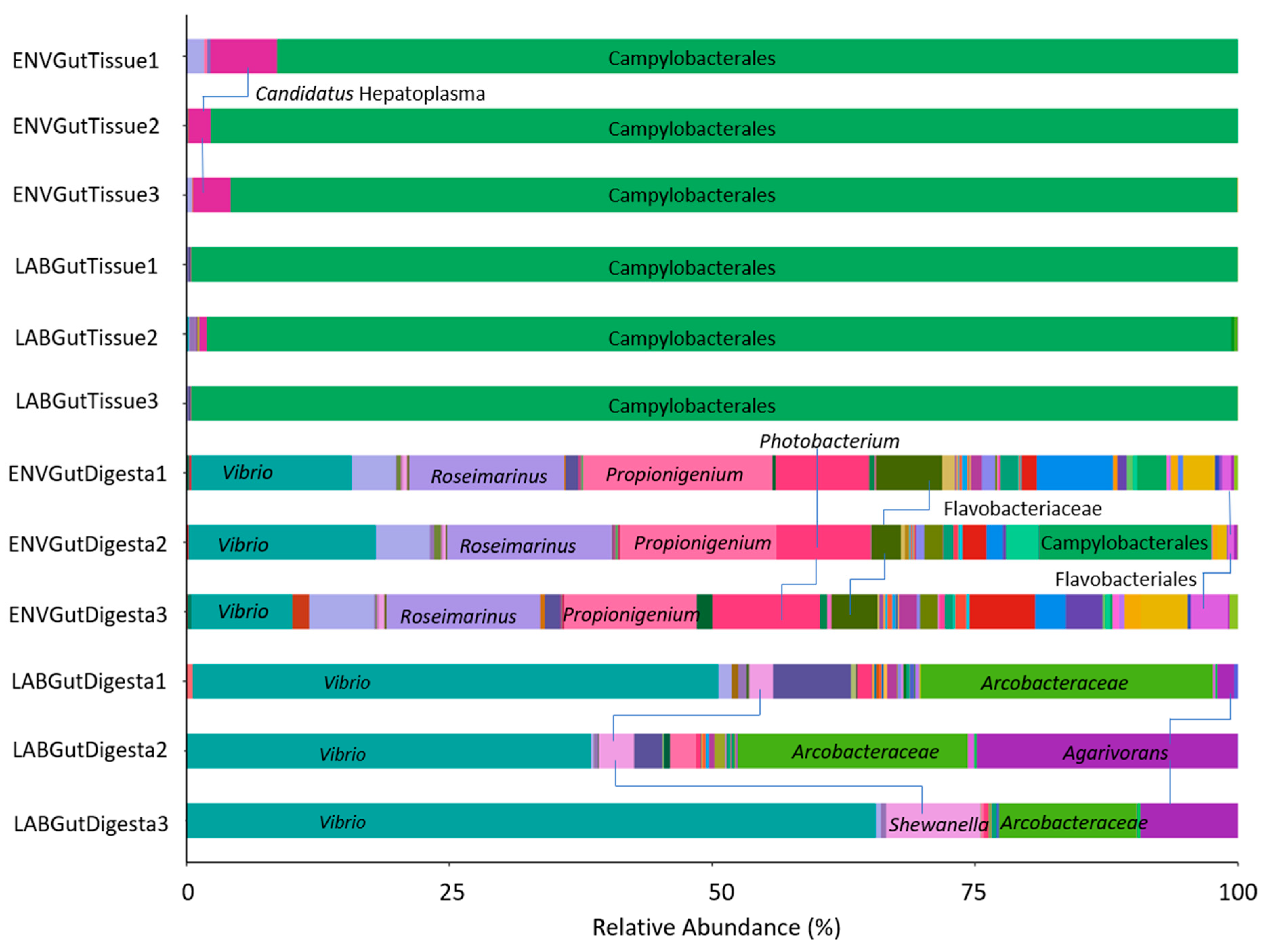
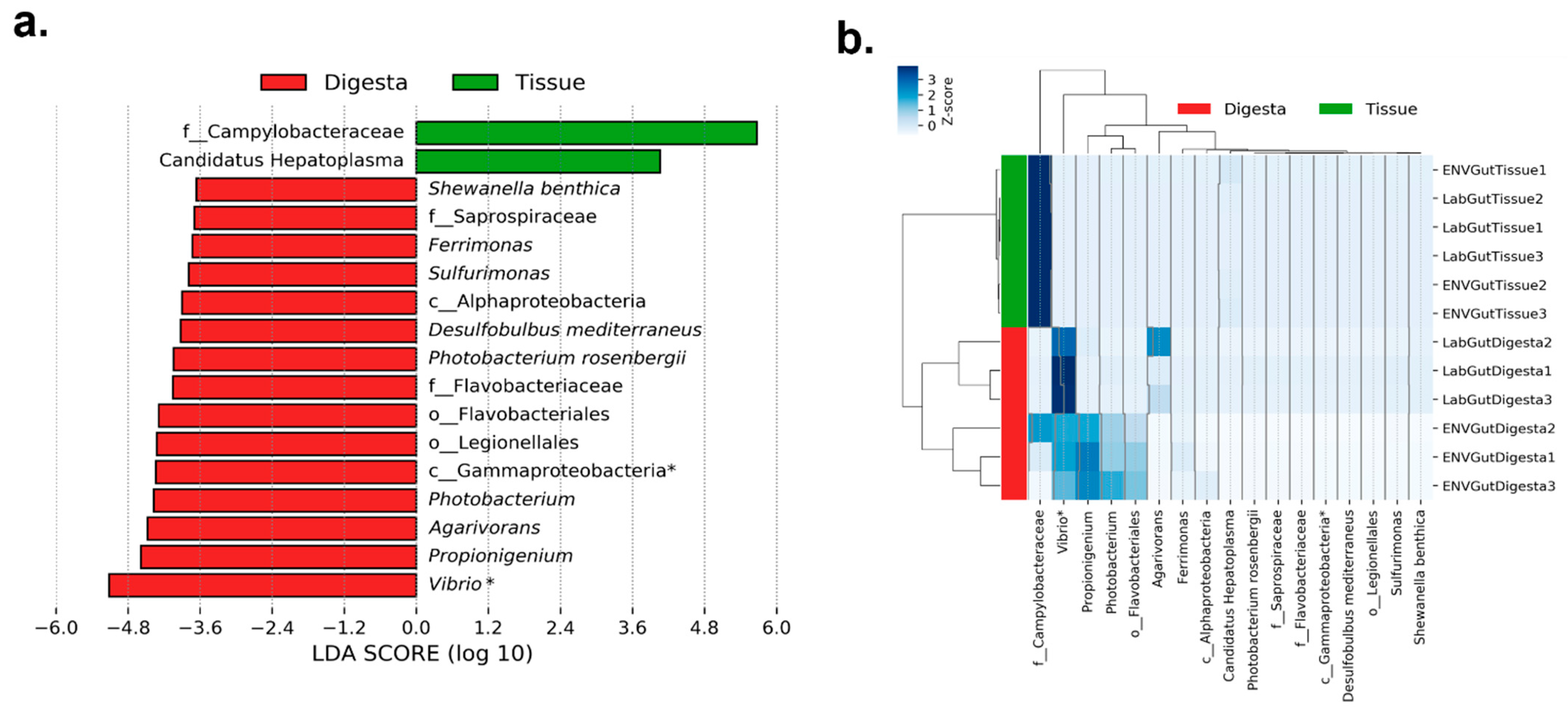
3.2. Taxonomic Distribution across Samples
3.3. Alpha Diversity
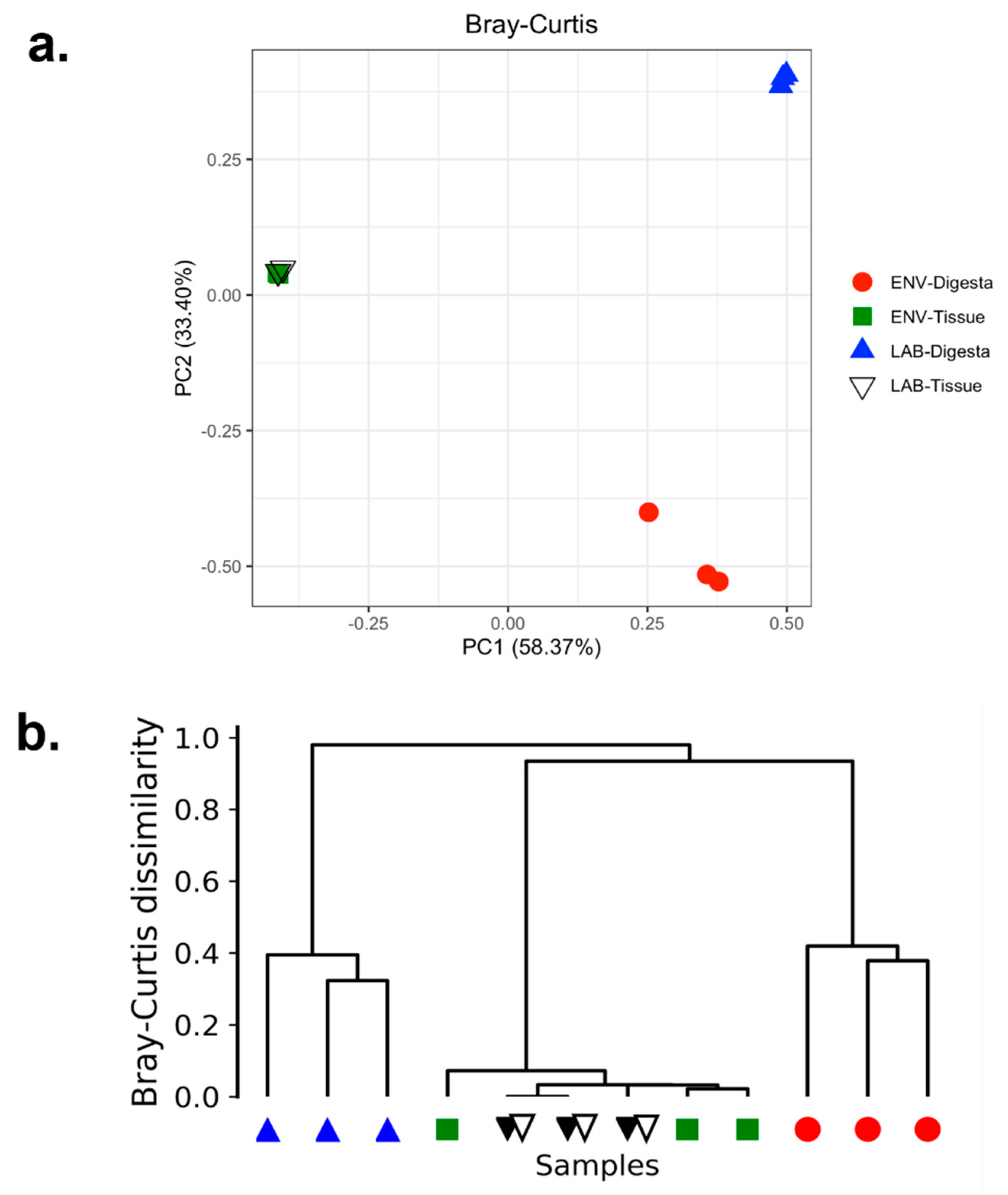
3.4. Beta Diversity
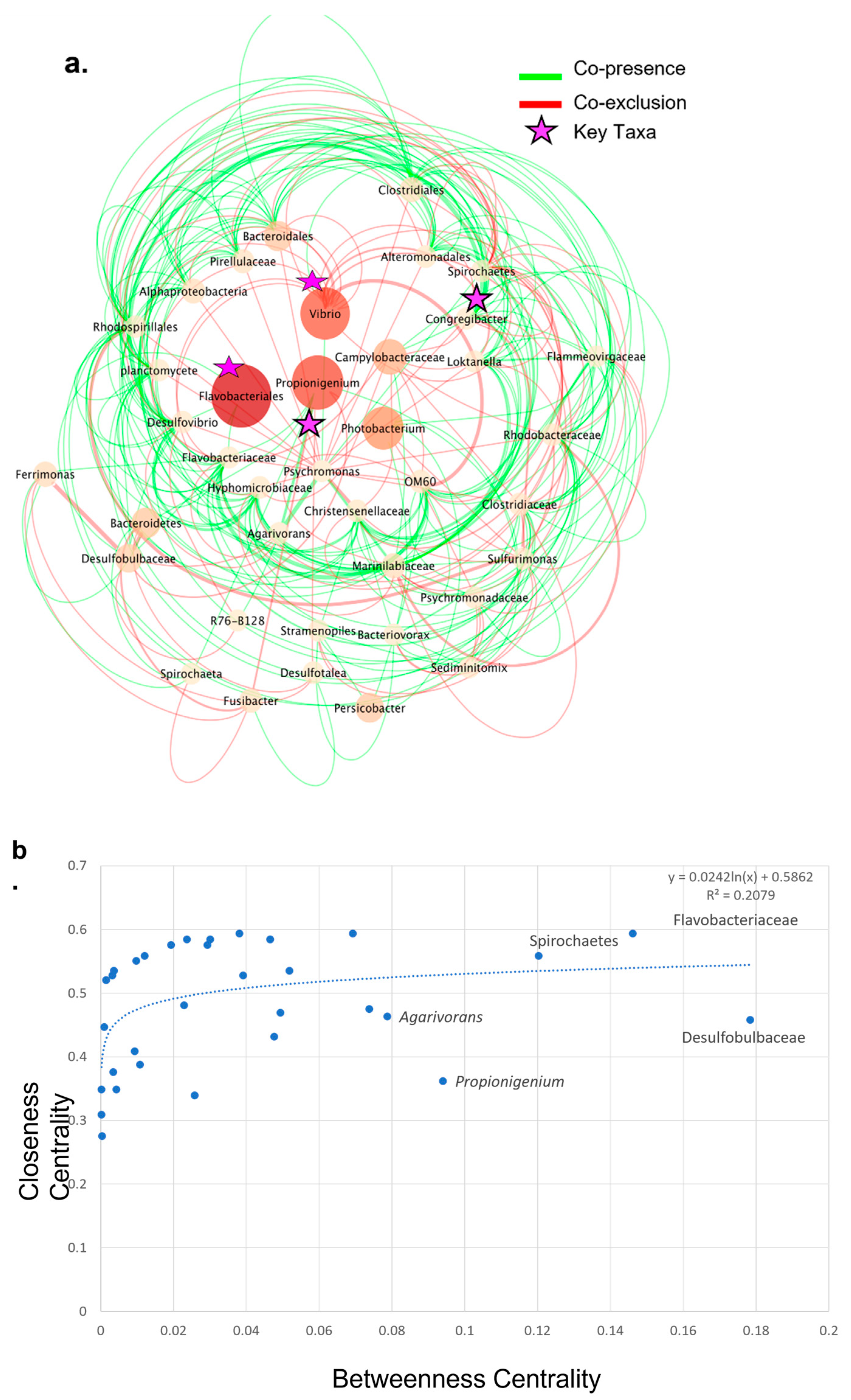
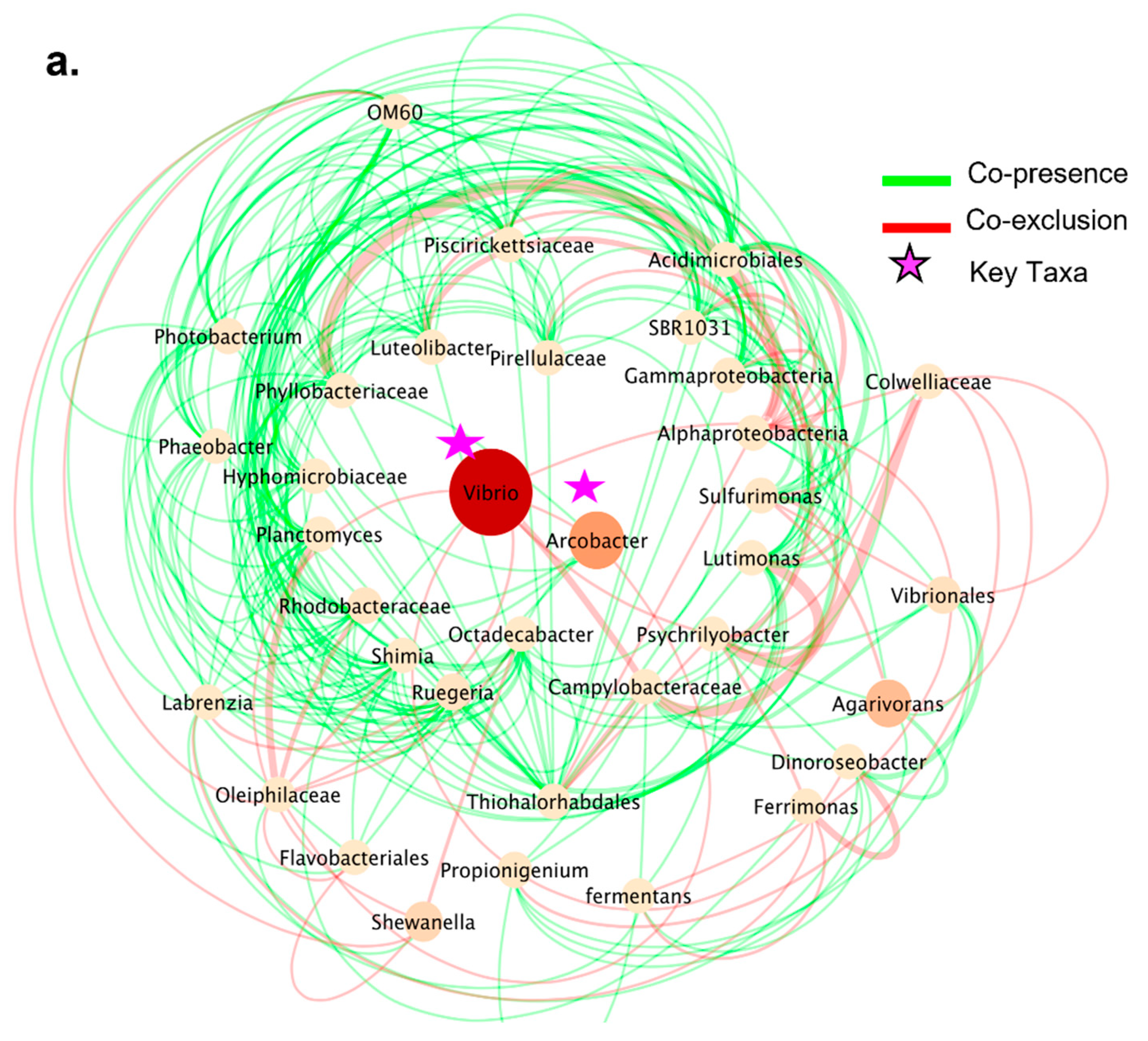
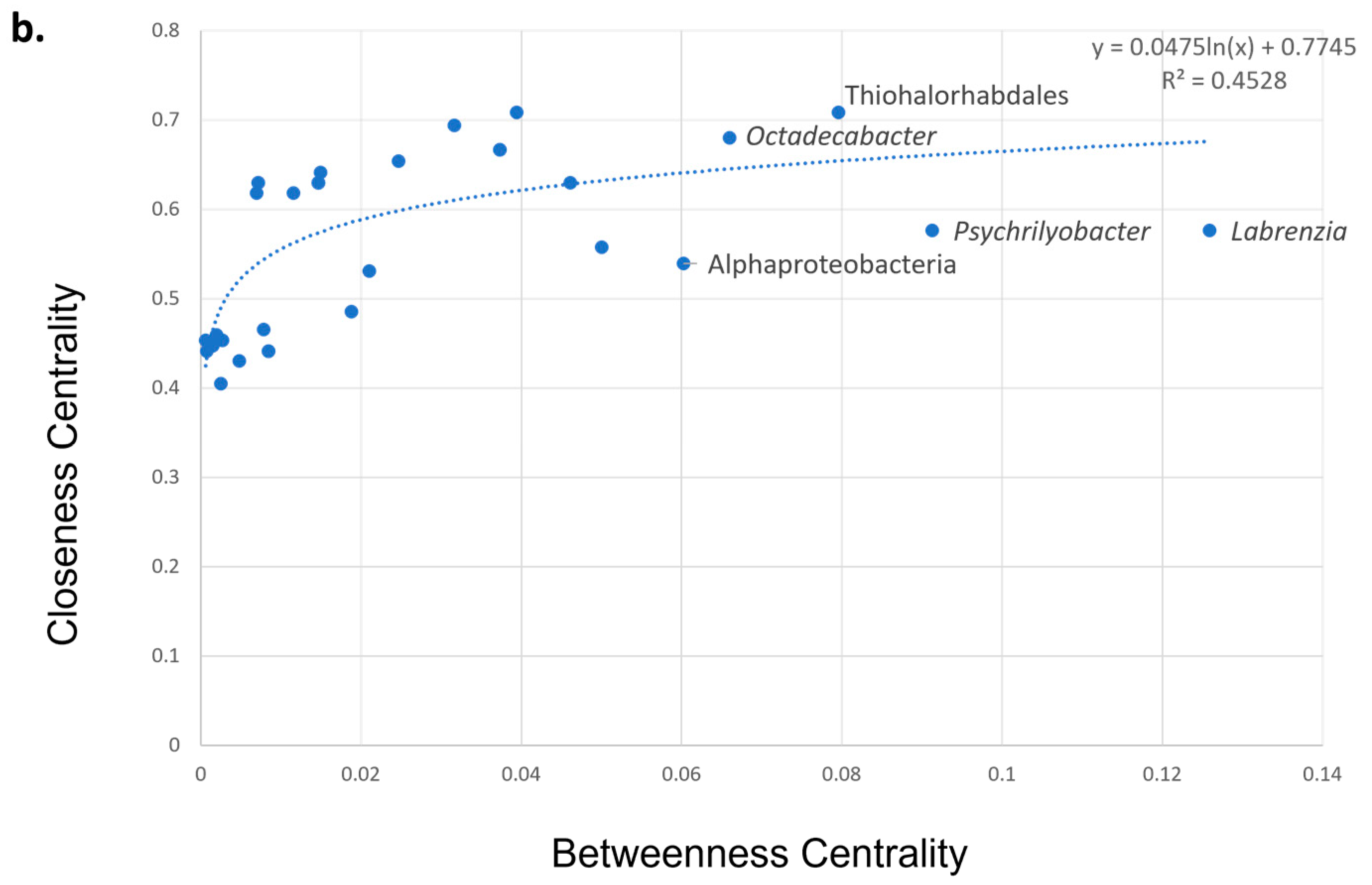
3.5. Co-Presence, Co-Exclusion, and Key Taxa in ENV and LAB Digesta
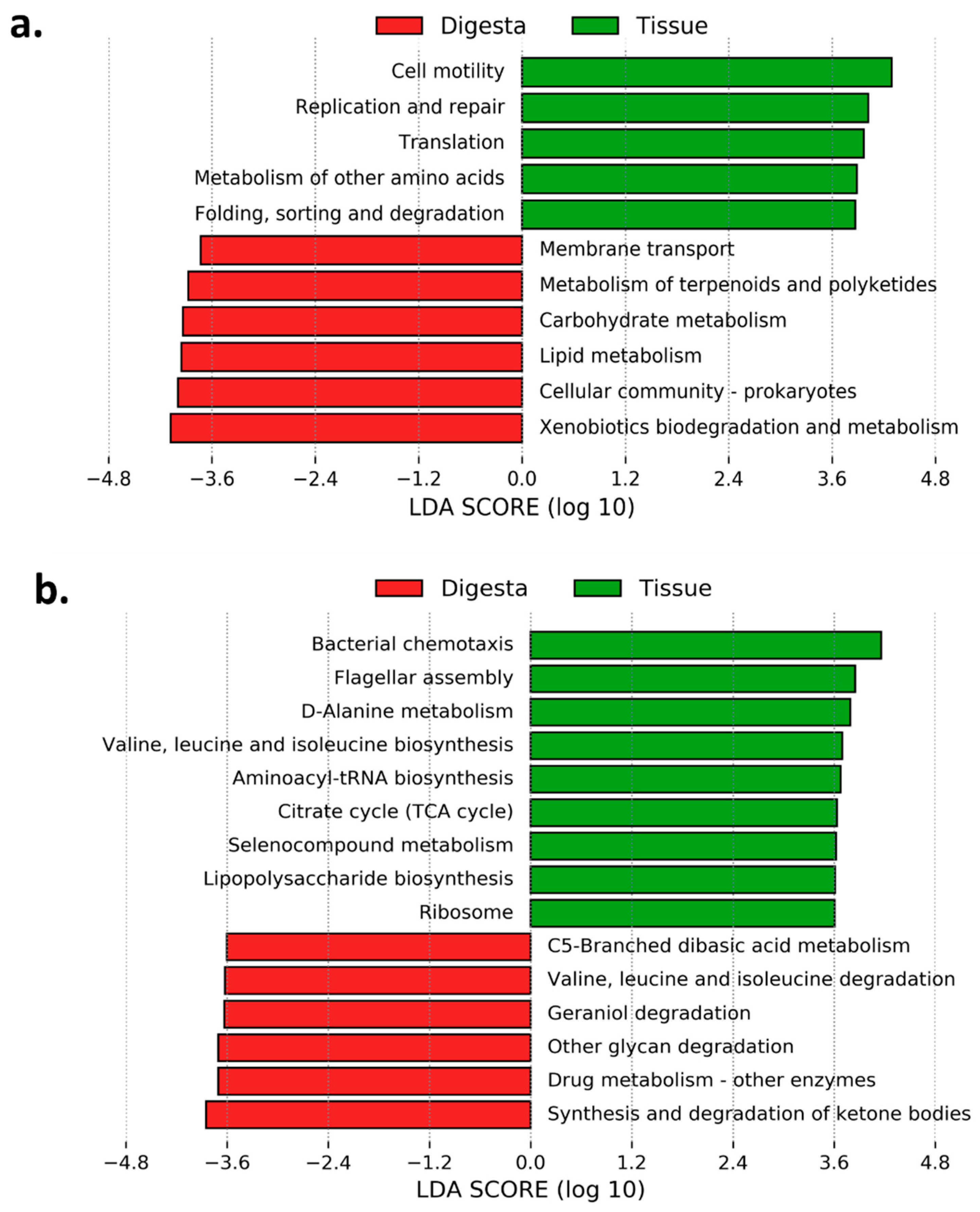
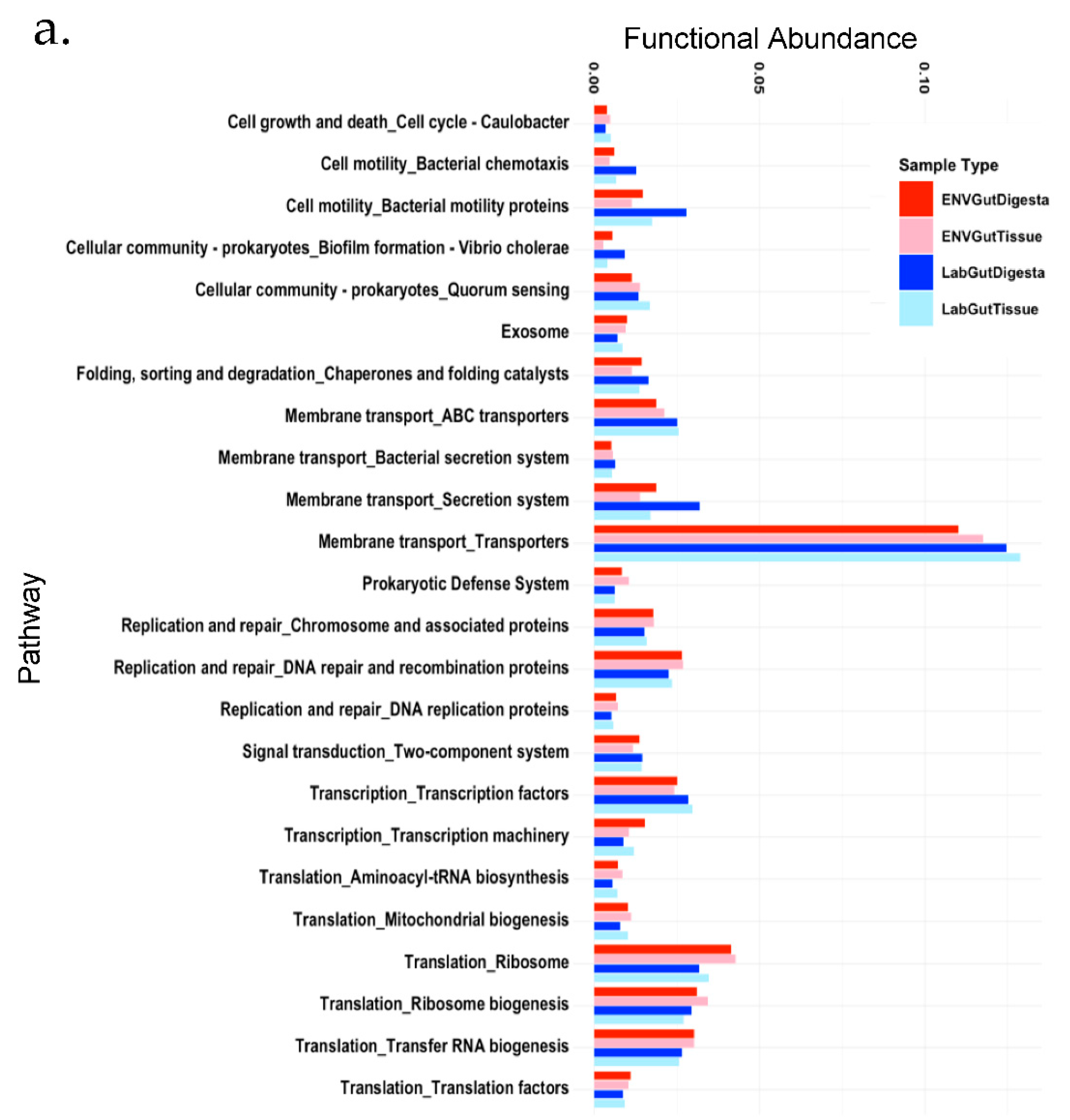
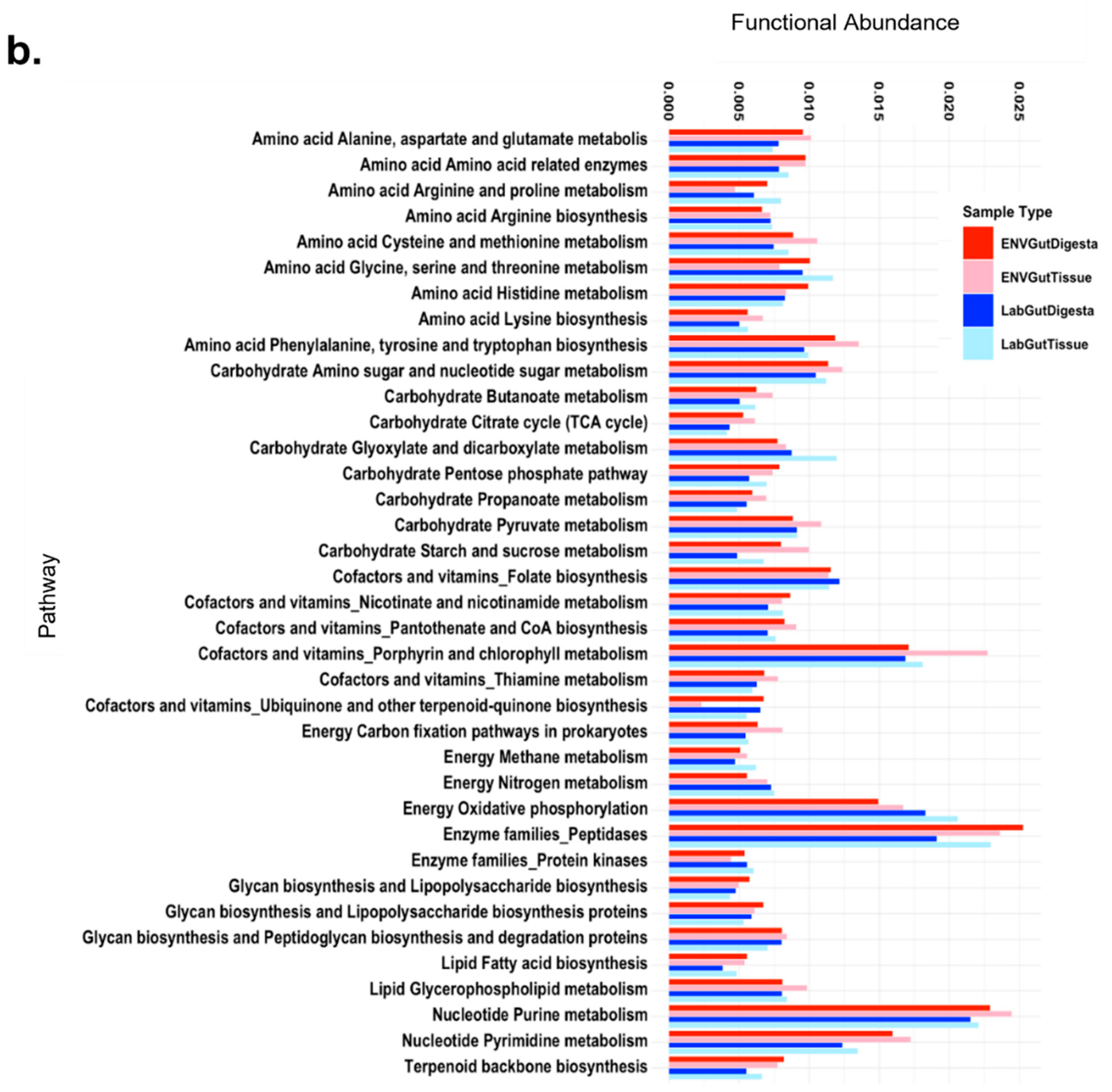
3.6. Predicted Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elmqvist, T.; Folke, C.; Nyström, M.; Peterson, G.; Bengtsson, J.; Walker, B.; Norberg, J. Response diversity, ecosystem change, and resilience. Front. Ecol. Environ. 2003, 1, 488–494. [Google Scholar] [CrossRef]
- Levin, S.A.; Lubchenco, J. Resilience, robustness, and marine ecosystem-based management. Bioscience 2008, 58, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Palumbi, S.R.; McLeod, K.L.; Grünbaum, D. Ecosystems in action: Lessons from marine ecology about recovery, resistance, and reversibility. BioScience 2008, 58, 33–42. [Google Scholar] [CrossRef]
- Rosshart, S.P.; Vassallo, B.G.; Angeletti, D.; Hutchinson, D.S.; Morgan, A.P.; Takeda, K.; Hickman, H.D.; McCulloch, J.A.; Badger, J.H.; Ajami, N.J. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 2017, 171, 1015–1028.e13. [Google Scholar] [CrossRef] [Green Version]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirksen, P.; Marsh, S.A.; Braker, I.; Heitland, N.; Wagner, S.; Nakad, R.; Mader, S.; Petersen, C.; Kowallik, V.; Rosenstiel, P. