
The Role of Surface in the Pathogenesis and Treatment of COVID-19


1
Department of Emergency Medicine, Camden Clark Medical Center, West Virginia University, Parkersburg, WV 26101, USA
2
Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
*
Author to whom correspondence should be addressed.
COVID 2021, 1(2), 465-471; https://0-doi-org.brum.beds.ac.uk/10.3390/covid1020040
Submission received: 6 September 2021
/
Revised: 29 September 2021
/
Accepted: 1 October 2021
/
Published: 3 October 2021
Abstract
:Recently, an inverse relationship between incidence of COVID-19 and seasonal aerosolization of mold spores was demonstrated. Analyses of that relationship suggested mold spores compete with SARS-CoV-2 virions for a receptor on the pulmonary epithelial surface. By inference, the operative receptor was proposed to be Toll-like receptor 4, with surface-localized virions being responsible for symptomatology. In this report, the pathogenesis of COVID-19 is further developed, with a focus on a role for surfactant protein D in the process. This developed proposal provides both mechanistic understanding and suggested treatments of COVID-19.
{kind=link}
{kind=link}
1. Background, Introduction, and Rationale
The SARS-CoV-2 pandemic has produced a broad spectrum of clinical presentations, from asymptomatic to fatally ill [1]. According to the model espoused here, symptomatic illness is a consequence of the complexation of virions with Toll-like receptor 4 (TLR4), a notion congruent with the findings that: (1) aerosolized mold spores, with which sharply seasonal respiratory viruses appear to compete, have, as their likely receptor, TLR4 [2], (2) TLR4-mediated signaling is upregulated in COVID-19 patients [3], (3) the SARS-CoV-2 spike protein induces pro-inflammatory responses in human leukocytes via TLR4 activation [4], (4) activated TLR4 may increase the expression of angiotensin-converting enzyme 2 (ACE2) [5], a membrane protein generally held responsible for SARS-CoV-2 entry [6], and (5) TLR4 activation is a determinant for viral entry and tropism in other seasonal respiratory viruses [7]. However, if the complexation of SARS-CoV-2 with TLR4 is responsible for symptomatic COVID-19, then, given the existence of so many asymptomatic persons, there must be an immune defense operating to prevent such complexation. And because humans are naïve to the SARS-CoV-2 virus [8], that defense must be innate.
TLR4 is expressed abundantly on the pneumocytes and alveolar macrophages that constitute the pulmonary epithelial surface [9,10]. That surface serves not only as a nidus of SARS-CoV-2 infection, but also—by means of cough—as a platform for viral transmission. In this regard, TLR4 potentiates the activity of transient receptor potential V1 (TRPV1) [11], a receptor implicated in the genesis of cough [12]. Because some seasonal respiratory viruses upregulate TRPV1 in the airways of diseased hosts [13], it seems likely that TLR4 activation benefits them by triggering cough, thereby maximizing the dissemination of viral progeny.
2. Hypothesis
Because: (1) engagement of epithelial TLR4 yields the systemic inflammation characteristic of SARS-CoV-2 infection [3], (2) TRPV1 activation can occur at the level of the pulmonary epithelium [12], and (3) the life cycle of SARS-CoV-2 necessitates persistence of the virus on the environmental face of the pulmonary tree, it appears that the pulmonary epithelial surface is central to COVID-19 pathogenesis. This notion is further supported by the targeting of ACE2, which is expressed primarily on ciliated cells of the respiratory tract [14]. Inasmuch as mucociliary clearance is responsible for removal of virions from the epithelial surface [15], its disruption ensures accumulation and—again by means of cough—maximal aerosolization and the transmission of infectious materials. Although the virus demonstrates tissue tropism for some organs [16], early reports suggested that most hospitalized patients did not have detectable levels of viral materials in their sera [17]. Although a more recent report has demonstrated viral materials in 50–60% of sera of moderate-to-critically ill patients, no association has yet been shown between serum detection and the development of multiorgan dysfunction syndrome, either prior to admission or during the first 24 h of intensive care [18]. Overall, the evidence suggests that viremia is not a requirement for either symptomatology or severe disease.
