
Metal Binding Proteins

Institute for Biological Instrumentation of the Russian Academy of Sciences, Federal Research Center Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences, Pushchino, 142290 Moscow, Russia
Encyclopedia 2021, 1(1), 261-292; https://0-doi-org.brum.beds.ac.uk/10.3390/encyclopedia1010024
Submission received: 20 January 2021
/
Revised: 17 February 2021
/
Accepted: 12 March 2021
/
Published: 15 March 2021
Definition
:Metal ions play several major roles in proteins: structural, regulatory, and enzymatic. The binding of some metal ions increase stability of proteins or protein domains. Some metal ions can regulate various cell processes being first, second, or third messengers. Some metal ions, especially transition metal ions, take part in catalysis in many enzymes. From ten to twelve metals are vitally important for activity of living organisms: sodium, potassium, magnesium, calcium, manganese, iron, cobalt, zinc, nickel, vanadium, molybdenum, and tungsten. This short review is devoted to structural, physical, chemical, and physiological properties of proteins, which specifically bind these metal cations.
1. Introduction
Metal ions play an extremely important role in functioning of all, without any exceptions, biological systems. Intracellular and extracellular fluids contain high concentrations of metal ions. Many biological events critically depend on metal ions. Metal ions interact with all charged and polar groups of biopolymers contacting with biological liquids, but their specific strong interactions with proteins play the most important role. More than a quarter of all the proteins contained in Protein Data Bank, possess bound metal ions. Most of all, these proteins contain bound zinc ions, followed by iron, calcium, and magnesium. In the modern literature, one can find quite a few reviews devoted to metal binding proteins (see, for example, [1,2,3]). This review aims to provide a general understanding of the structural, physicochemical, and functional properties of metal binding proteins.
Metal ions play three fundamental roles in proteins: structural, regulatory, and enzymatic. The binding of some metal ions stabilize proteins or protein domains. Some metal ions can take part in regulation of various cell processes being first, second, or third messengers. Some metal ions, especially transition metal ions, take part in catalysis in many enzymes. Between ten and twelve metals are vital for living organisms: sodium (Na), potassium (K), magnesium (Mg), calcium (Ca), manganese (Mn), iron (Fe), cobalt (Co), zinc (Zn), nickel (Ni), vanadium (V), molybdenum (Mb), and tungsten (W). Sometimes these metals are called “life metals”. Below (Table 1) is a fragment of Mendelleev periodic table of elements, which shows positions of “life metals”.
Most of the biologically significant metals are located in the fourth period of the periodic table, except sodium and magnesium, which are in the third period, and molybdenum and tungsten, which are in the fifth and sixth periods, respectively. The biologically significant metals are divided into two groups: nontransition elements (Na, K, Mg, Ca, Zn) and transition elements (Mn, Fe, Co, Cu, Mo, W). Nontransition elements have constant oxidation state (valency) and their ions have completely filled electron shells. In contrast, transition elements have variable oxidation state (valency) and their ions have incompletely filled electron shells. Incompletely filled electron shells are reflected in special physical and chemical properties (absorption bands in ultraviolet and visible spectral regions, paramagnetism, and so on). Sometimes such properties facilitate their studies. Zinc is not a transition element since it has completely filled d-orbitals, at the same time Zn properties are very different from the properties of the rest of the nontransition metal ions. Below (Table 2) is a table of ionic radii of biologically significant metals in various oxidation states.
Na and К possess one s-electron on the outer shell besides the electron structure of rare gas atoms ([Ne] 3s1 and [Ar] 4s1, respectively). For this reason, they possess low ionization potentials and rather large size of their ions (0.95 and 1.33 Å, respectively). Mg and Ca are characterized by completely filled electron shells (2s2 2p6 and 3s2 3p6); therefore, they have no preferences in respect to the direction of bonds formation. Their ionic radii are 0.65 and 0.99 Å, respectively.
Dipole water molecules, which surround metal ions in water solution, are oriented by the electric field of the ions (charge-dipole interaction). At least three layers of water molecules are influenced by the central ion. Geometry of the first hydration shell of calcium and monovalent metal cations can have various geometry. Some ions possess rather rigid and stable first hydration shell, which can have tetrahedral or octahedral (Mg2+, Co2+, Ni2+) geometry. Zn2+ ion can have both tetrahedral and octahedral geometry of the first hydration shell.
2. Structure of Metal Binding Sites
A structure consisting of a metal ion bonded to a group of surrounding molecules or groups is called coordination complex. Molecules or groups bound to the metal ion in the complex are called ligands. Ligands are usually negatively charged or polar molecules or groups. The central ion and bound ligands with negatively charged electron donor atoms form a coordination sphere. The number of donor atoms bound to the metal ion is called coordination number. A ligand with a single donor atom is called monodentate ligand. There exist ligands with two (bidentate ligand) or more donor atoms, which can be simultaneously coordinated by the metal ion (polydentate ligands or chelators).
In proteins, Ca2+, Mg2+, Na+, and K+, are practically always bound to “hard” oxygen ligands but not to nitrogen or sulfur ligands (hard ligands have more concentrated, less polarizable electron shells). This interaction is purely electrostatic without preferential directions. In contrast to this, Zn2+ and transition metal ions interact with nitrogen and sulfur ligands as well as with oxygen ligands. The bonds are formed in preferential directions because of their partially covalent character.
The interaction of metal ions with a protein is characterized by equilibrium dissociation constant, Kd (M), which is a ratio of dissociation (off) and association (on) rate constants koff/kon (s−1/M−1 s−1). Sometimes it is useful to use equilibrium association (binding) constant Ka = 1/Kd (M−1).
In the strongest calcium binding sites of proteins the coordinating oxygen atoms (provided by carboxylates of Asp and Glu amino acid residues and carbonyls of polypeptide chain) are located in vertexes of a slightly distorted pentagonal bipyramide or octahedron (reviewed in [1,2,4]). In pentagonal bipyramide five oxygen atoms are located in equatorial plane at a distance of 2.5 Å from each other. This is a rather tight packing when the oxygens try to push each other from equatorial plane.
Atomic radius of Mg2+ is 0.65 Å, while atomic radius of oxygen is 0.99 Å. For this reason, only six oxygen atoms can be located in the octahedral geometry in the vicinity of Mg2+. Due to essentially higher charge density of Mg2+ compared to Ca2+, the energy of the single Me-O bond seems to be higher for magnesium than for calcium. Water molecules dissociate from aquo-complexes of magnesium essentially slower compared to calcium. Upon Mg2+ binding to Ca2+ binding sites the protein should fit its six oxygen ligands to the vertexes of almost ideal octahedron. Mg2+ binding sites in proteins contain at least one carboxylate ligand coordinating Mg2+ ion in monodentate fashion. Since atomic radius of Ca2+ is larger, it has a tendency for bidentate binding of carboxylates but lower affinity to water.
Figure 1 shows the structure of the strong calcium binding site in human α-lactalbumin. Coordinating oxygen atoms are located in vertexes of a distorted pentagonal bipyramide and belong to side chains of aspartic acids and carbonyls of the skeletal polypeptide chain. Such arrangement is very typical for many calcium binding proteins.
In contrast to Mg2+ and Ca2+, Zn2+ prefers “more soft” ligands such as sulfur atoms of Cys and nitrogen atoms of His (soft ligands have less concentrated, more polarizable electron shells), though it can be coordinated also by oxygen atoms of Asp and Glu (reviewed in [4,5]). In such proteins as the proteins with zinc fingers and zinc-containing enzymes, Zn2+ is coordinated tertahedrically, but sometimes it can show 5- or 6-coordination geometry as well. Figure 2 shows the structure of the zinc binding site in Zn-finger protein. In this case zinc ion is coordinated by nitrogen atoms of imidazol rings of histidines and sulfur atoms of cysteines. Ions of transition metals also prefer “soft” ligands such as sulfur atoms of Cys and nitrogen atoms of His. Figure 3 demonstrates the structure of tetrahedral copper binding site in azurin, as an example of the binding sites of transition metal ions. Histidine, cysteine, and methionin residues take part in the binding of transition metal ions.
Metal binding sites are usually located in the environment with low dielectric constant inside cavities and clefts of protein molecules. Such location strengthens electrostatic interactions of metal ion and its ligands. Coordination numbers of metal ions in metalloproteins are within the region from 3 to 8, with 6 being the most usual.
Specificity of a binding site depends on ligands type, coordination number, interactions of side chains, and total charge and shape of the binding site cavity. In proteins, selectivity of a binding site to a metal ion is usually inversely proportional to the concentration of this ion in the environment in which the protein functions. For instance, Mg2+-binding sites are not very specific to Mg2+, concentration of which is very high in biological fluids.
Many enzymes require metal ions for their activity. Such enzymes are divided into two groups. In metalloenzymes metal ions serve as prosthetic groups, while in metal activated enzymes metal ions are used as cofactors. Cofactors perform functions similar to functions of prosthetic groups but bind in a transient, dissociable manner either to the enzyme or to a substrate. Some organic molecules functioning as cofactors can bind metal ions.
3. Calcium Binding Proteins
Calcium accounts for approximately 1.5% of total body weight. Furthermore, 99% of the calcium is located in bones and teeth, while the remaining 1% is distributed in other tissues.
The resting concentrations of intracellular free Ca2+ (~10−7 M) is 104 times lower than its extracellular concentration. In response to external stimuli, the intracellular concentration of Ca2+ can increase and in this case Ca2+ can act as a second messenger. Ca2+ moves into the intracellular milieu either from outside the cell (via plasma-membrane Ca2+ channels) or from intracellular stores. The Ca2+ is released in aliquots called sparks, puffs, or waves. After this short increase in intracellular Ca2+ concentration intracellular Ca2+-binding proteins and Ca2+ pumps immediately start to transport it to intracellular storage sites or outside the cell. For this reason, the cytosolic level of Ca2+ generally only raises 10-fold to a concentration of ~10−6 M and never approaches its extracellular level.
Calcium binding proteins of cytosol and membrane calcium binding proteins directed to cytosol are called calcium-modulated proteins since they take part in generation of calcium gradient and/or transfer external impulse information (reviewed in [6,7]). Some intracellular calcium binding proteins bind calcium and change their conformation, and this gives them the ability to interact with their target proteins. Such proteins are called calcium sensor proteins.
3.1. EF-Hand Proteins
EF-hand proteins contain EF-hand calcium binding domains (reviewed in [1,2,3]). These domains were first discovered by means of X-ray analysis in parvalbumin [8]. Parvalbumin molecule consists of six helices A to D. The loops between C and D helices and between E and F helices form calcium binding sites. The structure E-helix—calcium binding loop—F-helix was called EF-hand domain (Figure 4).
Calcium binding loop of canonical EF-hand domain consists of 12 residues, beginning with Asp and ending with Glu. In most EF-hand proteins Ca2+ ion is coordinated by seven oxygens arranged in pentagonal bipyramide. Such geometry can be achieved in several different ways: Ca2+-binding loop can have 11 residues (calpain, grancalcin, ALG-2) and 13 residues (SPARC, osteonectin, BM-40). The EF-hand domains occur in proteins usually in adjacent pairs. The pairs form compact globular structures. Most of the known EF-hand proteins consist of one to six EF-hand domain pairs. The only exceptions are the fifth domains of penta-EF-hand proteins and the AB-domain of parvalbumin.
Classical EF-hand calcium binding protein is parvalbumin (reviewed in [1,2,9,10]). Parvalbumin is supposed to play the role of a soluble relaxation factor in fast-twitch skeletal muscles; however, it was found also in other tissues including the central nervous system, testis, kidney, and several endocrine glands. It has very high affinity to Ca2+ (Kd 10−6–10−9 M). Parvalbumin is composed of three EF-hand domains, one of which, AB domain, is spoiled by a deletion in its loop and does not bind calcium.
Calmodulin is the most famous EF-hand calcium sensor protein (reviewed in [1,2]). Calmodulin is a highly conserved, soluble, intracellular, Ca2+-binding protein, which is present in all eucariotic cells. This protein activates many enzymes and regulates many cellular functions. Most of calmodulin interactions with its target proteins occur only in the presence of Ca2+ ions, but some of them take place only in the absence of Ca2+ ions. Calmodulin molecule resembles a dumbbell: two globular lobes connected by a long eight-turn α-helix [11]. Each globular lobe is composed of two EF-hand calcium binding domains (Figure 5).
Ca2+ binding causes a change in calmodulin conformation opening its both globular domains and exposing hydrophobic surfaces that form binding sites for the target enzymes [12,13].
Molecule of troponin C, the closest relative of calmodulin, has a similar dumbbell shape. Troponin C is a Ca2+ binding subunit of troponin complex, which triggers a change in the disposition of the troponin-tropomyosin complex on actin fibrils [14].
S100 proteins (this protein family consists of at least 25 members) also possess the EF-hand calcium binding domains (reviewed in [15,16]). S100 monomer has two EF-hand Ca2+ binding domains interconnected by a hinge region. Most of the S100 proteins form antiparallel packed homodimers (in some cases heterodimers). The S100 proteins are exclusively expressed in vertebrates. Many functional roles were proposed for S100 proteins and many human diseases are associated with their altered expression (cancer, neurodegenerative diseases, cardiomyopathies, inflammations, diabetes, and allergies).
One more family of EF-hand Ca2+-binding proteins is the neuronal calcium sensor (NCS) protein family (recoverin, frequenin, VILIPS, Kv-channel interacting protein, guanylate cyclase-activating protein) (reviewed in [7,17,18]). In contrast to the dumbbell structure of calmodulin and troponin C, the four EF hands of recoverin form a compact globule. Only EF hands 2 and 3 of recoverin can bind calcium ions, while the other two EF hands have structural features that prevent calcium binding. NCS proteins are expressed in retinal photoreceptors or neurons and neuroendocrine cells and this fact suggests that they seem to have specialized roles in these cell types (reviewed in [7,17,18]). For example, it has been revealed that at high Ca2+ levels recoverin inhibits rhodopsin kinase, which results in a prolongation the lifetime of photoexcited rhodopsin.
Among the subfamilies of the EF-hand family of proteins, there exist very unusual ones. One of them is the subfamily of photoproteins including Ca2+-activated photoprotein from jellyfish Aequorea salvatio, aequorin, and photoprotein from the hydroid Obelia and some other photoproteins (halistaurin, phialidin, clytin, mitrocomin) (reviewed in [19,20]). Aequorin has a covalently linked prosthetic group, coelenterazine, a substituted imidozolopyrazine. The binding of Ca2+ to three EF-hand domains of aequorin induces a conformational change resulting in the oxidation of coelenterazine to coelenteramide and the emission of blue light. It was suggested that bioluminescence may provide some advantages for the survival of marine organisms.