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biol. 2016, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Berg, M.; Dierking, K.; Félix, M.-A.; Shapira, M.; Samuel, B.S.; Schulenburg, H. Caenorhabditis elegans as a model for microbiome research. Front. Microbiol. 2017, 8, 485. [Google Scholar] [CrossRef]
- Staubach, F.; Baines, J.F.; Künzel, S.; Bik, E.M.; Petrov, D.A. Host species and environmental effects on bacterial communities associated with Drosophila in the laboratory and in the natural environment. PLoS ONE 2013, 8, e70749. [Google Scholar] [CrossRef] [Green Version]
- Hills, R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut microbiome: Profound implications for diet and disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Ericsson, A.C.; Franklin, C.L. The gut microbiome of laboratory mice: Considerations and best practices for translational research. Mamm. Genome 2021, 1–12. [Google Scholar]
- Turner, P.V. The role of the gut microbiota on animal model reproducibility. Anim. Models Exp. Med. 2018, 1, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M. Laboratory animal control diets: Very important, often neglected. Lab. Anim. 2015, 44, 240. [Google Scholar] [CrossRef]
- Xiang, Z.; Zhu, H.; Yang, B.; Fan, H.; Guo, J.; Liu, J.; Kong, Q.; Teng, Q.; Shang, H.; Su, L. A glance at the gut microbiota of five experimental animal species through fecal samples. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McBride, S.C. Sea urchin aquaculture. In Proceedings of the American Fisheries Society Symposium. Am. Fish. Soc. Symp. 2005, 46, 179–208. [Google Scholar]