Observations made early in the pandemic indicate that COVID-19 severity is a function of viral dose [19], an indication supported by an animal model [20] and elaborated upon by others [21]. Given both the dose dependence and the certain existence of an innate defense, symptomatic SARS-CoV-2 infection must be a consequence of a viral dose that exceeds temporally the capacity of the innate defense. Because available evidence suggests that symptomatic COVID-19 involves the engagement of TLR4 on pulmonary epithelium, the innate defense operating to prevent engagement likely involves an effector native to the pulmonary epithelial surface.
3. Discussion
Innate effectors active on pulmonary epithelium especially feature pulmonary surfactant, a lipoprotein complex comprised of 90% lipid and 10% protein. Although the biophysical function of pulmonary surfactant in preventing alveolar collapse is well-understood, the immunological function of it remains actively investigated. Essential to the operation of pulmonary surfactant are surfactant proteins A (SP-A) and D (SP-D), collagen-containing C-type lectins, or collectins, expressed constitutively by pneumocytes [22]. They are involved in the clearance of sharply seasonal respiratory viruses [23,24], ones proposed to elicit disease by engaging TLR4 [2]. More specifically, SP-A and -D have affinities for viral fusion proteins [25], e.g., the SARS-CoV-2 spike proteins [26,27].
Although both SP-A and SP-D have roles in defense against respiratory viruses, available evidence prioritizes SP-D in the defense against coronaviruses [28,29,30]. As examples of that priority, SP-D binds the SARS-CoV-1 spike protein with higher affinity than does SP-A [28]. That binding, in turn, promotes the recognition of SARS-CoV-1 by dendritic cells [28]. SP-D also binds the spike protein of SARS-CoV-2, preventing cellular entry and replication [29,30]. Importantly, SP-D binds TLR4 [31] and can alter its interaction with pathogen-derived ligands [32], including, perhaps, the SARS-CoV-2 spike protein [33]. Consistent with these findings, SP-D directs TLR4-mediated inflammation [34]. Finally, SP-D regulates the expression of pulmonary surfactant phospholipid [35], anionic species of which modulate interactions between pathogen-derived ligands and TLR4 [36]. Given the roles of SP-D in modulating pathogen-directed TLR4 signaling and the role, proposed here, of TLR4 activation in COVID-19, it comes as no surprise that lung biopsies of COVID-19 patients show hypertrophy and hyperplasia of type II pneumocytes [37], cells responsible for SP-D production and recycling.
Because SP-D is such a key player, it is reasonable to propose COVID-19 severity depends on SP-D availability relative to the load of SARS-CoV-2: a low dose of virion is readily neutralized, yielding the asymptomatic state, whilst a high dose of virion—one in excess of SP-D availability—is not neutralized, yielding the symptomatic state. Persons with low ambient levels of SP-D, i.e., smokers [38], the obese [39], or the pregnant [40], should be most susceptible to severe COVID-19 [41,42,43]. Moreover, because youths have larger intracellular and alveolar surfactant pools, as well as a much higher rate of SP-D recycling compared to the elderly [44], advanced age, too, should be a risk factor for disease severity [45].
Regarding SP-D availability, the use of steroids to treat COVID-19 [46] is also noteworthy: the critically ill benefit most, with a trend toward harm in those less sick [47]. Interestingly, steroids upregulate SP-D production [48]. Even as corticosteroid increases SP-D levels, insulin reduces them [48], providing rationale for diabetes as a risk factor for severe COVID-19 [49].