3.2. Annexins
Annexins form a family of ubiquitous intra- and extracellular calcium- and phospholipid-binding proteins (reviewed in [21,22]). Annexins have specific repeatable motifs, called “annexin folds”, which regulate calcium-dependent interactions with membrane lipids. Annexin molecule consists of a conserved core region formed by four (in human annexins A1–A5, A7, A8, A11 and A13) or eight (only in annexin A6, Figure 6) repeated amino acid sequences and a variable regulatory N-terminal region. The core region contains the calcium and phospholipid binding sites. Each repeat contains five (A–E) α-helices and usually has a characteristic calcium binding “type 2” motif with the sequence “GxGT-[38 residues]-D/E”.
Despite their structural similarities, annexins perform various physiological functions taking part in various cellular activities including vesicle trafficking, cell division, apoptosis, and growth regulation. The main feature of these proteins is their calcium-dependent interaction with membranes.
3.3. C2 Domain Containing Proteins
C2 domain is a Ca2+-dependent lipid binding domain. It is one of the most widely distributed calcium-binding motifs. There exist Ca2+-dependent and Ca2+-independent forms of C2 domains (reviewed in [23,24]). Most proteins with C2 domains take part in signal transduction (cytoplasmic phospholipase A2, phosphoinositide-specific phospholipase C, phosphatidyl inositol 3-kinases and so on) or membrane traffic (synaptotagmins, rabphillin-3 and so on).
C2 domain consists of a compact β-sandwich composed of two four-stranded β-sheets (Figure 7). The eight β-strands are connected by three loops at the top of the domain and by four loops at the bottom. Calcium binding sites are located exclusively at the top three loops. They are formed primarily by bidentate oxygen ligands of aspartate side chains and bind two or three Ca2+ ions.
3.4. Calcium ATPases
Calcium pumps, or Ca-ATPases, are membrane proteins located mainly in the plasma membrane or in the internal membranes of endoplasmic or sarcoplasmic reticulum. They transport two calcium ions per ATP hydrolyzed against a concentration gradient across membrane (reviewed in [25,26]). Cooperative binding of two calcium ions to transport sites of Ca-ATPase causes phosphoryl transfer from ATP (reviewed in [27]). Ca2+-ATPase from rabbit skeletal muscle sarcoplasmic reticulum consists of four basic domains (Figure 8). The transmembrane domain consists of 10 transmembrane helices M1-M10 interconnected by short loops on the outer and cytoplasmic surfaces. Four transmembrane helices extend into cytoplasm and form a stalk. The three cytoplasmic domains consist of two large cytoplasmic loops between transmembrane helices M2/M3 and M4/M5.
One calcium binding site in Ca2+-ATPase is formed by oxygen atoms of amino acid side chains and water molecules, and the other one is formed by side- and main-chain oxygen atoms. The Ca2+-ATPase pump has to discriminate between Ca2+ and the other metal cations present in any cell: Na+, K+, and Mg2+.
3.5. Extracellular Calcium Binding Proteins
The concentration of calcium ions outside cells is rather high (in millimolar region) and calcium affinity of extracellular proteins is usually low. Numerous proteins are associated with a very complex extracellular matrix. The extracellular Ca2+-binding modules can be divided into two groups: Ca2+ can either be bound to a single domain or can be bound between two independent domains. One can mention the following extracellular proteins: BM-40 (osteonectin or SPARC), matrix metalloproteinases, proteins with epidermal growth factor (EGF)-like domains, cadherins, integrins, blood clotting proteins, Ca2+-binding lectins, calsequestrin, Ca2+-binding hydrolytic enzymes, bone matrix proteins.
4. The Binding of Magnesium Ions to Proteins
Magnesium is essentially an intracellular ion: about 98% of nonskeletal magnesium is located inside cells. Ligands of magnesium ions are usually oxygen atoms but not nitrogen or sulfur atoms. In proteins, magnesium ion has usually six oxygen ligands in octahedral arrangement. Magnesium ions compete for most calcium binding sites (see above). Although calcium binding sites are subdivided into calcium-magnesium (they bind both Ca2+ and Mg2+) and calcium-specific (they do not bind Mg2+) sites, this classification is conditional. As a rule, while the Ca2+, Mg2+-sites bind calcium ions very tightly (dissociation constants 10−9–10−8 M), they bind magnesium ions 4 to 5 orders of magnitude weaker. The calcium specific sites bind calcium much less tightly (dissociation constants 10−7–10−6 M) and if they still bind magnesium 4 to 5 orders of magnitude worse, then it would be rather hard to measure such high dissociation constants. For this reason such binding sites are usually considered as “Ca2+-specific” ones.
Free calcium concentration in cells changes in very wide limits. In contrast to this, free magnesium concentration in cells is high and practically constant. Homeostasis of Mg2+ ions is maintained by Mg2+ transporters. One of the magnesium transporters, membrane protein CorA, is a homopentamer. Each monomer of CorA has two transmembrane domains and the central metal conducting pore [28]. The pore is formed by the TM1 transmembrane domain and surrounded from outside by the TM2 domain.
5. The Binding of Zinc Ions to Proteins
Zinc ion (Zn2+) contains a filled d orbital (d10) and therefore does not participate in redox reactions but rather functions as a Lewis acid. It accepts a pair of electrons in reactions such as proteolysis and the hydration of carbon dioxide.
There are three primary types of zinc binding sites: structural, catalytic, and cocatalytic (reviewed in [29,30]). The most common amino acids that provide nitrogen, oxygen, and sulfur ligands to Zn2+ are His, Glu, Asp, and Cys.
Zinc serves as a structural factor in many proteins including DNA-binding proteins containing the domain called “zinc finger”. Structural zinc sites consist of four protein ligands (Cys is the preferred ligand in such sites) without bound water molecules (Figure 2). Zinc serves as a cofactor for more than 300 various enzymes representing all the six main classes of enzymes. In catalytic sites zinc forms complexes with any three nitrogen, oxygen, and sulfur donors. Water is always present as a ligand in such sites. Cocatalytic sites contain two or three metals in close proximity with two of the metals bridged by a side chain moiety of a single amino acid residue, such as Asp, Glu, or His and sometimes a water molecule. No Cys ligands are found in such sites.
In the fourth type of zinc binding site, protein interface binding site, the ligands are provided by amino acid residues located at the interface surface of two proteins. Such zinc site usually has the coordination properties of a catalytic or structural site.
An interesting feature of all zinc sites is that the metal ion in them is surrounded by a shell of hydrophilic groups that is embedded to a larger shell of hydrophobic groups (reviewed in [31]). In addition, the amino acid side chains providing ligands to zinc in these structures often form hydrogen bonds with other residues.
The highly conserved catalytic zinc binding site of carboxypeptidase A consists of His69 (Nδ1), Glu72 (Oε1 and Oε2), His196 (Nδ1), and a water molecule (Figure 9). The first two ligands, separated by a short spacer of two amino acids, are located in a seven amino acid loop region between a β-sheet and an α-helix while His196 is the last residue in a β-pleated sheet.
Thermolysin-like mammalian zinc metalloendopeptidases hydrolyze peptide bonds in short substrates (less than 40 amino acids) (reviewed in [32,33]). These enzymes require a zinc ion in the active site and their amino acid sequences contain the classic HExxH motif (where x denotes any amino acid residue) typical of zinc metallopeptidases.
Members of astacin zinc protease family contain a unique 18-amino acid sequence, HExxHxxGFxHExxRxDR, which also begins with the HExxH zinc binding motif (reviewed in [34,35]). The members of the astacin family were found in species ranging from hydra to humans. These proteases take part in activation of growth factors, degradation of polypeptides, and processing of extracellular proteins.
Three independent classes of carbonic anhydrases, α, β, and γ, have been found. The active sites of all three classes possess a single zinc ion essential for catalysis (reviewed in [36,37]). Crystal structures of five different forms of the α-class carbonic anhydrase revealed that the catalytic zinc in these enzymes is located in a 15 Å deep active site cavity. The catalytic zinc is tetrahedrally coordinated by the Nε2 of His94 and His96 supplied from a β-strand, the Nδ1 of His119 from a second β-strand and a water molecule.
Two or more zinc (or other metal) atoms may operate in concert in active centers of multimetal enzymes (reviewed in [29]). Such cocatalytic sites are usually bridged by a side chain moiety of a single amino acid residue, usually Asp, Glu, and sometimes a water molecule. Asp and His residues predominate as ligands in the cocatalytic zinc sites. Sometimes these sites also contain unusual zinc ligands such as amide carbonyls provided by Asn, Gln, and the peptide backbone; hydroxyl groups from Ser, Thr, and Tyr, and the amine nitrogen of Lys or the N-terminal amino acid of the protein. Cysteine residues almost always are absent in these sites.
The copper/zinc enzymes, superoxide dismutases catalyze the dismutation of the superoxide anion into molecular oxygen and hydrogen peroxide (reviewed in [38,39]). Figure 10 shows ribbon model of X-ray structure of E. coli Cu,Zn superoxide dismutase. The cocatalytic copper/zinc site in superoxide dismutase has a bridging His residue (His63). His63 binds to the adjacent Zn2+ ions through the other deprotonated imidazole nitrogen. The zinc site is composed of three His residues (His 63, 71, 80, 83) and Zn2+ ion is coordinated by their Nδ1 nitrogens and the Oδ1 oxygen of Asp81 while the copper site is composed of four His residues (His 46, 48, 63, 120) and Cu2+ ion is coordinated by three Nε2 nitrogens and by one Nδ1 nitrogen (His46). All known superoxide dismutases use the redox active Cu in the active site in order to accomplish the catalytic breakdown of superoxide anion.
Some of the zinc enzymes that catalyze phosphomonoester hydrolysis also have cocatalytic zinc sites containing two or three metal atoms in close proximity (reviewed in [29]). E. coli alkaline phosphatase is probably the best studied representative of this group. Cocatalytic zinc sites in both of its subunits contain two zinc atoms and one magnesium atom.
Zinc binding to ligands, which belong to two different protein molecules, may result in formation of dimers or trimers of the same protein or link two different proteins (reviewed in [29]). His, Glu, and Asp primarily supply the ligands to these sites but Cys can also take part in these sites. The ligands are generally contributed by some form of secondary structure with β-sheets predominating.
In most cells, a total cellular zinc concentration is in the range of 0.1–0.5 mM, which is similar to that of iron. Most part of intracellular zinc is bound to proteins. Free zinc concentration in a cell seems to be in the range of 10−9 to 10−12 M. Zinc homeostasis in eukaryotic cells is controlled by intracellular zinc storing vesicles (“zincosomes”) (reviewed in [40]). The major zinc binding protein metallothionein regulates the cellular zinc level and the nuclear translocation of zinc in the course of the cell cycle and differentiation. Mammalian metallothionein consists of 62 amino acid residues folded into two domains. Only cysteine residues take part in the binding of metal ions. Metallothionein binds up to seven tetrahedrally coordinated atoms such as Zn (II) and Cd (II) or up to twelve of three-coordinated Cu (I) atoms in planar geometry.
Two families of zinc transporters have been found in eucaryotes. The proteins of ZIP (Zrt-, Irt-like Protein) family transport zinc from outside the cell into the cytoplasm and mobilize stored zinc by transporting it from within an intracellular compartment into the cytoplasm. A second group of transporters, the Cation Diffusion Facilitator (CDF) family, transports zinc in the direction opposite to that of the ZIP proteins.
6. The Binding of Copper Ions to Proteins
Copper is an essential element for all living organisms. Copper-binding proteins take part in various biological processes from electron transfer to oxidation of various substrates (reviewed in [41,42,43,44]). In many proteins, copper is used as a redox mediator, at the same time, stray copper ions can be harmful and living organisms are forced strictly regulate the distribution of copper ions.
Historically, copper atoms bound to proteins have been classified into three types (type 1, type 2, and type 3). The classification is based on their spectroscopic properties (reviewed in [44]). Type 1 (blue) copper shows absorption maximum around 610 nm (extinction coefficient 100 times higher than those for model copper complexes), and type 3 copper has absorption maximum around 330 nm. Type 1 and type 2 coppers are electron paramagnetic resonance (EPR) detectable, while the dinuclear type 3 coppers are EPR silent.
The type 1 copper-binding site consists of two histidines, one cysteine and one methionine. The two histidines and one cysteine tightly coordinate the copper ion in trigonal geometry and are essential for the blue copper-binding site, while the fourth residue, an axial methionine, is rather distant and its interaction with the copper is weaker. Moreover, sometimes this residue is replaced by such amino acids, as leucine or phenylalanine. Interdomain copper-binding sites consisting of only histidine residues are characteristic of multicopper blue proteins. It is of interest that sometimes, copper binding sites can contain tyrosine residues.
The term “multicopper blue proteins” is used for a group of copper-containing proteins including nitrite reductase and multicopper oxidases (reviewed in [45]). Multicopper blue proteins contain tandem repeats of a domain, which demonstrates distant homology to a cupredoxin fold domain. The single-domain cupredoxin fold protein family includes plastocyanin, azurin, pseudoazurin, rusticyanin, stellacyanin, and amicyanin.
The cupredoxin fold, an eight-stranded Greek key β-barrel, was first found in the small blue copper proteins, plastocyanin and azurin and later in the related proteins pseudoazurin, amicyanin, and cucumber basic protein. The cupredoxin-fold domain usually contains a type 1 copper-binding site, which is responsible for the characteristic blue color of these proteins. The single-domain cupredoxin-fold proteins and multicopper blue proteins together are often called blue copper proteins.
In multicopper blue proteins, the function of the blue copper is to accept an electron (oxidize) from a substrate, and the function of the interdomain copper is to donate the electron to reduce another substrate.
All blue multicopper oxidases have at least one type-1 copper and at least three additional copper ions: one type-2 and two type-3 copper ions, arranged in a trinuclear cluster (reviewed in [43,44]). The type-1 copper oxidizes substrates and the extracted electrons are transferred to the type-2/type-3 copper site via a strongly conserved His-Cys-His tripeptide motif. In this site molecular oxygen is reduced to water. Interdomain sites of some laccases contain one type-2 copper atoms and two type-3 copper atoms, coordinated by eight histidine residues.
Copper-containing nitrite reductases have a trimeric structure (reviewed in [46]) and contain both type-1 (type-1 Cu) and type-2 (type-2 Cu) copper centers. Each subunit (molecular mass of ~40 kDa) consists of two domains and binds six copper atoms. In addition, homotrimer of nitrite reductase has interdomain copper binding sites.