- Heflin, L.E.; Makowsky, R.; Taylor, J.C.; Williams, M.B.; Lawrence, A.L.; Watts, S.A. Production and economic optimization of dietary protein and carbohydrate in the culture of juvenile sea urchin Lytechinus variegatus. Aquaculture 2016, 463, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Albright, R.; Bland, C.; Gillette, P.; Serafy, J.E.; Langdon, C.; Capo, T.R. Juvenile growth of the tropical sea urchin Lytechinus variegatus exposed to near-future ocean acidification scenarios. J. Exp. Mar. Biol. Ecol. 2012, 426, 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendler, G. Sea Stars, Sea Urchins, and Allies: Echinoderms of Florida and the Caribbean; Smithsonian Institution Press: Washington, DC, USA, 1995; 390p. [Google Scholar]
- Watanabe, J.M.; Harrold, C. Destructive grazing by sea urchins Strongylocentrotus spp. in a central California kelp forest: Potential roles of recruitment, depth, and predation. Mar. Ecol. Prog. Ser. Oldendorf 1991, 71, 125–141. [Google Scholar] [CrossRef]
- Watts, S.A.; McClintock, J.B.; JLawrence, J.M. Lytechinus. In Developments in Aquaculture and Fisheries Science; Lawrence, J.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 43, pp. 661–680. [Google Scholar]
- Annunziata, R.; Perillo, M.; Andrikou, C.; Cole, A.G.; Martinez, P.; Arnone, M.I. Pattern and process during sea urchin gut morphogenesis: The regulatory landscape. Genesis 2014, 52, 251–268. [Google Scholar] [CrossRef]
- De Ridder, C.; Jangoux, M. Digestive systems: Echinoidea. In Echinoderm Nutrition; CRC Press: Boca Raton, FL, USA, 2020; pp. 213–234. [Google Scholar]