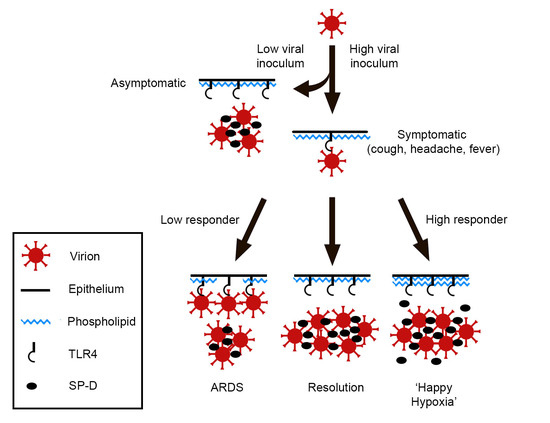
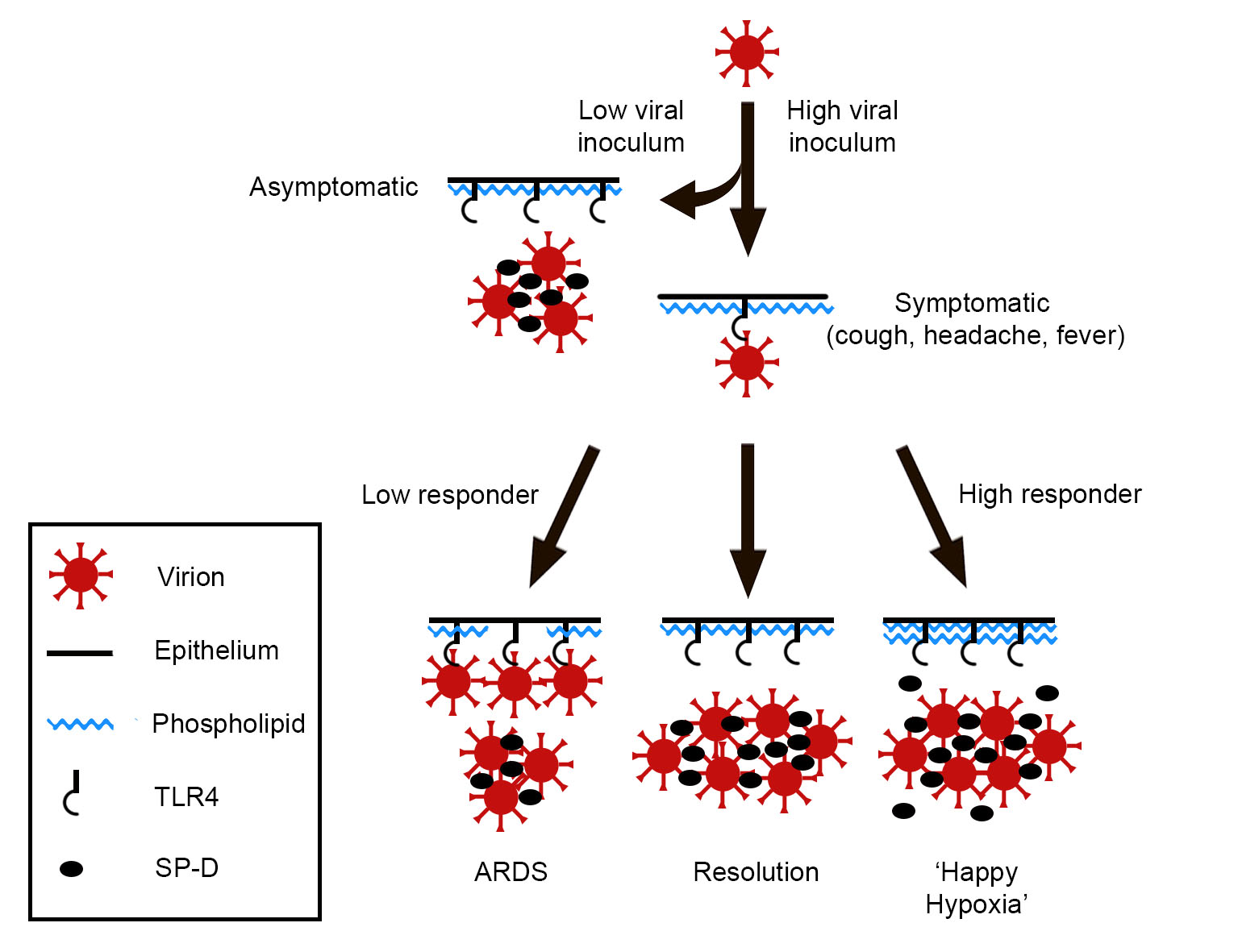
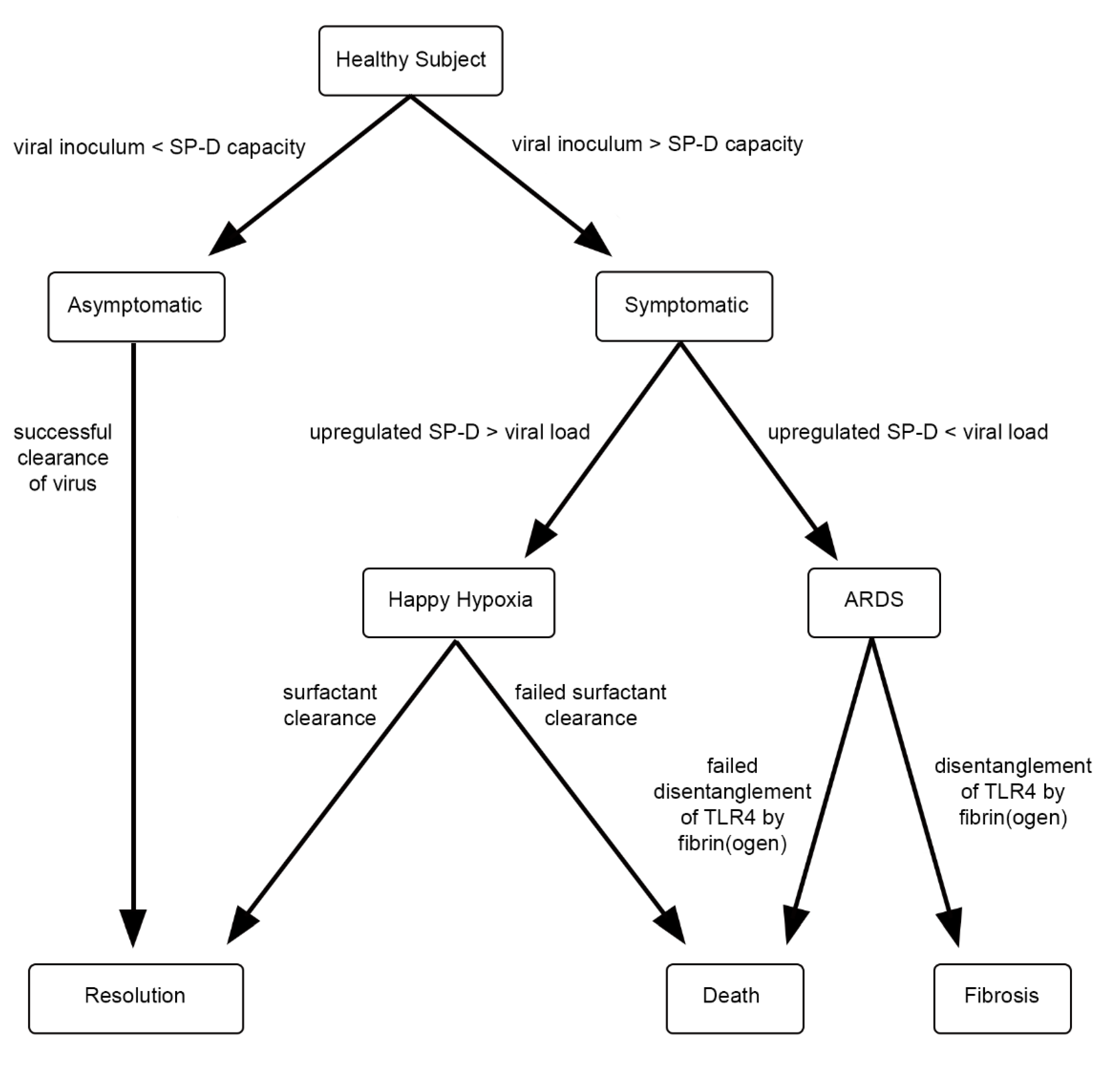
One can imagine that, under homeostatic conditions, the ambient level of SP-D accommodates both respiration and innate immune function. However, if, for example, a SARS-CoV-2 ‘dose’ is a large one, the availability of SP-D increases commensurately to accommodate the viral threat. The physiological response, then, is a function of both viral burden—exogenous and endogenous—and surfactant availability, Figure 1.
An abrupt increase of pulmonary surfactant within alveoli affects oxygenation, so symptomatic individuals tend toward ‘happy hypoxia’ if the compensatory response is overexuberant. In contrast, if the surfactant response is inadequate, viral activation of TLR4 continues unabated, resulting in cytokine storm and, ultimately, acute respiratory distress syndrome (ARDS). Interestingly, SARS-CoV-2-induced ARDS is attended by increased vascular permeability, with deposition of fibrin in alveolar spaces [50]. It is tempting to speculate that fibrin(ogen), a ligand of TLR4 [51], competes with virions for that receptor, thereby mitigating viral infectivity. However, because many SARS-CoV-2 patients in ARDS progress to pulmonary fibrosis [52,53], such a measure, if operative, is costly.