Tyrosinases are copper-containing enzymes present in plant and animal tissues that catalyze the production of melanin and other pigments oxidizing tyrosine. Tyrosinase active site contains a pair of antiferromagnetically coupled copper ions, CuA and CuB, which are coordinated by six histidine residues (reviewed in [47]). Each of the two copper ions is bound by three conserved histidine residues.
There exists a group of blue proteins with monocopper sites (reviewed in [48]), which contain two histidines, one cysteine, and one methionin in a coordination sphere with a distorted tetrahedral geometry (Figure 11).
The blue copper sites are characterized by an intense absorption band at 600 nm due to the ligand to metal charge transfer Cys− → Cu (II), which provides the blue colour to solutions of these proteins. Moreover, this site has a high redox potential and electron transfer causes its structural changes.
The cupredoxins (plastocyanins, azurin, stellacyanin, rustacyanin, amicyanin), members of monocopper blue proteins family, are small (10–20 kDa) soluble copper proteins. They transfer electrons, one by one, from a donor to an acceptor protein in the respiratory and photosynthetic chains of many bacteria and plants (reviewed in [49]).
The well-studied cupredoxin is plastocyanin, which is an important component of the photosynthetic electron transfer chain (reviewed in [50,51]). Figure 12 shows a model of the plastocyanin structure.
Plastocyanin has a distorted tetrahedral copper binding site at one end of an eight-stranded antiparallel β-barrel. The copper binding site is formed by His37, His87, Cys84, and Met92. The site can bind both Cu (I) and Cu (II), which facilitates electron transfer in this system.
Azurin structure is similar to the plastocyanin structure: one α-helix and eight antiparallel β-strands that fold into a β-barrel arranged in a double-wound Greek key topology (reviewed in [52]). The indole group of the single tryptophan residue in azurin is located in the center of the protein molecule. An environment of the indole group is extremally rigid and unpolar, which is reflected in unusually blue spectrum of tryptophan fluorescence with distinct vibrational structure [53].
Binuclear Cu proteins are divided into two groups based on the magnetic interaction between the two Cu atoms: proteins with coupled and proteins with noncoupled binuclear sites (reviewed in [54]). The coupled binuclear Cu proteins include, for example, hemocyanin, tyrosinase, and catechol oxidase. These proteins possess two Cu sites strongly magnetically coupled with each other through direct bridging ligands, which allows them to perform the 2-electron reduction of O2.
Most of copper in human adult is used as a cofactor of enzymes catalyzing oxidation-reduction reactions. Such enzymes include cytochrome-c oxidase, superoxide dismutase, ceruloplasmin and copper-thionein, protein-lysine 6-oxidase, dopamine-β-monooxygenase, monophenol monooxygynase, peptide-α-amidating monooxygenase, and some others.
Cytochrome с oxidase, which catalyzes the reduction of O2 to H2O during cellular respiration, contains binuclear CuA sites (reviewed in [55,56]). Binuclear CuA sites in cytochrome с oxidase contain a di-copper unit with a delocalized mixed valence in its resting state. Cu1.5Cu1.5 atoms are located at a distance about 2.5 Å in a distorted tetrahedron coordination sphere. The copper atoms are bound to two S (Cys), two N (His) and by two additional ligands in axial position (one S(Met) and one O (carbonyl of glutamate)) (Figure 13).
The copper-only N2O-reductases (reviewed in [57]) are composed of two identical subunits of about 65 kDa, containing a binuclear copper site, CuA, similar to the one revealed in cytochrome c oxidase. This site plays a role in intramolecular electron transfer. In addition, N2O-reductases possess a multinuclear copper site, CuZ, which is necessary for catalysis (Figure 14).
The catalytic site CuZ in N2O-reductase obtains electrons from CuA. Copper atoms in the unique cluster CuZ are coordinated by seven N atoms of histidine residues, one O atom (hydroxyl or water) and one sulfur atom S (inorganic sulfur). CuZ site is a μ4-sulfide bridged tetranuclear copper center. In CuZ site of Ps. nautica N2O-reductase, Cu-1 and Cu-2 are coordinated by two histidine residues (His270/His325 and His79/His128, respectively), Cu-3 is bound to His80, His376; Cu-4 is bound to a single protein ligand, His437. All Cu atoms are bridged by a sulfur atom.
The hemolymph of many species of arthropods and clams contain hemocyanins, which are the oxygen carrier glycoproteins (reviewed in [58]). The respiratory function of hemocyanin is similar to that of hemoglobin; at the same time, these proteins differ in molecular structure and mechanism of functioning. Hemoglobin carries its iron atoms using heme groups. Oxygen binding in hemocyanins is mediated by a pair of copper atoms, which are bound to the protein as prosthetic groups coordinated by six histidine residues.
Under normal conditions, the concentration of copper in plasma is regulated by strong homeostatic mechanisms. In most mammals, serum albumin has a high capacity for tight copper binding, but more than 70–80% of plasma copper is bound to the cuproprotein ceruloplasmin.
The cysteine-rich metallothioneins and phytochelatins and sulfide are the main sequestration molecules for transition metal ions, including Cu (I) and Cd (II) (reviewed in [59]). In these molecules, the metals are bound in polynuclear metal thiolate clusters. The cysteines in metallothioneins (20–30% of the amino acids) are arranged in repetitive Cys-Cys, Cys-Xaa-Cys and Cys-Xaa-Xaa-Cys motifs.
Disturbances in copper balances in humans result in serious diseases such as Menkes syndrome or Wilson disease. These diseases are characterized by disturbances in the distribution of copper in organs and tissues. Additionally, copper is implicated in neurodegenerative diseases such as familial amyotropic lateral sclerosis (FALS), Alzheimer′s disease, and prion diseases of neuronal spongiform encephalopathy.
7. Iron Binding Proteins
Iron has two stable oxidation states, Fe (II) and Fe (III), which easily interconvert to each other and this property allows iron to take part in many reduction-oxidation reactions in biology. Moreover, some spin states of iron ions are used in catalysis of the reactions with participation of molecular oxygen. For this reason, iron is an essential element in many important processes such as respiration, DNA synthesis, nitrogen fixation, and photosynthesis.
The interconversion of the two iron oxidation states creates two problems. On the one hand, Fe (II) activates dioxygen with the production of very reactive species, which can cause serious oxidative damage processes, and it prevents its use as an available iron source. On the other hand, Fe (III) has a low solubility under physiological conditions and this shows that living organisms must have efficient iron storage/transport/usage mechanisms.
Transferrin is a serum protein responsible for iron transport (reviewed in [60]). Transferrin has two Fe3+ binding sites with dissociation constants of the order of 10−22 M. At the same time, transferrin does not bind ferro-iron. The cells absorb iron ion via an interaction of transferrin with cell surface receptors. Phosphorylation of the receptor by protein kinase C initiates the entrance of the iron-transferrin-receptor complex to the cell. After the entrance, the iron is released.
Ferritin is a major protein used for intracellular storage of iron (reviewed in [61]). If the capacity of ferritin in the cell is exceeded, iron is deposited near the ferritin-iron complexes in the cell (amorphous iron hemosiderin).
Iron-containing heme-proteins are responsible for the transportation and storage of oxygen (hemoglobin and myoglobin), electron transfer (cytochromes), catalysis of the oxygen and hydrogen peroxide oxidation (oxidases and peroxidases), and catalysis of the hydrogen peroxide decomposition (catalases). About 70% of the total iron in humans is bound to hemoglobin. Heme, a derivative of porphyrin, the mandatory component of the hemoproteins, serves to bind iron. The structure of iron (II)-porphyrin is shown in Figure 15. In the structure, the iron atom is coordinated by four nitrogen ligands in a plane square configuration.
Myoglobin is a cytoplasmic hemoprotein consisting of a single polypeptide chain of 154 amino acid residues (reviewed, for example, in [62] and in many textbooks) (Figure 16). Like hemoglobin, myoglobin reversibly binds O2. It facilitates O2 transport from red blood cells to mitochondria during increased metabolic activity or serves as an O2 depot during hypoxic conditions. Unlike hemoglobin, however, monomeric myoglobin has a hyperbolic O2-saturation curve characteristic of normal Michaelis–Menten enzyme kinetics rather than the sigmoid-shaped curve typical for tetrameric hemoglobin.
Myoglobin consists of eight α-helices A-H and binds oxygen by its heme (reviewed in [62]). The heme prosthetic group is located between two histidine residues, His64 and His93. The iron ion interacts with six ligands, four of which are the N atoms provided by the four heme pyrols. The imidazole group of His93 provides the fifth N ligand. This interaction stabilizes the heme group and slightly displaces the iron ion away from the plane of the heme. The sixth ligand position, empty in deoxy-myoglobin, serves as the binding site for O2 or for other ligands such as CO or NO.
Myoglobin is perhaps best known as an O2-storage protein in muscle. This myoglobin function is especially evident in marine mammals and birds that undergo extended periods of apnea when diving. In the absence of inhaled O2, oxygen stored in oxy-myoglobin flows to the locomotor muscles involved in diving-related activities.
Hemoglobin is a tetrameric protein with molecular mass 64 kDa consisting of two pairs of identical subunits (α and β) (Figure 17) (reviewed in [63,64]). Each subunit contains heme and has a structure, which is similar to the structure of myoglobin.
A proximal histidine residue holds the heme group, which forms a penta-coordinated high spin ferrous complex (reviewed in [65]). The heme group provides four nitrogen ligands for iron atom. Like in myoglobin, oxygen binds at the vacant site of the heme ferrous complex to form a pseudo-octahedral low spin complex.
The oxygen saturation curve of hemoglobin is sigmoidal since the affinity of hemoglobin for O2 (or CO for ferro-hemoglobin and CN for met-hemoglobin) increases with the increase of O2 concentration. These properties make hemoglobin an efficient transporter of O2 from the lungs to the tissues and of CO from the tissues to the lungs.
Cytochromes are heme proteins involved to the electron transfer chain of mitochondria using the redox properties of the Fe (III)-Fe (II) pair. In addition, cytochromes take part in some stages of the nitrogen cycle and in some reactions in photosynthesis. Cytochrome c is an electron transfer protein: it accepts electron from cytochrom c1 and transfers it to cytochrome c oxidase. Moreover, it plays some moonlighting roles outside mitochondria triggering cell apoptosis (reviewed in [66,67,68]). Cytochromes c can contain one or several c-type hemes connected to the protein through thioether bonds involving the SH groups of two cysteines. The heme iron ion in cytochromes c is always axially coordinated by nitrogen of a histidine side chain.
Four classes of cytochromes c are identified, depending on the number of hemes, the type and the position of the axial iron ligands, and the redox potential. Mono-heme cytochrome c contains a single CxxCH sequence in which the two cysteins are connected to the porphyrin macrocycle, while the single histidine is connected to the bound iron.
Many heme-containing enzymes use redox properties of heme in order to activate relatively stable oxygen compounds such as hydrogen peroxide (H2O2) or molecular oxygen (O2). Peroxidases are heme proteins catalyzing the reactions of oxidation by hydrogen peroxide (reviewed in [69]). Catalases catalyze the disproportionation of hydrogen peroxide and can be considered as a special example of the peroxidase activity when the substrate oxidizable by hydrogen peroxide is another hydrogen peroxide molecule (reviewed in [70]). For this reason, the properties of peroxidase and catalase are similar.
Free iron ions induce formation of free radicals, which cause oxidative damages of tissues. Transferrins are a group of iron-binding proteins that control the levels of iron in the body fluids of vertebrates due to their ability to bind two Fe3+ and two CO32− (reviewed in [60]). The primary role of transferrin is therefore to transport iron safely around the body to supply growing cells.
The transferrin molecule consists of 679 amino acid residues and has a molecular weight of ~79 kD. 19 intra-chain disulfide bonds stabilize the transferrin molecule. Transferrin possesses three carbohydrate side chains attached to nitrogen of Asn413 and Asn611 and to oxygen of Ser32 (reviewed in [60,71]). The transferrin molecule consists of the N-lobe (336 amino acids) and C-lobe (343 amino acids), which are linked by a short spacer sequence. The distorted octahedral iron binding site in both the N- and C-terminal lobes consists of four conserved amino acids including two Tyr, one Asp, and one His. Transferrin bound iron is released during receptor-mediated endocytosis. In this process, the iron-loaded transferrin binds to the cell surface receptors. After internalization, it releases its iron to the endosome and returns to the cell surface for more rounds of binding and transport (reviewed [72]).
Ferritins are a broad superfamily of iron-storage proteins, widespread in the three kingdoms of life, in aerobic or anaerobic organisms (reviewed in [61,73]). Ferritins are large protein cages (100–120 Å), that provide the reversible formation of iron-oxy biominerals in a gigantic cavity. When ferritin contains no minerals, its cavity is filled with liquid from the surrounding solution. The basic physiological role of ferritins is to supply cells with the necessary iron, keeping its effective concentrations in living cells in the range of 10−3–10−5 M. They can be also involved in cell redox-stress resistance.
Ferritins and bacteroferritins have essentially the same architecture, assembling in a 24-mer cluster to form a hollow, roughly spherical structure with a diameter ~120 Å (reviewed [73,74]) (Figure 18).
In general, the iron storage cavity of ferritin has a diameter of ~80 Å and can accommodate up to 4500 iron ions. Each subunit (~20 kDa) is a bundle of four α-helices capped by a shorter helix at the C-terminal end. The 24 subunits are related by 4-, 3-, and 2-fold symmetry axes (432-point symmetry). Ferritin cages are self-assembled from multiple four α-helix bundle subunits. Maxi-ferritins have 24 subunits with 432 symmetry and mini-ferritins have 12 subunits with 32 symmetry.
Fe2+ enters the ferritin protein through its pores. After that, it rapidly (in milliseconds) reaches the ferroxidase site located within each H-ferritin subunit, which oxidizes Fe2+ and forms diferric-oxo mineral precursors (Fe3+-O-Fe3+) which are released into the interior of the protein. The gated pores in ferritins that control iron exit are related by both structure and function to ion channel proteins in cell membranes.
Iron-sulfur [Fe-S] clusters are ubiquitous prosthetic groups that are required for fundamental life processes (reviewed in [75]). The simplest Fe-S proteins contain a single iron tetrahedrally coordinated by four cysteinyl thiolates. Clusters of higher nuclearity are formed by the addition of inorganic sulfide to bridge iron atoms to form, for example, 2Fe-2S clusters, cubane and linear 3Fe-4S clusters, and cubane 4Fe-4S clusters (reviewed in [76]). (Fe-S) clusters participate in electron transfer, substrate binding/activation, iron/sulfur storage, regulation of gene expression, and enzyme activity.