- Hakim, J.A.; Koo, H.; Dennis, L.N.; Kumar, R.; Ptacek, T.; Morrow, C.D.; Lefkowitz, E.J.; Powell, M.L.; Bej, A.K.; Watts, S.A. An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin, Lytechinus variegatus. Front. Microbiol. 2015, 6, 1047. [Google Scholar] [CrossRef]
- Hakim, J.A.; Koo, H.; Kumar, R.; Lefkowitz, E.J.; Morrow, C.D.; Powell, M.L.; Watts, S.A.; Bej, A.K. The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles. FEMS Microbiol. Ecol. 2016, 92, fiw146. [Google Scholar] [CrossRef] [Green Version]
- Holland, N.D.; Ghiselin, M.T. A comparative study of gut mucous cells in thirty-seven species of the class Echinoidea (Echinodermata). Biol. Bull. 1970, 138, 286–305. [Google Scholar] [CrossRef]
- Lasker, R.; Giese, A.C. Nutrition of the sea urchin, Strongylocentrotus purpuratus. Biol. Bull. 1954, 106, 328–340. [Google Scholar] [CrossRef]
- Beleneva, I.; Kukhlevskii, A. Characterization of Vibrio gigantis and Vibrio pomeroyi isolated from invertebrates of Peter the Great Bay, Sea of Japan. Microbiology 2010, 79, 402–407. [Google Scholar] [CrossRef]
- Guerinot, M.L.; Patriquin, D. The association of N 2-fixing bacteria with sea urchins. Mar. Biol. 1981, 62, 197–207. [Google Scholar] [CrossRef]
- Guerinot, M.; Patriquin, D. N2-fixing vibrios isolated from the gastrointestinal tract of sea urchins. Can. J. Microbiol. 1981, 27, 311–317. [Google Scholar] [CrossRef]
- Brothers, C.J.; Van Der Pol, W.J.; Morrow, C.D.; Hakim, J.A.; Koo, H.; McClintock, J.B. Ocean warming alters predicted microbiome functionality in a common sea urchin. Proc. R. Soc. B 2018, 285, 20180340. [Google Scholar] [CrossRef] [PubMed]