Not only serologic, but also radiographic, findings implicate abnormal levels of pulmonary surfactant in severe COVID-19. The rare entity, pulmonary alveolar proteinosis (PAP), involves the impaired clearance of surfactant from pulmonary epithelium [54]. A characteristic feature is the computed tomographic finding of interlobular septal thickening in a ‘crazy paving’ pattern [54]. This finding is also characteristic of COVID-19 [55]. Although the mechanisms responsible may be different, the net result in both cases is the increased deposition of surfactant on pulmonary epithelium. That increased deposition, in turn, affects surface tension, the consequences of which are consolidation and decreased oxygenation, manifesting as dyspnea and hypoxia.
4. Closing
Just as does treatment of PAP, treatment of severe COVID-19 should address disease pathology where it occurs, on the environmental-facing surface of pulmonary epithelium. Symptomatic COVID-19 due to unmitigated TLR4 activation might be best treated using nebulized materials, e.g., recombinant surfactant proteins or, perhaps, TLR4 antagonists or even the C-terminus of the fibrinogen γ-chain [56,57,58,59]. Because severe hypoxia associated with COVID-19 may be derived from either of two mechanisms, therapy should be tailored accordingly. For the non-inflammatory hypoxic state, due to upregulated surfactant production and the accumulation of virion–surfactant protein aggregates, serial whole lung lavage might prove therapeutic. Although such therapy has not yet been standardized for PAP, various protocols are well-tolerated and routine treatment for the condition [54]. Because critically ill COVID-19 patients might not tolerate whole lung lavage, their therapy could proceed incrementally, via the sequential decontamination of individual lobes. Those whose pulmonary function has already been circumvented by extracorporeal membrane oxygenation might be suited for more aggressive lavage therapy. For the inflammatory hypoxic state involving unmitigated TLR4 activation and ARDS, lavage fluid could be supplemented with viral binding agents or TLR4 antagonists. Differentiating between the two states should be possible by quantifying downstream markers of TLR4 activation, such as IL-6 [60], the plasma levels of which have prognostic value [61]. Because the pathophysiology of other sharply seasonal viruses relates to TLR4 engagement, their therapies might exploit similar approaches.
Author Contributions
Conceptualization, A.C.R.; Formal analysis, A.C.R. and G.S.R.; Visualization, A.C.R.; Writing—original draft, A.C.R.; Writing—review and editing, G.S.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by departmental funds from the Department of Pathology, Feinberg School of Medicine, Northwestern University.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eythorsson, E.; Helgason, D.; Ingvarsson, R.F.; Bjornsson, H.K.; Olafsdottir, L.B.; Bjarnadottir, V.; Runolfsdottir, H.L.; Bjarnadottir, S.; Agustsson, A.S.; Oskarsdottir, K.; et al. Clinical spectrum of coronavirus disease 2019 in Iceland: Population based cohort study. BMJ 2020, 371, m4529. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.B.; Shah, R.D.; Retzinger, D.G.; Retzinger, A.C.; Retzinger, D.A.; Retzinger, G.S. Competing bioaerosols may influence the seasonality of influenza-like illnesses, including COVID-19. The Chicago experience. Pathogens 2021, 10, 1204. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.M.; Lee, S.-G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J. Korean Med Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41 [published correction appears in Cell Res. 27 April 2021]. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-Like Receptor 4-Mediated Activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Armstrong, L.; Medford, A.; Uppington, K.M.; Robertson, J.; Witherden, I.R.; Tetley, T.D.; Millar, A.B. Expression of functional toll-like receptor-2 and -4 on alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Juarez, E.; Nuñez, C.; Sada, E.; Ellner, J.J.; Schwander, S.K.; Torres, M. Differential expression of Toll-like receptors on human alveolar macrophages and autologous peripheral monocytes. Respir. Res. 2010, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.; Cho, W.