Delocalization of electron density over both Fe and S atoms in (Fe-S) clusters makes them ideally suited for biological electron transport (reviewed in [75,76]). (Fe-S) clusters are necessary components in the photosynthetic and respiratory electron transport chains. They transport electrons in membrane-bound and soluble redox enzymes and serve as redox-active centers in ferredoxins. Clusters involved in electron transfer contain (2Fe-2S), (3Fe-4S), (4Fe-4S), or (8Fe-7S) core units. In each site Fe is tetrahedrally coordinated by S atoms of cysteinates. Aspartate, histidine, serine, or backbone amide ligands at a unique Fe site are occasionally encountered in the clusters.
In eukaryotic cells, most Fe-S proteins are located within the mitochondria. These proteins include aconitase of the citric acid cycle, subunits of the respiratory chain complexes I, II and III and biotin synthase (reviewed in [77]). Mitochondrial electron transport chain contains up to 12 different Fe-S clusters.
Nitrogenase catalyzes the reductive breakage of the very strong triple bond of N2 to generate NH3 in the process of biological nitrogen fixation. Nitrogenase can also catalyze the reduction of protons to dihydrogen, so nitrogenase is also a hydrogenase. Nitrogenase consists of two component proteins, the iron (Fe) protein and the molybdenum-iron (MoFe) protein (reviewed in [76,78]). The Fe-protein is an electron donor, and the MoFe-protein is responsible for dinitrogen reduction. The Fe-protein is a dimer of two identical subunits (60 kDa) that symmetrically coordinate a single (4Fe-4S) cluster (Figure 19). The Fe-protein binds two molecules of nucleotides (MgATP or MgADP) per dimer and serves for the coupling of ATP hydrolysis to electron transfer.
Metalloenzymes hydrogenases (H2ases) catalyze the interconversion of molecular hydrogen and protons and electrons (reviewed in [78]). Their metal sites belong to two main types. One of them consists of the classical (2Fe-2S), (3Fe-4S), and (4Fe-4S) iron-sulfur clusters. The second type may be in two varieties, the (NiFe) and (Fe) active sites. The majority of hydrogenases are NiFe-containing enzymes.
Ferredoxins are small soluble iron-sulfur (Fe-S) proteins, which serve as low-potential electron carriers (reviewed in [79,80,81]). All ferredoxins have the same structure: five-stranded β-sheet, 2 to 3 α-helices and a long loop containing 3 to 4 cysteine ligands for Fe atoms. All the plant-type ferredoxins have a conservative sequence Cx4Cx2Cx22-33C to hold (2Fe-2S) cluster. Two Fe ions are coordinated tetraherically by sulfide ions and cysteine residues.
Biotin synthase catalyzes the final step in the biotin biosynthsis, the conversion of dethiobiotin to biotin (reviewed in [82]). Biotin synthase has a conserved CxxxCxxC sequence motif that coordinates (4Fe-4S) cluster. The enzyme uses S-adenosyl-methionine for radical generation. Escherichia coli biotin synthase is a 76.8 kDa homodimer, which contains one (2Fe-2S)2+ cluster per monomer. Under mild conditions, the protein is reconstituted to contain an additional (4Fe-4S)2+ cluster per monomer (Figure 20). The fold of each monomer is (αβ)8 barrel, with two additional helices at the N terminus and a disordered region at the C terminus. Both substrates and FeS clusters are located in the core of the barrel.
8. Molybdenum and Tungsten Containing Proteins
Tungsten and molybdenum have equal ionic (0.68 Å) radii and similar electronegativity. Both metals can be in various oxidation states (from +2 to +6) and are able to form complexes with polynucleotides.
More than 50 enzymes contain Mo and most of them are found in bacteria while in eukaryotes only six were revealed (reviewed in [83,84]). On the one hand, if an organism takes up too high amounts of Mo, toxicity symptoms are observed. On the other hand, unavailability of Mo is lethal for the organism. Organisms take up Mo in the form of its molybdate anion.
Both molybdenum and tungsten are redox-active under physiological conditions (oxidation states VI to IV). The physiological roles of the molybdenum and tungsten containing enzymes are fundamental and include the catalysis of key steps in carbon, nitrogen, and sulfur metabolism.
Biologically active Mo is usually complexed by a pterin compound forming the molybdenum cofactor (Moco) (Figure 21). Moco is the active compound at the catalytic site of Mo-enzymes.
In general, reactions catalyzed by Mo-enzymes are characterized by the transfer of an oxygen atom to or from a substrate in a two-electron redox reaction, in which the oxidation state of Mo changes between IV and VI.
Molybdenum containing enzymes are divided into three families, each with a distinct active-site structure and type of reaction catalyzed (reviewed in [84,85]). The first family (for example, xanthine oxidase from cow′s milk) has an L Mo (VI) OS (OH) core in the oxidized state, with one equivalent of the pterin cofactor (designated L) coordinated to Mo. These enzymes typically catalyze the hydroxylation of carbon centers. The second family includes sulfite oxidase and nitrate reductases. Again, the (oxidized) metal center has a single equivalent of the pterin cofactor, but as part of an L Mo (VI) O2 (S-Cys) core, with a cysteine ligand provided by the polypeptide. Members of the second family catalyze the transfer of an oxygen atom either to or from a lone pair of electrons on the substrate, respectively. The third family have two equivalents of the pterin cofactor bound to the metal. The molybdenum coordination sphere is usually completed by a single Mo = O group and a sixth ligand in an L2 Mo (VI) O (x) core. The sixth ligand, x, can be a serine, a cysteine or a selenocysteine, or a hydroxide and/or water molecule. The reactions catalyzed by members of this last family frequently involve oxygen-atom transfer, but dehydrogenation reactions also occur.
In eukaryotes, the most prominent Mo-enzymes are (1) sulfite oxidase, which catalyzes the final step in the degradation of sulfur-containing amino acids and is involved in detoxifying excess sulfite; (2) xanthine dehydrogenase, which is involved in purine catabolism and reactive oxygen production; (3) aldehyde oxidase, which oxidizes aldehydes and takes part in the biosynthesis of abscisic acid; (4) nitrate reductase, which catalyzes the key step in inorganic nitrogen assimilation. All Mo-enzymes, except plant sulfite oxidase, need at least one more redox active center, many of them using iron in electron transfer.
Xanthine oxidase catalyzes the oxidation of xanthine to uric acid. Oxygen is included to the final product of this reaction not from O2 but from water. Actually, xanthine oxidase and xanthine dehydrogenase are two different forms of the same enzyme. These α2 homodimer enzymes differ in the chemical nature of the oxidizing substrate: O2 for xanthine oxidase and NAD+ for xanthine dehydrogenase. The homodimers are composed of two identical subunits 150 kDa and each subunit is subdivided into three distinct domains: an N-terminal domain 20 kDa for binding of two [2Fe-2S] clusters, a 40 kDa domain with a FAD-binding site, and a C-terminal domain for Moco-binding and dimerization (reviewed in [83]) (Figure 22).
Sulfite oxidase, a member of the second Mo enzyme family, has a b-type cytochrome in addition to the Mo center (reviewed in [86]). The enzyme is a homodimer 101–110 kDa. Each monomer of the homodimeric chicken sulfite oxidase contains a small N-terminal b5-type cytochrome domain, a large central Mo-binding domain, and a large C-terminal interface domain. The Mo domain and the b5-type cytochrome domain are linked by a flexible loop of 10 amino acids. The Mo-binding domain has three β sheets and nine α helices, with the Mo center located in the middle.
The member of the third family of Mo-containing enzymes, DMSO reductase from R. sphaeroides or R. capsulatus is monomeric and lacks other redox centers. At the same time, the membrane-bound DMSO reductase from Escherichia coli has two additional subunits, one of which has four different [4Fe-4S] iron–sulfur centers.
The tungsten-containing aldehyde:ferredoxin and formaldehyde:ferredoxin oxidoreductases of the first family of tungsten-containing enzymes both possess [4Fe-4S] iron–sulfur clusters in addition to their tungsten centers (reviewed in [84]). These enzymes are composed of a single type of subunit—they are α2 and α4 oligomers.
9. Proteins Containing Nickel and Cobalt
Nickel, Ni, is contained naturally in only a few classes of proteins: urease, carbon monoxide dehydrogenase, S-methyl coenzyme-M reductase and hydrogenase (reviewed in [87,88]). Nickel is an essential component of enzymes involved in energy and nitrogen metabolism, in detoxification processes and in pathogenesis (urease, (NiFe)-hydrogenase and Ni-superoxide dismutase).
Cobalt is an important cofactor in vitamin-B12-dependent enzymes (reviewed in [89]). Vitamin B12 contains cobalt in a substituted corrin macrocycle (a porphyrin relative). The B12 coenzyme possesses an axial Co (III)-alkyl (5′-deoxyadenosine or methyl) group. Much effort has been concentrated on studies of the corrin cobalt. In contrast, only a few proteins containing noncorrin cobalt have been characterized.
Nickel and cobalt ions are necessary for many cellular functions, at the same time they require strict regulation to avoid toxic side reactions or incorporation into non-native binding sites. Thus, the concentrations of free Ni and Co ions should be under strict control appear to be exceedingly low in cells. The MerR family of transcription factors responds to many metal ions, including Hg (MerR), Zn (ZntR), Cu (CueR), Pb (PbrR), Cd (CadR) and Co (CoaR), and to oxidative stress (SoxR) and drugs (BmrR). Proteins of this family have homologous N-terminal DNA-binding domains but different C-terminal sensor domains that enable them to bind specific coactivator molecules (reviewed in [90]).
All forms of methionine aminopeptidase are activated by, or are sensitive to, metals such as Mn2+, Co2,+ and Zn2+ (reviewed in [89]). The enzyme from Salmonella typhimurium is stimulated only by Co2+, not by Mg2+, Mn2+, or Zn2+ and is inhibited by metal ion chelator EDTA. E. coli methionine aminopeptidase is a monomeric protein of 29 kDa consisting of 263 residues that possesses two Co2+ ions in its active site (Figure 23).
Superoxide dismutases are ubiquitous enzymes that protect biological molecules from oxidative damage. The catalytic center of nickel-containing superoxide dismutase from Streptomyces seoulensis is located in the N-terminal loop, where a Ni3+ ion is coordinated by the amino group of His1, the amide group of Cys2, two thiolate groups of Cys2 and Cys6, and the nitrogen of His1 as axial ligand that is lost in the chemically reduced state [91].
Methionine aminopeptidase has several weak absorption bands between 550 and 700 nm, which arise from d → d transitions of the d7 Co (II) ions. The two Co2+ ions are coordinated by Asp97, Asp108, His171, Glu204, and Glu235 and located between two double β-antiparallel sheets that constitute part of the active site of the enzyme. The geometry of each cobalt site is approximately octahedral.
10. Manganese-Containing Proteins
The ionic radius of Mn2+ (0.80 Å) in aqueous solutions lies between that of Mg2+ (0.65 Å) and Ca2+ (0.99 Å) and is close to that of Fe2+ (0.76 Å) and several other transition metal ions. It is therefore not surprising that Mn2+ and other cations may be interchangeable in the metal binding sites of many proteins.
Mn2+ ions prefer “hard” ligands such as oxygen atoms of Asp and Glu, as well as oxygen atoms of water molecules (reviewed in [92]). Sometimes N atoms of histidines also can be Mn2+ ligands in metalloenzymes, while sulfur atoms of cysteins have not been found so far in the Mn netalloenzymes. Coordination geometry of Mn2+ in manganese enzymes is usually square pyramid or trigonal pyramid with coordination number 5 or octahedral with coordination number 6.
The oxidation of H2O to O2 by the Mn-containing oxygen-evolving complex in the chloroplasts, and the reduction of O2 to H2O by cytochrome c oxidase in the mitochondria form the important cycle of dioxygen metabolism that is essential to both plant and animal life on Earth (2H2O ↔ O2 + 4e− + 4H+). Oxygen is relatively abundant in the atmosphere primarily due to its constant regeneration by photosynthetic water oxidation by the oxygen-evolving complex.
The oxygen-evolving complex is the site of water oxidation. It consists of the Mn4 cluster, YZ, and Ca2+, and Cl− cofactors (reviewed in [93,94]). The oxygen-evolving complex cycles through a series of intermediate states, Si (i = 04), where i represents the number of oxidizing equivalents stored on the oxygen-evolving complex. This process uses the energy of four successive photons absorbed by the pigment P680 of the photosystem II (PS II) reaction center. The Mn complex in the oxygen-evolving complex couples the four electrons oxidation of water with the one-electron photochemical process occurring at the PS II reaction center. The actual oxidation state of Mn in the manganese cluster in the different S states is still uncertain. Two combinations of spin states were found to have comparable stability in the S1 resting state: 2Mn (IV)2Mn (III) and Mn (IV)2Mn (III)Mn (IV). It is unknown whether Mn is oxidized in the S2 to S3 transition. At the same time, it was suggested that S2 state is 3Mn (III)Mn (IV).
A number of enzymes, including Mn-superoxide dismutase, Mn-catalase and arginase, specifically require Mn2+ for activity. In addition, glycolysis cannot proceed fully without 3-phosphoglycerate mutase, which, in several Gram-positive bacteria, is active only with bound Mn2+.
One of the examples of the Mn-containing enzymes is arginase. It is a trimeric enzyme that catalyzes the hydrolysis of the arginine side chain to form ornithine and urea (reviewed in [92,95]). Liver arginase is a 105 kDa homotrimer and each 35 kDa subunit contains a binuclear Mn (II) center that is critical for catalytic activity (Figure 24). The overall fold of each subunit consists of a parallel, 8 stranded β-sheet flanked on both sides by numerous α-helices.
The Mn2+-Mn2+ cluster is located on one edge of the central β-sheet (Figure 25). The metal ion that is located deep in the active site cleft is designated Mn2+A and is coordinated by His101, Asp124, Asp128, Asp232, and a bridging hydroxide ion with square pyramidal geometry.
A limited number of prokaryotes, especially hyperthermophiles, express tetrameric Mn- or Fe-superoxide dismutase enzymes. Eukaryotes in general have only Mn-superoxide dismutase as a tetramer.
11. Sodium- and Potassium-Binding Proteins
Alkali metal ions Na+ and K+ are required for living organisms to maintain the balance of the body fluids and for nerve and muscle functioning. Na+ ions are most abundant outside the cells (~10 mM in mammalian cell and ~100 mM in blood plasma), while K+ ions are present in high concentration inside the cells (~100 mM in mammalian cell and ~4 mM in blood plasma). Concentration gradients of these ions across the cell membrane provide an energy source for action potential created by the opening and closing of the Na+ and K+ channels and for movements of various substances and other ions through the membranes by means of transporters.
The Na+ and K+ sites both contain oxygen atoms, mostly with partial negative charges. A very important factor distinguishing Na+ and K+ sites is the size of their cavity (reviewed in [96]).
Depending on the activation mechanism, the ion channels are divided into voltage gated channels and ligand gated channels. The voltage gated K+ channel conducts K+ ions selectively across the cell membrane, down the electrochemical gradient. It is a 4-fold symmetric tetramer resembling a teepee that surrounds a central pore (Figure 26) (reviewed in [96,97,98]). The pore is 12 Å long and lined exclusively by main chain carbonyl oxygens. Four voltage sensing helices S1–S4 surround the pore domain and control its gates. Each of the four subunits of the channel are capable of coordinating completely dehydrated K+ but not the smaller Na+ ions. The assembly of the channel-forming subunits is controlled by an intracellular N-terminal tetramerization domain (T1), which also serves as a scaffold to bind accessory β subunits (Kvβs).