- Hammer, H.; Hammer, B.; Watts, S.; Lawrence, A.; Lawrence, J. The effect of dietary protein and carbohydrate concentration on the biochemical composition and gametogenic condition of the sea urchin Lytechinus variegatus. J. Exp. Mar. Biol. Ecol. 2006, 334, 109–121. [Google Scholar] [CrossRef]
- Beddingfield, S.D.; McClintock, J.B. Food Resource Utilization in the Sea Urchin Lytechinus variegatus in Contrasting Shallow-Water Microhabits of Saint Joseph Bay, Florida. Gulf Mex. Sci. 1999, 17, 3. [Google Scholar] [CrossRef]
- Deming, J.W.; Carpenter, S.D. Factors influencing benthic bacterial abundance, biomass, and activity on the northern continental margin and deep basin of the Gulf of Mexico. Deep Sea Res. Part II Top. Stud. Oceanogr. 2008, 55, 2597–2606. [Google Scholar] [CrossRef]
- Beddingfield, S.D.; McClintock, J.B. Demographic characteristics of Lytechinus variegatus (Echinoidea: Echinodermata) from three habitats in a North Florida Bay, Gulf of Mexico. Mar. Ecol. 2000, 21, 17–40. [Google Scholar] [CrossRef]
- Erwin, P.M.; Olson, J.B.; Thacker, R.W. Phylogenetic diversity, host-specificity and community profiling of sponge-associated bacteria in the northern Gulf of Mexico. PLoS ONE 2011, 6, e26806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felder, D.L.; Camp, D.K. Gulf of Mexico Origin, Waters, and Biota: Biodiversity; Texas A&M University Press: College Station, TX, USA, 2009. [Google Scholar]
- Kellogg, C.A.; Lisle, J.T.; Galkiewicz, J.P. Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the northeastern Gulf of Mexico. Appl. Environ. Microbiol. 