-H.; Lee, H.; Choi, B.; Kim, Y.-J.; Lee, H.; Joo, Y.; Jung, S.J.; Choi, S.-Y.; Lee, S.; et al. Association of TRPV1 and TLR4 through the TIR domain potentiates TRPV1 activity by blocking activation-induced desensitization. Mol. Pain 2018, 14, 1744806918812636. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H. The emerging role of TRPV1 in airway inflammation. Allergy Asthma Immunol. Res. 2018, 10, 187–188. [Google Scholar] [CrossRef] [Green Version]
- Omar, S.; Clarke, R.; Abdullah, H.; Brady, C.; Corry, J.; Wintrt, H.; Touzelet, O.; Power, U.F.; Lundy, F.; McGarvey, L.P.A.; et al. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE 2017, 12, e0171681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.T.; Nakayama, T.; Wu, C.-T.; Goltsev, Y.; Jiang, S.; Gall, P.A.; Liao, C.-K.; Shih, L.-C.; Schürch, C.M.; McIlwain, D.R.; et al. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Kuek, L.E.; Lee, R.J. First contact: The role of respiratory cilia in host-pathogen interactions in the airways. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L603–L619. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Serrano, D.A.; Roy-Vallejo, E.; Cruz, N.D.Z.; Ramírez, A.M.; Rodríguez-García, S.C.; Arevalillo-Fernández, N.; Galván-Román, J.M.; García-Rodrigo, L.F.; Vega-Piris, L.; Llano, M.C.; et al. Detection of SARS-CoV-2 RNA in serum is associated with increased mortality risk in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guallar, M.P.; Meiriño, R.; Donat-Vargas, C.; Corral, O.; Jouvé, N.; Soriano, V. Inoculum at the time of SARS-CoV-2 exposure and risk of disease severity. Int. J. Infect. Dis. 2020, 97, 290–292. [Google Scholar] [CrossRef]
- Ryan, K.A.; Bewley, K.R.; Fotheringham, S.A.; Slack, G.S.; Brown, P.; Hall, Y.; Wand, N.I.; Marriott, A.C.; Cavell, B.E.; Tree, J.A.; et al. Dose-dependent response to infection with SARS-CoV-2 in the ferret model and evidence of protective immunity. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Van Damme, W.; Dahake, R.; van de Pas, R.; Vanham, G.; Assefa, Y. COVID-19: Does the infectious inoculum dose-response relationship contribute to understanding heterogeneity in disease severity and transmission dynamics? Med Hypotheses 2020, 146, 110431. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Mallampalli, R.K. The role of surfactant in lung disease and host defense against pulmonary infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.M.; Whitsett, J.A.; Hartshorn, K.; Crouch, E.C.; Korfhagen, T.R. Surfactant protein D enhances clearance of influenza a virus from the lung in vivo. J. Immunol. 2001, 167, 5868–5873. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.M.; Elliott, J.; Whitsett, J.A.; Srikiatkhachorn, A.; Crouch, E.; DeSilva, N.; Korfhagen, T. Surfactant protein-D enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2004, 31, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Phipps, M.J.; Clark, H.W.; Skylaris, C.-K.; Madsen, J. Surfactant proteins A and D: Trimerized innate immunity proteins with an affinity for viral fusion proteins. J. Innate Immun. 2018, 11, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Leth-Larsen, R.; Zhong, F.; Chow, V.; Holmskov, U.; Lu, J. The SARS coronavirus spike glycoprotein is selectively recognized by lung surfactant protein D and activates macrophages. Immunobiology 2007, 212, 201–211. [Google Scholar] [CrossRef]
- Arroyo, R.; Grant, S.; Kingma, P. Late breaking abstract—The innate immune collectin surfactant protein SP-D binds to SARS-CoV-2 spike-protein. Eur. Resp. J. 2020, 56, 1055. [Google Scholar]