The pore region of all the voltage-gated Na+, K+, and Ca2+ channels includes a common structural motif consisting of two transmembrane helices separated by a loop (P-loop) forming the selectivity filter (reviewed in [99]). The selectivity filter is a relatively narrow region that creates a barrier for the permeation of organic ligands. The filter is located at the narrowest part of the pore and its role is to ensure K+ selectivity. This is achieved due to the presence of a ring of oxygen atoms contributed by the amino acid residues of the peptide backbone at the region of the selectivity filter. These oxygens serve as binding sites for “bare” K+ ions for a short period of time free of water molecules. This transient dehydration is thought to be energetically much less favorable for Na+.
The sodium pump, also known as the Na+/K+-ATPase, establishes and maintains electrochemical gradient in animal cells (reviewed in [100]). Na+/K+-ATPase transforms chemical energy of ATP to osmotic work and maintains electrochemical Na+ and K+ gradients across cell membranes. This enzyme is located in the plasma membrane and transports Na+ and K+ using ATP hydrolysis energy. For every molecule of ATP hydrolyzed, three Na+ ions from the intracellular space and two K+ ions from the external medium are exchanged. Thus, the Na+/K+-ATPase ensures the maintenance of the membrane potential of the cell, provides the basis for neuronal communication, and contributes to the osmotic regulation of the cell volume.
Enzymes activated by monovalent cations were found both in plants and animals (reviewed in [101]). Because the concentration of Na+ and K+ is strictly controlled in vivo, these metal cations do not function as regulators of enzyme activity. Rather, they facilitate substrate binding and catalysis by lowering energy barriers in the ground and/or transition states (reviewed in [102]). Enzymes activated by Me+ take advantage of the large availability of Na+ outside the cell and K+ inside the cell. A strong correlation exists between the preference for K+ or Na+ and the intracellular or extracellular localization of such enzymes.
Me+ complexation results in selective stabilization of one conformation of the enzyme and may produce local and even long-range changes of the enzyme structure. The mechanism of Me+ activation can be seen from crystal structures of the enzymes as cofactor-like (type I) or allosteric (type II) (reviewed in [102]). In the former case, substrate binding to the enzyme active site is mediated by Me+, often acting in tandem with a divalent cation like Mg2+. In the latter case, Me+ binding enhances enzyme activity due to conformational changes triggered upon its binding. In this case, the cation makes no direct contact with substrate.
Diol and glycerol dehydratases provide the simplest example of Type I enzymes. Diol dehydratase is a coenzyme B12-dependent enzyme with an absolute requirement for K+. In these proteins K+ ion is coordinated by five protein ligands and also by two hydroxyl oxygens from substrate (Figure 27) (reviewed in [101,102]). In the substrate-free form, the two substrate ligands are substituted by water molecules.
Some kinases belong to Type II of K+-activated enzymes because in this case K+ does not contact ATP directly (reviewed in [101]). In this structure, K+ activates the enzyme indirectly changing the conformation of active site residues.
Na+ binding is very important for function of such Me+-activated enzymes as chymotrypsin-like proteases (reviewed in [102]). This large family of enzymes catalyzes the hydrolytic cleavage of peptide bonds to facilitate various physiological and cellular functions. Na+ coordination near the S1 binding pocket activates trypsin-like enzymes, and binding is allosterically linked with distant and distinct regions on the protease surface (reviewed in [103]).
12. Interactions of Proteins with Toxic Metal Ions
Ions of mercury (Hg), cadmium (Cd), arsenic (As), chromium (Cr), thallium (Tl), and lead (Pb) are considered very toxic. They can interact with calcium and zinc binding sites in proteins disrupting their functioning. The three most dangerous pollutant heavy metals are mercury, lead, and cadmium. Heavy metal poisoning could result, for instance, from drinking-water contamination or intake via the food chain.
Mercury has a number of effects on humans: disruption of the nervous system; damage of brain functions; DNA damage; allergic reactions; negative reproductive effects, birth defects, and so on. Mercuric ions prefer to bind to reduced sulfur atoms belonging to thiol-containing molecules, such as glutathione, cysteine, homocysteine, N-acetylcysteine, metallothionein, and albumin. The affinity constant for mercury binding to thiolate anions is as high as 1015 to 1020 M−1. In contrast, the affinity constants for mercury binding to oxygen- or nitrogen-containing ligands (e.g., carbonyl or amino groups) are about 10 orders of magnitude lower.
In humans, the kidneys are the primary targets of mercuric ions. Chelation agents such as the dithiols sodium 2,3-dimercaptopropanesulfate (DMPS) and meso-2,3-dimercaptosuccinic acid (DMSA) are used for treatments of mercury poisoning. Alpha-lipoic acid (ALA), a disulfide, and its metabolite dihydrolipoic acid (DHLA), a dithiol, have also been used for such treatment.
Bacterial resistance to mercury compounds is one of the most widely observed phenomenon in eubacteria (reviewed in [104]). The genes encoding the proteins of mercury resistance occur naturally on chromosomes, plasmids, and transposable elements. The regulator MerR and the major detoxification enzyme, MerA, the mercuric ion reductase play distinct but related roles in prokaryotic physiology. MerA, the mercuric reductase, a cytosolic flavin disulfide oxidoreductase (homodimer 120 kDa) uses NADPH as a reductant [104]. In Gram-negative bacteria MerA has a flexible N-terminal domain, which is homologous to MerP, a small periplasmic mercury binding protein. MerT, the 116-residue (12.4 kDa) inner membrane protein, is predicted to have three transmembrane helices, the first of which has a cysteine pair located within the first hydrophobic helix and is accessible from the periplasmic side. The second pair of cysteines in MerT is predicted to lie on the cytoplasmic face of the inner membrane between the second and third transmembrane helices. Hg2+ may be transferred from the N-terminal proximal cysteine pair to form a di-coordinate protein complex with these cysteines and then transferred to cytoplasmic low-molecular mass thiols and/or directly to MerA.
Lead is one of the metals that has the most damaging effects on human health. The interaction of lead with proteins represents a fundamental mechanism by which Pb exerts toxicity (reviewed in [105]). Pb interacts with enzyme functional groups and high-affinity metal-binding sites of various proteins, such as metallothioneins. One of the best documented targets for lead is the second enzyme in the heme biosynthetic pathway, a zinc enzyme called aminolevulinic acid dehydratase (ALAD) or porphobilinogen synthase (PBS) [106]. ALAD contains an unusual Zn2+-Cys3 (i.e., Zn2+-S3) active site and Pb2+ binds to this site in a trigonal pyramidal geometry. It has been concluded that lead avoids four-coordination in sulfur-rich sites and prefers binding as trigonal pyramidal Pb2+-S3 or as Pb2+-S5-8. In the structural zinc binding protein sites, lead binds in a three-coordinate mode, in a geometry that is fundamentally different from the natural coordination of zinc in these sites, and it explains why lead disrupts the structure of these proteins and provides a detailed molecular mechanism of toxicity of lead in this case.
The intracellular concentration of lead at low dosage levels in major target organs such as the kidney and brain is explained by Pb2+ complexation by a group of low molecular weight proteins, which are rich in aspartic and glutamic amino acids [107]. These proteins possess dissociation constant values for Pb2+ of the order of 10−8 M and normally bind zinc. Zawia et al. [108] found that zinc fingers of transcription factors could be potential targets for perturbation by Pb2+. Pb2+ ions interfere with the DNA-binding properties of transcription factors Sp1 and Egr-1, both in vivo and in vitro.
The nervous system is the primary target for the Pb2+ exposure and the developing brain appears to be especially vulnerable to Pb2+ neurotoxicity. Pb2+ has high affinity for free SH groups in enzymes and its binding can alter their function. This may be the reason for the observed inhibition of acetylcholinesterase activity and consequent increase in acetylcholine level. It has been found that the N-methyl-D-aspartate type of excitatory amino acid receptor (NMDAR) is a direct target for Pb2+ in the brain [109].
The most dangerous characteristic of cadmium is that it accumulates throughout a lifetime. Cadmium accumulates mostly in the liver and kidney and has a long biological half-life of 17 to 30 years in humans. Cadmium ions are taken up through calcium channels of the plasma membrane of various cell types, and cadmium is accumulated intracellularly due to its binding to cytoplasmic and nuclear material. It has been found that the intestinal transporter of nonheme iron, divalent metal transporter 1 (DMT1), mediates the transport of Pb2+ and Cd2+ [110]. Himeno et al. [111] found that the transport system for Mn2+ is used for Cd2+ uptake in mammalian cells.
At the cellular level, cadmium affects proliferation, differentiation, and causes apoptosis [112]. Indirect effects of cadmium provoke generation of reactive oxygen species and DNA damage. Once absorbed, cadmium rapidly leaves the blood and concentrates in hepatic and renal tissues, which may be due to their ability to produce large amounts of metallothionein [113]. The presence of metallothionein within cells markedly decreases cadmium toxicity. At elevated cytotoxic concentrations, cadmium inhibits the biosyntheses of DNA, RNA, and protein, and it induces lipid peroxidation, DNA strand breaks, and chromosome aberrations [114]. Two mechanisms that play an important role in cadmium mutagenicity were suggested: (1) induction of reactive oxygen species; and (2) inhibition of DNA repair [115].
p53 protein is a multifunction nuclear factor that is activated in response to multiple forms of stress and controls the proliferation, survival, DNA repair and differentiation of cells exposed to potentially genotoxic DNA damage. p53 protein activity is crucially dependent on the availability of Zn2+ ions and is impaired by exposure to Cd2+, a metal which readily substitutes for Zn2+ in a number of transcription factors. Inactivation by Cd2+ suppresses the p53-dependent responses to DNA damage [116].
In plants, Cd2+ cause various effects, such as inhibition of photosynthesis, respiration, nitrogen metabolism as well as the decrease of water and mineral uptake [117]. Cd2+ is thought to enter plant cells through cation transporters with broad substrate specificity. In cytosol, Cd2+ is bound by chelators, such as phytochelatins and citric acid and then sequestered in the vacuole. Cd2+ causes both the perturbation of intracellular calcium level and interfere with calcium signaling by substituting Ca2+ in calmodulin regulation. Cd2+ can substitute for zinc in coordination complex of zinc-finger motif of transcription factors.
13. Concluding Remarks
The data presented in this review show that metal ions play very important roles in all biological systems and processes. At present, we know many but not all details of their functioning. We know three dimensional structures and metal binding properties of many proteins in vitro. However, we still do not know what their state is in vivo and often even with what metal ion they are complexed in vivo. Moreover, the relationship between metal complexation of proteins and their physiological functions is unknown or known only presumably. Biological systems contain many physiologically significant monovalent, divalent, and trivalent metal cations, which can compete with each other for the same binding sites in proteins or which can bind to separate binding sites. The proteins binding sites can be independent or they can interact with each other (binding cooperativity). Concentrations of these metal cations in different parts of the cell sarcoplasm and in different cell compartments are known badly and the attempts to measure these concentrations experimentally most often disturb the existing ionic equilibria. Just for these reasons, metalloproteomics, i.e., the study of structural, physicochemical, and functional properties of metal binding proteins in their complicated interplay with all metal cations in the cell, is at the beginning of its development.
Funding
This work received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Entry Link on the Encyclopedia Platform
The link to the entry published on the encyclopedia platform: https://encyclopedia.pub/8852.
References
- Permyakov, E.A. Metalloproteomics; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Permyakov, E.A.; Kretsinger, R.H. Calcium Binding Proteins; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Kretsinger, R.H.; Uversky, V.N.; Permyakov, E.A. Encyclopedia of Metalloproteins; Springer: New York, NY, USA, 2013. [Google Scholar]
- Dudev, T.; Lim, C. Principles Governing Mg, Ca, and Zn Binding and Selectivity in Proteins. Chem. Rev. 2003, 103, 773–788. [Google Scholar] [CrossRef]
- Christianson, D.W. Structural Biology of Zinc. Protein Simul. 1991, 42, 281–355. [Google Scholar] [CrossRef]
- Tang, S.; Deng, X.; Jiang, J.; Kirberger, M.; Yang, J.J. Design of Calcium-Binding Proteins to Sense Calcium. Molecular 2020, 25, 2148. [Google Scholar] [CrossRef] [PubMed]
- Elíes, J.; Yáñez, M.; Pereira, T.M.C.; Gil-Longo, J.; MacDougall, D.A.; Campos-Toimil, M. An Update to calcium binding pro-teins. Adv. Exp. Med. Biol. 2020, 1131, 183–213. [Google Scholar] [PubMed]
- Kretsinger, R.H.; Nockolds, C.E. Carp muscle calcium binding protein. II. Structural determination and general description. J. Biol. Chem. 1973, 248, 3313–3326. [Google Scholar] [CrossRef]
- Permyakov, E.A. Parvalbumin; Nova Science Publishers: New York, NY, USA, 2006. [Google Scholar]
- Permyakov, E.A.; Uversky, V.N.; Permyakov, S.E. Parvalbumin as a Pleomorphic Protein. Curr. Protein Pept. Sci. 2017, 18, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Babu, Y.S.; Sack, J.S.; Greenhough, T.J.; Bugg, C.E.; Means, A.R.; Cook, W.J. Three-dimensional structure of calmodulin. Nat. Cell Biol. 1985, 315, 37–40. [Google Scholar] [CrossRef]
- Dürvanger, Z.; Harmat, V. Structural Diversity in Calmodulin—Peptide Interactions. Curr. Protein Pept. Sci. 2019, 20, 1102–1111. [Google Scholar] [CrossRef]
- Sharma, R.K.; Parameswaran, S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. [Google Scholar] [CrossRef]
- Haiech, J.; Moreau, M.; Leclerc, C.; Kilhoffer, M.-C. Facts and conjectures on calmodulin and its cousin proteins, parvalbumin and troponin C. Biochim. Biophys. Acta (BBA)-Bioenerg. 2019, 1866, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Geczy, C.L.; Weber, D.J. S100 proteins. In Encyclopedia of Metalloproteins; Kretsinger, R.H., Uversky, V.N., Permyakov., E.A., Eds.; Springer: New York, NY, USA, 2013; pp. 1863–1874. [Google Scholar]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2020, 1867, 118677. [Google Scholar] [CrossRef]
- Philippov, P.; Koch, K. Neuronal Calcium Sensor Proteins; Nova Science Publishers: New York, NY, USA, 2006. [Google Scholar]
- Burgoyne, R.D.; Helassa, N.; McCue, H.V.; Haynes, L.P. Calcium Sensors in Neuronal Function and Dysfunction. Cold Spring Harb. Perspect. Biol. 2019, 11, a035154. [Google Scholar] [CrossRef]
- Vysotski, E.S.; Lee, J. Ca2+-Regulated Photoproteins: Structural Insight into the Bioluminescence Mechanism. Accounts Chem. Res. 2004, 37, 405–415. [Google Scholar] [CrossRef]
- Eremeeva, E.V.; Vysotski, E.S. Exploring Bioluminescence Function of the Ca2+-regulated Photoproteins with Site-directed Mutagenesis. Photochem. Photobiol. 2018, 95, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Schloer, S.; Pajonczyk, D.; Rescher, U. Annexins in Translational Research: Hidden Treasures to Be Found. Int. J. Mol. Sci. 2018, 19, 1781. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.E.; Morgan, R.O. The annexins. Genome Biol. 2004, 5, 1–8. [Google Scholar] [CrossRef]