2009, 75, 2294–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.; Mojib, N.; Thacker, R.W.; Bej, A.K. Comparative analysis of bacterial community-metagenomics in coastal Gulf of Mexico sediment microcosms following exposure to Macondo oil (MC252). Antonie Leeuwenhoek 2014, 106, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Skoog, A.; Biddanda, B.; Benner, R. Bacterial utilization of dissolved glucose in the upper water column of the Gulf of Mexico. Limnol. Oceanogr. 1999, 44, 1625–1633. [Google Scholar] [CrossRef] [Green Version]
- Hakim, J.A.; Schram, J.B.; Galloway, A.W.; Morrow, C.D.; Crowley, M.R.; Watts, S.A.; Bej, A.K. The purple sea urchin Strongylocentrotus purpuratus demonstrates a compartmentalization of gut bacterial microbiota, predictive functional attributes, and taxonomic co-occurrence. Microorganisms 2019, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Eipers, P.; Little, R.B.; Crowley, M.; Crossman, D.K.; Lefkowitz, E.J.; Morrow, C.D. Getting started with microbiome analysis: Sample acquisition to bioinformatics. Curr. Protoc. Hum. Genet. 2014, 82, 18.8.1–18.8.29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [Green Version]
- Hakim, J.A.; Morrow, C.D.; Watts, S.A.; Bej, A.K. High-throughput amplicon sequencing datasets of the metacommunity DNA of the gut microbiota of naturally occurring and laboratory aquaculture green sea urchins Lytechinus variegatus. Data Brief 2019, 26, 104405. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.J.; Walsh, D.C. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.; O’hara, R.; Simpson, G.; Solymos, P.; Stevens, M.; Wagner, H. Vegan: Community Ecology Package. R Package Version. 2.0-10. CRAN. 2013. Available online: https://www.researchgate.net/publication/258996451_Vegan_Community_Ecology_Package_R_Package_Version_20-10 (accessed on 14 July 2021).