- Wu, Y.P.; Liu, Z.H.; Wei, R.; Pan, S.D.; Mao, N.Y.; Chen, B.; Han, J.J.; Zhang, F.S.; Holmskov, U.; Xia, Z.L.; et al. Elevated Plasma Surfactant Protein D (SP-D) levels and a direct correlation with anti-severe acute respiratory syndrome coronavirus-specific IgG antibody in SARS patients. Scand. J. Immunol. 2009, 69, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.H.; Beirag, N.; Murugaiah, V.; Chou, Y.C.; Kuo, W.S.; Kao, H.F.; Madan, T.; Kishore, U.; Wang, J.Y. Human Surfactant Protein D Binds Spike Protein and Acts as an Entry Inhibitor of SARS-CoV-2 Pseudotyped Viral Particles. Front Immunol. 2021, 12, 641360. [Google Scholar] [CrossRef]
- Madan, T.; Biswas, B.; Varghese, P.M.; Subedi, R.; Pandit, H.; Idicula-Thomas, S.; Kundu, I.; Rooge, S.; Agarwal, R.; Tripathi, D.M.; et al. A Recombinant Fragment of Human Surfactant Protein D Binds Spike Protein and Inhibits Infectivity and Replication of SARS-CoV-2 in Clinical Samples. Am J Respir Cell Mol Biol. 2021, 65, 41–53. [Google Scholar] [CrossRef]
- Ohya, M.; Nishitani, C.; Sano, H.; Yamada, C.; Mitsuzawa, H.; Shimizu, T.; Saito, T.; Smith, K.; Crouch, E.; Kuroki, Y. Human pulmonary surfactant protein D binds the extracellular domains of toll-like receptors 2 and 4 through the Carbohydrate Recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry 2006, 45, 8657–8664. [Google Scholar] [CrossRef]
- Yamazoe, M.; Nishitani, C.; Takahashi, M.; Katoh, T.; Ariki, S.; Shimizu, T.; Mitsuzawa, H.; Sawada, K.; Voelker, D.R.; Takahashi, H.; et al. Pulmonary Surfactant Protein D Inhibits Lipopolysaccharide (LPS)-induced Inflammatory cell responses by altering lps binding to its receptors. J. Biol. Chem. 2008, 283, 35878–35888. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Saka, R.; Wakimoto, T.; Nishiumi, F.; Sasaki, T.; Nose, S.; Fukuzawa, M.; Oue, T.; Yanagihara, I.; Okuyama, H. Surfactant protein-D attenuates the lipopolysaccharide-induced inflammation in human intestinal cells overexpressing toll-like receptor 4. Pediatr. Surg. Int. 2015, 32, 59–63. [Google Scholar] [CrossRef]
- Korfhagen, T.R.; Sheftelyevich, V.; Burhans, M.S.; Bruno, M.D.; Ross, G.F.; Wert, S.E.; Stahlman, M.T.; Jobe, A.H.; Ikegami, M.; Whitsett, J.A.; et al. Surfactant protein-D regulates surfactant phospholipid homeostasis in vivo. J. Biol. Chem. 1998, 273, 28438–28443. [Google Scholar] [CrossRef] [Green Version]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef] [Green Version]
- Borczuk, A.C.; Salvatore, S.P.; Seshan, S.V.; Patel, S.S.; Bussel, J.B.; Mostyka, M.; Elsoukkary, S.; He, B.; DEL Vecchio, C.; Fortarezza, F.; et al. COVID-19 pulmonary pathology: A multi-institutional autopsy cohort from Italy and New York City. Mod. Pathol. 2020, 33, 1–13. [Google Scholar] [CrossRef]
- Winkler, C.; Atochina-Vasserman, E.N.; Holz, O.; Beers, M.F.; Erpenbeck, V.J.; Krug, N.; Roepcke, S.; Lauer, G.; Elmlinger, M.; Hohlfeld, J.M. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir. Res. 2011, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawed, S.; Mannan, N.; Qureshi, M.A. Association of surfactant protein-D with obesity. J. Ayub. Med. Coll. Abbottabad JAMC 2017, 28, 489–492. [Google Scholar]
- Kale, K.; Vishwekar, P.; Balsarkar, G.; Jassawalla, M.J.; Alkahtani, S.; Kishore, U.; Sawant, G.; Madan, T. Serum levels of collectins are sustained during pregnancy: Surfactant protein D levels are dysregulated prior to missed abortion. Reprod. Sci. 2020, 27, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Alberca, R.W.; Lima, J.C.; de Oliveira, E.A.; Gozzi-Silva, S.C.; Ramos, Y.L.; Andrade, M.M.D.S.; Beserra, D.R.; Oliveira, L.D.M.; Branco, A.C.C.C.; Pietrobon, A.J.; et al. COVID-19 Disease course in former smokers, smokers and COPD patients. Front. Physiol. 2021, 11, 637627. [Google Scholar] [CrossRef]
- Ellington, S.; Strid, P.; Tong, V.T.; Woodworth, K.; Galang, R.R.; Zambrano, L.D.; Nahabedian, J.; Anderson, K.; Gilboa, S.M. Characteristics of women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–June 7, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 769–775. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, T.; Wang, Y.; Xia, L. The centrality of obesity in the course of severe COVID-19. Front. Endocrinol. 2021, 12, 620566. [Google Scholar] [CrossRef]