- Rizo, J.; Südhof, T.C. C2-domains, Structure and Function of a Universal Ca2+-binding Domain. J. Biol. Chem. 1998, 273, 15879–15882. [Google Scholar] [CrossRef]
- Corbalan-Garcia, S.; Gómez-Fernández, J.C. Signaling through C2 domains: More than one lipid target. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1536–1547. [Google Scholar] [CrossRef]
- Young, H.; Stokes, D. The Mechanics of Calcium Transport. J. Membr. Biol. 2004, 198, 55–63. [Google Scholar] [CrossRef]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepúlveda, M.R.; Vangheluwe, P. Primary Active Ca2+Transport Systems in Health and Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035113. [Google Scholar] [CrossRef]
- Gouaux, E.; MacKinnon, R. Principles of selective ion transport in channels and pumps. Science 2005, 310, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Lunin, V.V.; Dobrovetsky, E.; Khutoreskaya, G.; Zhang, R.; Joachimiak, A.; Doyle, D.A.; Bochkarev, A.; Maguire, M.E.; Edwards, A.M.; Koth, C.M. Crystal structure of the CorA Mg2+ transporter. Nat. Cell Biol. 2006, 440, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S. Zinc coordination sphere in biochemical zinc sites. BioMetals 2001, 14, 271–313. [Google Scholar] [CrossRef]
- Kochańczyk, T.; Drozd, A.; Krężel, A. Relationship between the architecture of zinc coordination and zinc binding affinity in proteins—Insights into zinc regulation. Metallomics 2015, 7, 244–257. [Google Scholar] [CrossRef]
- McCall, K.A.; Huang, C.-C.; Fierke, C.A. Function and Mechanism of Zinc Metalloenzymes. J. Nutr. 2000, 130, 1437S–1446S. [Google Scholar] [CrossRef] [PubMed]
- Shrimpton, C.N.; Smith, A.I.; Lew, R.A. Soluble Metalloendopeptidases and Neuroendocrine Signaling. Endocr. Rev. 2002, 23, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef]
- Bond, J.S.; Beynon, R.J. The astacin family of metalloendopeptidases. Protein Sci. 1995, 4, 1247–1261. [Google Scholar] [CrossRef]
- Bond, J.S. Proteases: History, discovery, and roles in health and disease. J. Biol. Chem. 2019, 294, 1643–1651. [Google Scholar] [CrossRef]
- Tripp, B.C.; Smith, K.; Ferry, J.G. Carbonic anhydrase: New insights for an ancient enzyme. J. Biol. Chem. 2001, 276, 48615–48618. [Google Scholar] [CrossRef]
- Di Fiore, A.; Supuran, C.T.; Scaloni, A.; De Simone, G. Human carbonic anhydrases and post-translational modifications: A hidden world possibly affecting protein properties and functions. J. Enzym. Inhib. Med. Chem. 2020, 35, 1450–1461. [Google Scholar] [CrossRef]
- Culotta, V.C.; Yanga, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Bio-chim. Biophys. Acta. 2006, 1763, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxid. Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. BioMetals 2001, 14, 331–341. [Google Scholar] [CrossRef]
- Nakamura, K.; Go, N. Function and molecular evolution of multicopper blue proteins. Cell. Mol. Life Sci. 2005, 62, 2050–2066. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M.; Kaplan, J.H. Copper transporters and copper chaperones: Roles in cardiovascular physiology and disease. Am. J. Physiol. Physiol. 2018, 315, C186–C201. [Google Scholar] [CrossRef]
- Ariöz, C.; Wittung-Stafshede, P.Q. Folding of copper proteins: Role of the metal? Rev. Biophys. 2018, 51, e4. [Google Scholar] [CrossRef]
- Warren, J.J.; Lancaster, K.M.; Richards, J.H.; Gray, H.B. Inner- and outer-sphere metal coordination in blue copper proteins. J. Inorg. Biochem. 2012, 115, 119–126. [Google Scholar] [CrossRef]
- Pérez-Henarejos, S.A.; Alcaraz, L.A.; Donaire, A. Blue Copper Proteins: A rigid machine for efficient electron transfer, a flexible device for metal uptake. Arch. Biochem. Biophys. 2015, 584, 134–148. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, I.S.; Murphy, M.E.P. Type-2 copper-containing enzymes. Cell. Mol. Life Sci. 2007, 64, 2887–2899. [Google Scholar] [CrossRef]
- Claus, H.; Decker, H. Bacterial tyrosinases. Syst. Appl. Microbiol. 2006, 29, 3–14. [Google Scholar] [CrossRef]
- Gray, H.B.; Malmström, B.G.; Williams, R. Copper coordination in blue proteins. JBIC J. Biol. Inorg. Chem. 2000, 5, 551–559. [Google Scholar] [CrossRef]
- De Rienzo, F.; Gabdoulline, R.R.; Wade, R.C.; Sola, M.; Menziani, M.C. Computational approaches to structural and functional analysis of plastocyanin and other blue copper proteins. Cell. Mol. Life Sci. 2004, 61, 1123–1142. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Davidson, V.L. Cupredoxins—A study of how proteins may evolve to use metals for bioenergetic processes. Metallomics 2011, 3, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R.; Yeates, T.O.; Merchant, S. Plastocyanin: Structural and functional analysis. J. Bioenerg. Biomembr. 1994, 26, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Wittung-Stafshede, P. Role of Cofactors in Folding of the Blue-Copper Protein Azurin. Inorg. Chem. 2004, 43, 7926–7933. [Google Scholar] [CrossRef]
- Burstein, E.; Permyakov, E.; Yashin, V.; Burkhanov, S.; Agrò, A.F. The fine structure of luminescence spectra of azurin. Biochim. Biophys. Acta (BBA)-Protein Struct. 1977, 491, 155–159. [Google Scholar] [CrossRef]
- Chen, P.; Solomon, E.I. O2 activation by binuclear Cu sites: Noncoupled versus exchange coupled reaction mechanisms. Proc. Natl. Acad. Sci. USA 2004, 101, 13105–13110. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, G.; Palese, L.L.; Papa, F.; Papa, S. Allosteric Cooperativity in Proton Energy Conversion in A1-Type Cytochrome c Oxidase. J. Mol. Biol. 2020, 432, 534–551. [Google Scholar] [CrossRef]
- Llases, M.E.; Morgada, M.N.; Vila, A.J. Biochemistry of copper site assembly in heme-copper oxidases: A theme with varia-tions. Int. J. Mol. Sci. 2019, 20, 3830. [Google Scholar] [CrossRef]
- Tavares, P.; Pereira, A.S.; Moura, J.; Moura, I. Metalloenzymes of the denitrification pathway. J. Inorg. Biochem. 2006, 100, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Coates, C.J.; Costa-Paiva, E.M. Multifunctional Roles of Hemocyanins. Regul. Proteolysis Microorg. 2020, 94, 233–250. [Google Scholar] [CrossRef]
- Dameron, C.T.; Harrison, M.D. Mechanisms for protection against copper toxicity. Am. J. Clin. Nutr. 1998, 67, 1091S–1097S. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Hanck-Silva, G.; Baveloni, F.G.; Junior, J.A.O.; De Lima, F.T.; Eloy, J.O.; Chorilli, M. A Review of Properties, Delivery Systems and Analytical Methods for the Characterization of Monomeric Glycoprotein Transferrin. Crit. Rev. Anal. Chem. 2020, 1–12. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, X.; Zhao, G. Ferritin Nanocage: A Versatile Nanocarrier Utilized in the Field of Food, Nutrition, and Medicine. Nanomaterials 2020, 10, 1894. [Google Scholar] [CrossRef]
- Ordway, G.A.; Garry, D.J. Myoglobin: An essential hemoprotein in striated muscle. J. Exp. Biol. 2004, 207, 3441–3446. [Google Scholar] [CrossRef]
- Linberg, R.; Conover, C.D.; Shum, K.L.; Shorr, R.G. Hemoglobin based oxygen carriers: How much methemoglobin is too much? Artif. Cells Blood Substit. Immobil. Biotechnol. 1998, 26, 133–148. [Google Scholar]
- Olson, J.S. Lessons Learned from 50 Years of Hemoglobin Research: Unstirred and Cell-Free Layers, Electrostatics, Baseball Gloves, and Molten Globules. Antioxid. Redox Signal. 2020, 32, 228–246. [Google Scholar] [CrossRef]
- Brittain, T. Root effect hemoglobins. J. Inorg. Biochem. 2005, 99, 120–129. [Google Scholar] [CrossRef]
- González-Arzola, K.; Velázquez-Cruz, A.; Guerra-Castellano, A.; Casado-Combreras, M.Á.; Pérez-Mejías, G.; Díaz-Quintana, A.; Díaz-Moreno, I.; De la Rosa, M.Á. New moonlighting functions of mitochondrial cytochrome c in the cytoplasm and nu-cleus. FEBS Lett. 2019, 593, 3101–3119. [Google Scholar] [CrossRef]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef]
- Bertini, I.; Cavallaro, G.; Rosato, A. Cytochrome c: Occurrence and Functions. Chem. Rev. 2006, 37, 90–115. [Google Scholar] [CrossRef] [PubMed]
- Zeida, A.; Trujillo, M.; Ferrer-Sueta, G.; DeNicola, A.; Estrin, D.A.; Radi, R. Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols. Chem. Rev. 2019, 119, 10829–10855. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gomme, P.T.; McCann, K.B.; Bertolini, J. Transferrin: Structure, function and potential therapeutic actions. Drug Discov. Today 2005, 10, 267–273. [Google Scholar] [CrossRef]
- Baker, E.N.; Baker, H.M.; Kidd, R.D. Lactoferrin and transferrin: Functional variations on a common structural framework. Biochem. Cell Biol. 2002, 80, 27–34. [Google Scholar] [CrossRef]
- Carrondo, M.A. New Embo Member’s Review: Ferritins, iron uptake and storage from the bacterioferritin viewpoint. EMBO J. 2003, 22, 1959–1968. [Google Scholar] [CrossRef]
- Theil, E.C. Ferritin. In Handbook of Metalloproteins; Messerschmidt, A., Poulos, H.R., Weighardt, K., Eds.; Wiley: Chichester, UK, 2001; Volume 2, pp. 771–781. [Google Scholar]
- Braymer, J.J.; Freibert, S.A.; Rakwalska-Bange, M.; Lill, R. Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochim. Biophys. Acta (BBA)-Bioenerg. 2021, 1868, 118863. [Google Scholar] [CrossRef]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, Function, and Formation of Biological Iron-Sulfur Clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.; Lill, R. Biogenesis of iron-sulfur proteins in eukaryotes: Components, mechanism and pathology. Mitochondrion 2002, 2, 71–86. [Google Scholar] [CrossRef]
- Rees, D.C. Great Metalloclusters in Enzymology. Annu. Rev. Biochem. 2002, 71, 221–246. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J. Ferredoxins of the third kind. FEBS Lett. 2001, 509, 1–5. [Google Scholar] [CrossRef]
- Sticht, H.; Rösch, P. The structure of iron-sulfur proteins. Prog. Biophys. Mol. Biol. 1998, 70, 95–136. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.A. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Jarrett, J.T. The novel structure and chemistry of iron–sulfur clusters in the adenosylmethionine-dependent radical enzyme biotin synthase. Arch. Biochem. Biophys. 2005, 433, 312–321. [Google Scholar] [CrossRef]
- Mendel, R.R.; Bittner, F. Cell biology of molybdenum. Biochim. Biophys. Acta (BBA)-Bioenerg. 2006, 1763, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Hille, R. Molybdenum and tungsten in biology. Trends Biochem. Sci. 2002, 27, 360–367. [Google Scholar] [CrossRef]
- Brondino, C.D.; Romão, M.J.; Moura, I.; Moura, J.J. Molybdenum and tungsten enzymes: The xanthine oxidase family. Curr. Opin. Chem. Biol. 2006, 10, 109–114. [Google Scholar] [CrossRef]
- Feng, C.; Tollin, G.; Enemark, J.H. Sulfite oxidizing enzymes. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2007, 1774, 527–539. [Google Scholar] [CrossRef]
- Zambelli, B.; Uversky, V.N.; Ciurli, S. Nickel impact on human health: An intrinsic disorder perspective. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2016, 1864, 1714–1731. [Google Scholar] [CrossRef]
- Volbeda, A.; Fontecilla-Camps, J.C.; Frey, M. Novel metal sites in protein structures. Curr. Opin. Struct. Biol. 1996, 6, 804–812. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimizu, S. Cobalt proteins. JBIC J. Biol. Inorg. Chem. 1999, 261, 1–9. [Google Scholar] [CrossRef]
- Barondeau, D.P.; Getzoff, E.D. Structural insights into protein–metal ion partnerships. Curr. Opin. Struct. Biol. 2004, 14, 765–774. [Google Scholar] [CrossRef]
- Wuerges, J.; Lee, J.-W.; Yim, Y.-I.; Yim, H.-S.; Kang, S.-O.; Carugo, K.D. Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc. Natl. Acad. Sci. USA 2004, 101, 8569–8574. [Google Scholar] [CrossRef]
- Christianson, D.W.; Cox, J.D. Catalysis By Metal-Activated Hydroxide in Zinc and Manganese Metalloenzymes. Annu. Rev. Biochem. 1999, 68, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, E.M.; Gascón, J.A.; McEvoy, J.P.; Brudvig, G.W.; Batista, V.S. Quantum mechanics/molecular mechanics structural models of the oxygen-evolving complex of photosystem II. Curr. Opin. Struct. Biol. 2007, 17, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, S.; Höller, S.; Meier, B.; Peiter, E. Manganese in Plants: From Acquisition to Subcellular Allocation. Front. Plant Sci. 2020, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Siddappa, S.; Marathe, G.K. What we know about plant arginases? Plant Physiol. Biochem. 2020, 156, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Roux, B. Ion channels and ion selectivity. Essays Biochem. 2017, 61, 201–209. [Google Scholar]
- Burdette, S.C.; Lippard, S.J. Meeting of the minds: Metalloneurochemistry. Proc. Natl. Acad. Sci. USA 2003, 100, 3605–3610. [Google Scholar] [CrossRef]
- Robertson, J.L.; Roux, B. One Channel: Open and Closed. Structure 2005, 13, 1398–1400. [Google Scholar] [CrossRef] [PubMed]
- Zhorov, B.S.; Tikhonov, D.B. Potassium, sodium, calcium and glutamate-gated channels: Pore architecture and ligand action. J. Neurochem. 2004, 88, 782–799. [Google Scholar] [CrossRef]
- Rakowski, R.F.; Sagar, S. Found: Na+ and K+ Binding Sites of the Sodium Pump. News Physiol. Sci. 2003, 18, 164–168. [Google Scholar] [CrossRef]
- Di Cera, E. A Structural Perspective on Enzymes Activated by Monovalent Cations. J. Biol. Chem. 2006, 281, 1305–1308. [Google Scholar] [CrossRef]
- Page, M.J.; Di Cera, E. Role of Na+and K+in Enzyme Function. Physiol. Rev. 2006, 86, 1049–1092. [Google Scholar] [CrossRef]
- Krem, M.M.; Di Cera, E. Molecular markers of serine protease evolution. EMBO J. 2001, 20, 3036–3045. [Google Scholar] [CrossRef]
- Barkay, T.; Miller, S.M.; Summers, A.O. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol. Rev. 2003, 27, 355–384. [Google Scholar] [CrossRef]
- Goering, P.L. Lead-protein interactions as a basis for lead toxicity. NeuroToxicol. 1993, 14, 45–60. [Google Scholar]
- Magyar, J.S.; Weng, T.C.; Stern, C.M.; Dye, D.F.; Rous, B.W.; Payne, J.C.; Bridgewater, B.M.; Mijovilovich, A.; Parkin, G.; Zaleski, J.M.; et al. Reexamination of lead(II) coordination preferences in sulfur-rich sites: Implications for a critical mechanism of lead poisoning. J. Am. Chem. Soc. 2005, 127, 9495–9505. [Google Scholar] [CrossRef]
- Fowler, B.A. Roles of lead-binding proteins in mediating lead bioavailability. Environ. Health Perspect. 1998, 106, 1585–1587. [Google Scholar] [CrossRef] [PubMed]
- Zawia, N.H.; Crumpton, T.; Brydie, M.; Reddy, G.R.; Razmiafshari, M. Disruption of the zinc finger domain: A common target that underlies many of the effects of lead. Neurotoxicology 2000, 21, 1069–1080. [Google Scholar] [PubMed]
- Toscano, C.D.; Guilarte, T.R. Lead neurotoxicity: From exposure to molecular effects. Brain Res. Rev. 2005, 49, 529–554. [Google Scholar] [CrossRef] [PubMed]
- Bressler, J.P.; Olivi, L.; Cheong, J.H.; Kim, Y.; Bannona, D. Divalent Metal Transporter 1 in Lead and Cadmium Transport. Ann. N. Y. Acad. Sci. 2004, 1012, 142–152. [Google Scholar] [CrossRef]
- Himeno, S.; Yanagiya, T.; Enomoto, S.; Kondo, Y.; Imura, N. Cellular Cadmium Uptake Mediated by the Transport System for Manganese. Tohoku J. Exp. Med. 2002, 196, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Bertin, G.; Averbeck, D. Cadmium: Cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochime 2006, 88, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Liu, J.; Choudhuri, S. Metallothionein: An Intracellular Protein to Protect Against Cadmium Toxicity. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 267–294. [Google Scholar] [CrossRef]
- Beyersmann, D.; Hechtenberg, S. Cadmium, Gene Regulation, and Cellular Signalling in Mammalian Cells. Toxicol. Appl. Pharmacol. 1997, 144, 247–261. [Google Scholar] [CrossRef]
- Filipič, M.; Fatur, T.; Vudrag, M. Molecular mechanisms of cadmium induced mutagenicity. Hum. Exp. Toxicol. 2006, 25, 67–77. [Google Scholar] [CrossRef]
- Méplan, C.; Verhaegh, G.; Richard, M.-J.; Hainaut, P. Metal ions as regulators of the conformation and function of the tumour suppressor protein p53: Implications for carcinogenesis. Proc. Nutr. Soc. 1999, 58, 565–571. [Google Scholar] [CrossRef]
- Deckert, J. Cadmium Toxicity in Plants: Is There any Analogy to its Carcinogenic Effect in Mammalian Cells? BioMetals 2005, 18, 475–481. [Google Scholar] [CrossRef]
Figure 1.
Ribbon model of X-ray structure of Ca2+-bound bovine α-lactalbumin (PDB ID 1hfz). Gray sphere represents bound calcium ion; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.
Figure 1.
Ribbon model of X-ray structure of Ca2+-bound bovine α-lactalbumin (PDB ID 1hfz). Gray sphere represents bound calcium ion; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.