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual comparisons by ranking methods. In Breakthroughs in Statistics; Springer: Berlin/Heidelberg, Germany, 1992; pp. 196–202. [Google Scholar]
- Fisher, R.A. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936, 7, 179–188. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef]
- Choma, M.; Bárta, J.; Šantrůčková, H.; Urich, T. Low abundance of Archaeorhizomycetes among fungi in soil metatranscriptomes. Sci. Rep. 2016, 6, 1–6. [Google Scholar] [CrossRef]
- Pearson, K. Notes on Regression and Inheritance in the Case of Two Parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar]
- Spearman, C. The Proof and Measurement of Association Between Two Things. In Studies in Individual Differences: The Search for Intelligence; Jenkins, J.J., Paterson, D.G., Eds.; Appleton-Century-Crofts: Norwalk, CT, USA, 1961; pp. 45–58. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Cover, T.M.; Thomas, J.A. Information theory and statistics. Elem. Inf. Theory 1991, 1, 279–335. [Google Scholar]
- Wiese, R.; Eiglsperger, M.; Kaufmann, M. yFiles—Visualization and automatic layout of graphs. In Graph Drawing Software; Springer: Berlin/Heidelberg, Germany, 2004; pp. 173–191. [Google Scholar]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Wang, H.; Dsouza, M.; Lou, J.; He, Y.; Dai, Z.; Brookes, P.C.; Xu, J.; Gilbert, J.A. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 2016, 10, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sheng, H.-F.; He, Y.; Wu, J.-Y.; Jiang, Y.-X.; Tam, N.F.-Y.; Zhou, H.-W. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 2012, 78, 8264–8271. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- McNally, K.; Hogg, A.; Loizou, G. A computational workflow for probabilistic quantitative in vitro to in vivo extrapolation. Front. Pharmacol. 2018, 9, 508. [Google Scholar] [CrossRef] [Green Version]
- Zidorn, C. Secondary metabolites of seagrasses (Alismatales and Potamogetonales; Alismatidae): Chemical diversity, bioactivity, and ecological function. Phytochemistry 2016, 124, 5–28. [Google Scholar] [CrossRef]
- Windyaswari, A.; Purba, J.; Nurrahmah, S.; Ayu, I.; Imran, Z.; Amin, A.; Kurniawan, F.; Pratiwi, N.; Iswantari, A. Phytochemical profile of sea grass extract (Enhalus acoroides): A new marine source from Ekas Bay, East Lombok. In Proceedings of the IOP Conference Series: Earth and Environmental Science; 2019; Volume 278, p. 012081. [Google Scholar] [CrossRef]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, D.; Blackwell, N.; Langarica-Fuentes, A.; Peltzer, A.; Nahnsen, S.; Kleindienst, S. Interpretations of environmental microbial community studies are biased by the selected 16S rRNA (Gene) amplicon sequencing pipeline. Front. Microbiol. 2020, 11, 2652. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.; Blair, B.; Murdock, C.; Meade, M.; Watts, S.; Lawrence, A.L. Molecular Analysis of gut microflora in captive-raised sea urchins (Lytechinus variegatus). J. World Aquac. Soc. 2010, 41, 807–815. [Google Scholar] [CrossRef]
- Nakagawa, S.; Saito, H.; Tame, A.; Hirai, M.; Yamaguchi, H.; Sunata, T.; Aida, M.; Muto, H.; Sawayama, S.; Takaki, Y. Microbiota in the coelomic fluid of two common coastal starfish species and characterization of an abundant Helicobacter-related taxon. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, M.; Nakagawa, S.; Sawabe, T. Microbial communities associated with holothurians: Presence of unique bacteria in the coelomic fluid. Microbes Environ. 2009, 27, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.A.; Suárez, R.; Godoy, M. Microbial Diversity of the Red Sea Urchin Loxechinus albus during Controlled Farming in Puerto Montt, Chile, Using 16S rRNA Gene Amplicon Sequencing. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwob, G.; Cabrol, L.; Poulin, E.; Orlando, J. Characterization of the gut microbiota of the Antarctic heart urchin (Spatangoida) Abatus agassizii. Front. Microbiol. 2020, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Sawabe, T.; Oda, Y.; Shiomi, Y.; Ezura, Y. Alginate degradation by bacteria isolated from the gut of sea urchins and abalones. Microb. Ecol. 1995, 30, 193–202. [Google Scholar] [CrossRef]
- Unkles, S. Bacterial flora of the sea urchin Echinus esculentus. Appl. Environ. Microbiol. 1977, 34, 347–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Baik, K.S.; Hwang, Y.S.; Choi, J.-S.; Kwon, J.; Seong, C.N. Vibriohemicentroti sp. nov., an alginate lyase-producing bacterium, isolated from the gut microflora of sea urchin (Hemicentrotus pulcherrimus). Int. J. Syst. Evol. Microbiol. 2013, 63, 3697–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meziti, A.; Kormas, K.A.; Pancucci-Papadopoulou, M.-A.; Thessalou-Legaki, M. Bacterial phylotypes associated with the digestive tract of the sea urchin Paracentrotus lividus and the ascidian Microcosmus sp. Russ. J. Mar. Biol. 2007, 33, 84–91. [Google Scholar] [CrossRef]
- Farmer, J., III; Hickman-Brenner, F. The Genera Vibrio and Photobacterium; The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackenbrandt, E., Eds.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Gomez-Gil, B.; Roque, A.; Rotllant, G.; Peinado, L.; Romalde, J.L.; Doce, A.; Cabanillas-Beltran, H.; Chimetto, L.A.; Thompson, F.L. Photobacterium swingsii sp. nov., isolated from marine organisms. Int. J. Syst. Evol. Microbiol. 2011, 61, 315–319. [Google Scholar] [CrossRef]
- Seo, H.J.; Bae, S.S.; Lee, J.-H.; Kim, S.-J. Photobacterium frigidiphilum sp. nov., a psychrophilic, lipolytic bacterium isolated from deep-sea sediments of Edison Seamount. Int. J. Syst. Evol. Microbiol. 2005, 55, 1661–1666. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; Lee, J.-K.; Kim, Y.-O.; Oh, T.-K. Photobacterium lipolyticum sp. nov., a bacterium with lipolytic activity isolated from the Yellow Sea in Korea. Int. J. Syst. Evol. Microbiol. 2005, 55, 335–339. [Google Scholar] [CrossRef] [Green Version]
- De Vadder, F.; Kovatcheva-Datchary, P.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab. 2016, 24, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; Leitch, C.M.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014, 8, 1323–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schink, B.; Pfennig, N. Propionigenium modestum gen. nov. sp. nov. a new strictly anaerobic, nonsporing bacterium growing on succinate. Arch. Microbiol. 1982, 133, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Apprill, A. Marine animal microbiomes: Toward understanding host–microbiome interactions in a changing ocean. Front. Mar. Sci. 2017, 4, 222. [Google Scholar] [CrossRef] [Green Version]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The gut microbiota of rural papua new guineans: Composition, diversity patterns, and ecological processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellend, M. Conceptual synthesis in community ecology. Q. Rev. Biol. 2010, 85, 183–206. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Reese, A.T.; Dunn, R.R. Drivers of microbiome biodiversity: A review of general rules, feces, and ignorance. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Gall, C.A.; Scoles, G.A.; Magori, K.; Mason, K.L.; Brayton, K.A. Laboratory colonization stabilizes the naturally dynamic microbiome composition of field collected Dermacentor andersoni ticks. Microbiome 2017, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.H.; Stat, M.; Bunce, M.; Simmons, L.W. The influence of diet and environment on the gut microbial community of field crickets. Ecol. Evol. 2018, 8, 4704–4720. [Google Scholar] [CrossRef]