- Notter, R.H. Lung Surfactants: Basic Science and Clinical Applications; Dekker, M., Ed.; CRC Press: New York, NY, USA, 2008; pp. 140–142. [Google Scholar]
- Jiang, N.; Liu, Y.N.; Bao, J.; Li, R.; Ni, W.-T.; Tan, X.-Y.; Xu, Y.; Peng, L.-P.; Wang, X.-R.; Zeng, Y.-M.; et al. Clinical features and risk factors associated with severe COVID-19 patients in China [published online ahead of print, 1 April 2021. Chin. Med. J. 2021, 134, 944. [Google Scholar] [CrossRef]
- Sterne, J.A.C.; Murthy, S.; Diaz, S.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; Cavalcanti, A.B.; et al. WHO rapid evidence appraisal for COVID-19 therapies (REACT) working group. Association between administration of systemic corticosteroids and mortality among critically Ill patients with COVID-19: A meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef]
- The RECOVERY Collaborative Group dexamethasone in hospitalized patients with Covid-19. New. Engl. J. Med. 2021, 384, 693–704. [CrossRef]
- Sorensen, G.L. Surfactant protein D in respiratory and non-respiratory diseases. Front. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.; Lim, M.A.; Pranata, R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumonia—A systematic review, meta-analysis, and meta-regression. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.S.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris, J.S.M.; Chu, C.M.; Cheng, V.; Chan, K.; Hung, I.F.N.; Poon, L.; Law, K.; Tang, B.; Hon, T.; Chan, C.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive factors and risk reduction strategies. Pulm. Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Salvaterra, E.; Campo, I. Pulmonary alveolar proteinosis: From classification to therapy. Breathe 2020, 16, 200018. [Google Scholar] [CrossRef]
- Gillespie, M.; Flannery, P.; Schumann, J.A.; Dincher, N.; Mills, R.; Can, A. Crazy-Paving: A computed tomographic finding of coronavirus disease 2019. Clin. Pr. Cases Emerg. Med. 2020, 4, 461–463. [Google Scholar] [CrossRef]
- Kawamoto, T.; Ii, M.; Kitazaki, T.; Iizawa, Y.; Kimura, H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 2008, 584, 40–48. [Google Scholar] [CrossRef]
- Raja, S.G.; Dreyfus, G.D. Eritoran: The evidence of its therapeutic potential in sepsis. Core Évid. 2008, 2, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Neal, M.D.; Jia, H.; Eyer, B.; Good, M.; Guerriero, C.; Sodhi, C.P.; Afrazi, A.; Prindle, T., Jr.; Ma, C.; Branca, M.; et al. Discovery and Validation of a New Class of Small Molecule Toll-Like Receptor 4 (TLR4) Inhibitors. PLoS ONE 2013, 8, e65779. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, R.F.; McNamara, K.; Lin, K. Correlating structure and function during the evolution of fibrinogen-related domains. Protein Sci. 2012, 21, 1808–1823. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Wardill, H.R.; Bowen, J.M. Role of toll-like receptor 4 (TLR4)-mediated interleukin-6 (IL-6) production in chemotherapy-induced mucositis. Cancer Chemother. Pharmacol. 2018, 82, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.S.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Gil Castro, A.; Silvestre, R. Interleukin-6 is a biomarker for the development of fatal severe acute respiratory syndrome coronavirus 2 pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Proposed progression of COVID-19 pulmonary disease.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Retzinger, A.C.; Retzinger, G.S. The Role of Surface in the Pathogenesis and Treatment of COVID-19. COVID 2021, 1, 465-471. https://0-doi-org.brum.beds.ac.uk/10.3390/covid1020040
AMA Style
Retzinger AC, Retzinger GS. The Role of Surface in the Pathogenesis and Treatment of COVID-19. COVID. 2021; 1(2):465-471. https://0-doi-org.brum.beds.ac.uk/10.3390/covid1020040
Chicago/Turabian StyleRetzinger, Andrew C., and Gregory S. Retzinger. 2021. "The Role of Surface in the Pathogenesis and Treatment of COVID-19" COVID 1, no. 2: 465-471. https://0-doi-org.brum.beds.ac.uk/10.3390/covid1020040