Figure 2.
Ribbon model of X-ray structure of a single Zn-finger (PDB ID 3cnf) in a human enhancer binding protein. Gray sphere represents bound Zn ion; amino acid residues taking part in its coordination are represented by stick model; nitrogen atoms are shown in blue, while sulfur atoms are shown in yellow. New Molsoft Molecular Browser.
Figure 2.
Ribbon model of X-ray structure of a single Zn-finger (PDB ID 3cnf) in a human enhancer binding protein. Gray sphere represents bound Zn ion; amino acid residues taking part in its coordination are represented by stick model; nitrogen atoms are shown in blue, while sulfur atoms are shown in yellow. New Molsoft Molecular Browser.

Figure 3.
Ribbon model of X-ray structure of Pseudomonas aeruginosa azurin (PDB ID 3fsz). Gray sphere represents bound Cu ion, and amino acid residues taking part in its coordination are represented by stick model; nitrogen atoms are shown in blue, while sulfur atoms are shown in yellow. New Molsoft Molecular Browser.
Figure 3.
Ribbon model of X-ray structure of Pseudomonas aeruginosa azurin (PDB ID 3fsz). Gray sphere represents bound Cu ion, and amino acid residues taking part in its coordination are represented by stick model; nitrogen atoms are shown in blue, while sulfur atoms are shown in yellow. New Molsoft Molecular Browser.