- Tsukayama, P.; Boolchandani, M.; Patel, S.; Pehrsson, E.C.; Gibson, M.K.; Chiou, K.L.; Jolly, C.J.; Rogers, J.; Phillips-Conroy, J.E.; Dantas, G. Characterization of wild and captive baboon gut microbiota and their antibiotic resistomes. Msystems 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraune, S.; Bosch, T.C. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl. Acad. Sci. USA 2007, 104, 13146–13151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, C.R.; Gilmore, M.S. Native microbial colonization of Drosophila melanogaster and its use as a model of Enterococcus faecalis pathogenesis. Infect. Immun. 2007, 75, 1565–1576. [Google Scholar] [CrossRef] [Green Version]
- Hird, S.M. Evolutionary biology needs wild microbiomes. Front. Microbiol. 2017, 8, 725. [Google Scholar] [CrossRef]
- Scupham, A.J.; Patton, T.G.; Bent, E.; Bayles, D.O. Comparison of the cecal microbiota of domestic and wild turkeys. Microb. Ecol. 2008, 56, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Xenoulis, P.G.; Gray, P.L.; Brightsmith, D.; Palculict, B.; Hoppes, S.; Steiner, J.M.; Tizard, I.; Suchodolski, J.S. Molecular characterization of the cloacal microbiota of wild and captive parrots. Vet. Microbiol. 2010, 146, 320–325. [Google Scholar] [CrossRef]
- Schäfer, G. Membrane-Associated Energy Transduction in Bacteria and Archaea; Encyclopedia of Biological Chemistry, 2nd ed.; Elsevier: Amsterdam, The Nethrlands, 2013; pp. 28–35. [Google Scholar]
- Roalkvam, I.; Drønen, K.; Stokke, R.; Daae, F.L.; Dahle, H.; Steen, I.H. Physiological and genomic characterization of Arcobacter anaerophilus IR-1 reveals new metabolic features in Epsilonproteobacteria. Front. Microbiol. 2015, 6, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, S.; Lünsdorf, H.; Fuchs, B.M.; Tindall, B.J. The photosynthetic apparatus and its regulation in the aerobic gammaproteobacterium Congregibacter litoralis gen. nov., sp. nov. PLoS ONE 2009, 4, e4866. [Google Scholar] [CrossRef] [Green Version]
- Morrow, K.M.; Tedford, A.R.; Pankey, M.S.; Lesser, M.P. A member of the Roseobacter clade, Octadecabacter sp., is the dominant symbiont in the brittle star Amphipholis squamata. FEMS Microbiol. Ecol. 2018, 94, fiy030. [Google Scholar] [CrossRef] [Green Version]
- Vollmers, J.; Voget, S.; Dietrich, S.; Gollnow, K.; Smits, M.; Meyer, K.; Brinkhoff, T.; Simon, M.; Daniel, R. Poles apart: Arctic and Antarctic Octadecabacter strains share high genome plasticity and a new type of xanthorhodopsin. PLoS ONE 2013, 8, e63422. [Google Scholar] [CrossRef] [Green Version]
- Sauchyn, L.K.; Lauzon-Guay, J.-S.; Scheibling, R.E. Sea urchin fecal production and accumulation in a rocky subtidal ecosystem. Aquat. Biol. 2011, 13, 215–223. [Google Scholar] [CrossRef]
- Sauchyn, L.K.; Scheibling, R.E. Fecal production by sea urchins in native and invaded algal beds. Mar. Ecol. Prog. Ser. 2009, 396, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Sauchyn, L.K.; Scheibling, R.E. Degradation of sea urchin feces in a rocky subtidal ecosystem: Implications for nutrient cycling and energy flow. Aquat. Biol. 2009, 6, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Becker, P.T.; Samadi, S.; Zbinden, M.; Hoyoux, C.; Compère, P.; De Ridder, C. First insights into the gut microflora associated with an echinoid from wood falls environments. Cah. Biol. Mar. 2009, 50, 343. [Google Scholar]
- Tanrattanapitak, N.; Pairohakul, S. Bacterial Community in Gut Contents of the Sea Urchin Diadema setosum (Leske, 1778) and the Ambient Sediments from Sichang Island using Metagenomics Approaches. NU Int. J. Sci. 2018, 15, 117–125. [Google Scholar]
- Wirsen, C.O.; Sievert, S.M.; Cavanaugh, C.M.; Molyneaux, S.J.; Ahmad, A.; Taylor, L.; DeLong, E.; Taylor, C.D. Characterization of an autotrophic sulfide-oxidizing marine Arcobacter sp. that produces filamentous sulfur. Appl. Environ. Microbiol. 2002, 68, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Pati, A.; Gronow, S.; Lapidus, A.; Copeland, A.; Del Rio, T.G.; Nolan, M.; Lucas, S.; Tice, H.; Cheng, J.-F.; Han, C. Complete genome sequence of Arcobacter nitrofigilis type strain (CI T). Stand. Genom. Sci. 2010, 2, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Defoirdt, T. Quorum sensing positively regulates flagellar motility in pathogenic V ibrio harveyi. Environ. Microbiol. 2015, 17, 960–968. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar]
- Bell, A.; Juge, N. Mucosal glycan degradation of the host by the gut microbiota. Glycobiology 2020. [Google Scholar] [CrossRef]
- Martens, E.C.; Chiang, H.C.; Gordon, J.I. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 2008, 4, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Observed ASVs | Shannon Diversity | Simpson Diversity |
|---|---|---|---|
| ENV.Gut.Digesta.1 | 250 | 5.487767954 | 0.944642074 |
| ENV.Gut.Digesta.2 | 152 | 4.841885921 | 0.930314741 |
| ENV.Gut.Digesta.3 | 295 | 6.061862678 | 0.962199539 |
| ENV.Gut.Tissue.1 | 15 | 0.571363207 | 0.160769415 |
| ENV.Gut.Tissue.2 | 14 | 0.189163468 | 0.047561491 |
| ENV.Gut.Tissue.3 | 11 | 0.302996014 | 0.082618331 |
| LAB.Gut.Digesta.1 | 207 | 3.967739212 | 0.859951252 |
| LAB.Gut.Digesta.2 | 161 | 3.728789803 | 0.866174131 |
| LAB.Gut.Digesta.3 | 54 | 3.109402559 | 0.841666322 |
| LAB.Gut.Tissue.1 | 29 | 0.057624124 | 0.012002203 |
| LAB.Gut.Tissue.2 | 74 | 0.395491696 | 0.069834258 |
| LAB.Gut.Tissue.3 | 28 | 0.076786117 | 0.012002203 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Green, G.B.H.; Hakim, J.A.; Chen, J.-W.; Koo, H.; Morrow, C.D.; Watts, S.A.; Bej, A.K. The Gut Microbiota of Naturally Occurring and Laboratory Aquaculture Lytechinus variegatus Revealed Differences in the Community Composition, Taxonomic Co-Occurrence, and Predicted Functional Attributes. Appl. Microbiol. 2021, 1, 201-224. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1020016
Green GBH, Hakim JA, Chen J-W, Koo H, Morrow CD, Watts SA, Bej AK. The Gut Microbiota of Naturally Occurring and Laboratory Aquaculture Lytechinus variegatus Revealed Differences in the Community Composition, Taxonomic Co-Occurrence, and Predicted Functional Attributes. Applied Microbiology. 2021; 1(2):201-224. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1020016
Chicago/Turabian StyleGreen, George B. H., Joseph A. Hakim, Jiung-Wen Chen, Hyunmin Koo, Casey D. Morrow, Stephen A. Watts, and Asim K. Bej. 2021. "The Gut Microbiota of Naturally Occurring and Laboratory Aquaculture Lytechinus variegatus Revealed Differences in the Community Composition, Taxonomic Co-Occurrence, and Predicted Functional Attributes" Applied Microbiology 1, no. 2: 201-224. https://0-doi-org.brum.beds.ac.uk/10.3390/applmicrobiol1020016