Figure 4.
Ribbon model of X-ray structure of Ca2+-bound EF-domain of carp parvalbumin (PDB ID 4cpv). Gray sphere represents bound calcium ion; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.
Figure 4.
Ribbon model of X-ray structure of Ca2+-bound EF-domain of carp parvalbumin (PDB ID 4cpv). Gray sphere represents bound calcium ion; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.

Figure 5.
Ribbon model of X-ray structure of Ca2+-bound Drosophila melanogaster calmodulin (PDB ID 4cln). Gray spheres represent bound calcium ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.
Figure 5.
Ribbon model of X-ray structure of Ca2+-bound Drosophila melanogaster calmodulin (PDB ID 4cln). Gray spheres represent bound calcium ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.

Figure 6.
Ribbon model of X-ray structure of Ca2+-bound bovine annexin 6 (PDB ID 1avc). Gray spheres represent bound calcium ions. New Molsoft Molecular Browser.
Figure 6.
Ribbon model of X-ray structure of Ca2+-bound bovine annexin 6 (PDB ID 1avc). Gray spheres represent bound calcium ions. New Molsoft Molecular Browser.

Figure 7.
Ribbon model of X-ray structure of Ca2+-bound C2 domain (PDB ID 3w57). Gray spheres represent bound calcium ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.
Figure 7.
Ribbon model of X-ray structure of Ca2+-bound C2 domain (PDB ID 3w57). Gray spheres represent bound calcium ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red. New Molsoft Molecular Browser.

Figure 8.
Ribbon model of X-ray structure of sarcoplasmic reticulum Ca2+-ATPase of pig heart (PDB ID 6HXB). Gray spheres represent bound calcium ions. New Molsoft Molecular Browser.
Figure 8.
Ribbon model of X-ray structure of sarcoplasmic reticulum Ca2+-ATPase of pig heart (PDB ID 6HXB). Gray spheres represent bound calcium ions. New Molsoft Molecular Browser.

Figure 9.
Ribbon model of X-ray structure of Bacillus subtilis carboxypeptidase (PDB ID 3hq2). Black spheres represent bound zinc ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red; nitrogen atoms are shown in blue. New Molsoft Molecular Browser.
Figure 9.
Ribbon model of X-ray structure of Bacillus subtilis carboxypeptidase (PDB ID 3hq2). Black spheres represent bound zinc ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red; nitrogen atoms are shown in blue. New Molsoft Molecular Browser.

Figure 10.
Ribbon model of X-ray structure of E. coli Cu,Zn superoxide dismutase (PDB ID 2gbt). Black spheres represent bound zinc (upper sphere) and copper ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red; nitrogen atoms are shown in blue. New Molsoft Molecular Browser.
Figure 10.
Ribbon model of X-ray structure of E. coli Cu,Zn superoxide dismutase (PDB ID 2gbt). Black spheres represent bound zinc (upper sphere) and copper ions; amino acid residues taking part in its coordination are represented by stick model; oxygen atoms are shown in red; nitrogen atoms are shown in blue. New Molsoft Molecular Browser.

Figure 11.
The structure of copper binding site in plastocyanin (PDB ID 1jxd). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.
Figure 11.
The structure of copper binding site in plastocyanin (PDB ID 1jxd). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.

Figure 12.
Ribbon model of X-ray structure of plastocyanin (PDB ID 1jxd). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.
Figure 12.
Ribbon model of X-ray structure of plastocyanin (PDB ID 1jxd). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.

Figure 13.
Binuclear copper binding site in cytochrome с oxidase (PDB ID 6pty). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.
Figure 13.
Binuclear copper binding site in cytochrome с oxidase (PDB ID 6pty). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.

Figure 14.
Cluster of four copper and one sulfur (yellow) atoms in N2O-reductase (PDB ID 7apy). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.
Figure 14.
Cluster of four copper and one sulfur (yellow) atoms in N2O-reductase (PDB ID 7apy). Nitrogen atoms are shown in blue; sulfur atoms are shown in yellow. New Molsoft Molecular Browser.

Figure 15.
Structure of Fe-porphyrin and its complex with O2.

Figure 16.
Ribbon model of X-ray structure of sperm whale myoglobin with bound heme (yellow) (PDB ID 5hlq). New Molsoft Molecular Browser.
Figure 16.
Ribbon model of X-ray structure of sperm whale myoglobin with bound heme (yellow) (PDB ID 5hlq). New Molsoft Molecular Browser.

Figure 17.
Ribbon model of X-ray structure of human oxy T state hemoglobin with bound heme molecules (PDB ID 1gzx). New Molsoft Molecular Browser.
Figure 17.
Ribbon model of X-ray structure of human oxy T state hemoglobin with bound heme molecules (PDB ID 1gzx). New Molsoft Molecular Browser.

Figure 18.
Ribbon model of X-ray structure of Lithobates catesbeianus ferritin (PDB ID 1mfr). New Molsoft Molecular Browser.
Figure 18.
Ribbon model of X-ray structure of Lithobates catesbeianus ferritin (PDB ID 1mfr). New Molsoft Molecular Browser.

Figure 19.
Ribbon model of X-ray structure of Methanosarcina acetivorans nitrogenase with (4Fe-4S) cluster (PDB ID 6nzj). Fe atoms are shown in red. New Molsoft Molecular Browser.
Figure 19.
Ribbon model of X-ray structure of Methanosarcina acetivorans nitrogenase with (4Fe-4S) cluster (PDB ID 6nzj). Fe atoms are shown in red. New Molsoft Molecular Browser.

Figure 20.
(4Fe-4S) and (2Fe-S2) clusters in E. coli biotin synthase (PDB ID 1r30). New Molsoft Molecular Browser.
Figure 20.
(4Fe-4S) and (2Fe-S2) clusters in E. coli biotin synthase (PDB ID 1r30). New Molsoft Molecular Browser.

Figure 21.
Molybdenum cofactor: complex of pterin with molybdenum.

Figure 22.
Ribbon model of X-ray structure of bovine xanthine oxidase with [2Fe-2S] cluster and bound Mo (PDB ID 1fiq). New Molsoft Molecular Browser.
Figure 22.
Ribbon model of X-ray structure of bovine xanthine oxidase with [2Fe-2S] cluster and bound Mo (PDB ID 1fiq). New Molsoft Molecular Browser.

Figure 23.
Ribbon model of X-ray structure of E. coli methionine aminopeptidase with bound Co (black) and Na (blue) (PDB ID 1c21). New Molsoft Molecular Browser.
Figure 23.
Ribbon model of X-ray structure of E. coli methionine aminopeptidase with bound Co (black) and Na (blue) (PDB ID 1c21). New Molsoft Molecular Browser.

Figure 24.
Ribbon model of X-ray structure of rat arginase with bound Mn2+ (PDB ID 1t4p). New Molsoft Molecular Browser.
Figure 24.
Ribbon model of X-ray structure of rat arginase with bound Mn2+ (PDB ID 1t4p). New Molsoft Molecular Browser.

Figure 25.
Coordination of Mn in rat arginase (PDB ID 1t4p). Oxygen atoms are shown in red; nitrogen atoms are shown in blue. New Molsoft Molecular Browser.
Figure 25.
Coordination of Mn in rat arginase (PDB ID 1t4p). Oxygen atoms are shown in red; nitrogen atoms are shown in blue. New Molsoft Molecular Browser.

Figure 26.
Ribbon model of X-ray structure of potassium channel with three potassium ions inside (PDB ID 1bl8). New Molsoft Molecular Browser.
Figure 26.
Ribbon model of X-ray structure of potassium channel with three potassium ions inside (PDB ID 1bl8). New Molsoft Molecular Browser.

Figure 27.
Ribbon model of X-ray structure of α-chain of diol dehydratase with two bound potassium ions (PDB ID 5ysh)). Oxygen atoms are shown in red. New Molsoft Molecular Browser.
Figure 27.
Ribbon model of X-ray structure of α-chain of diol dehydratase with two bound potassium ions (PDB ID 5ysh)). Oxygen atoms are shown in red. New Molsoft Molecular Browser.


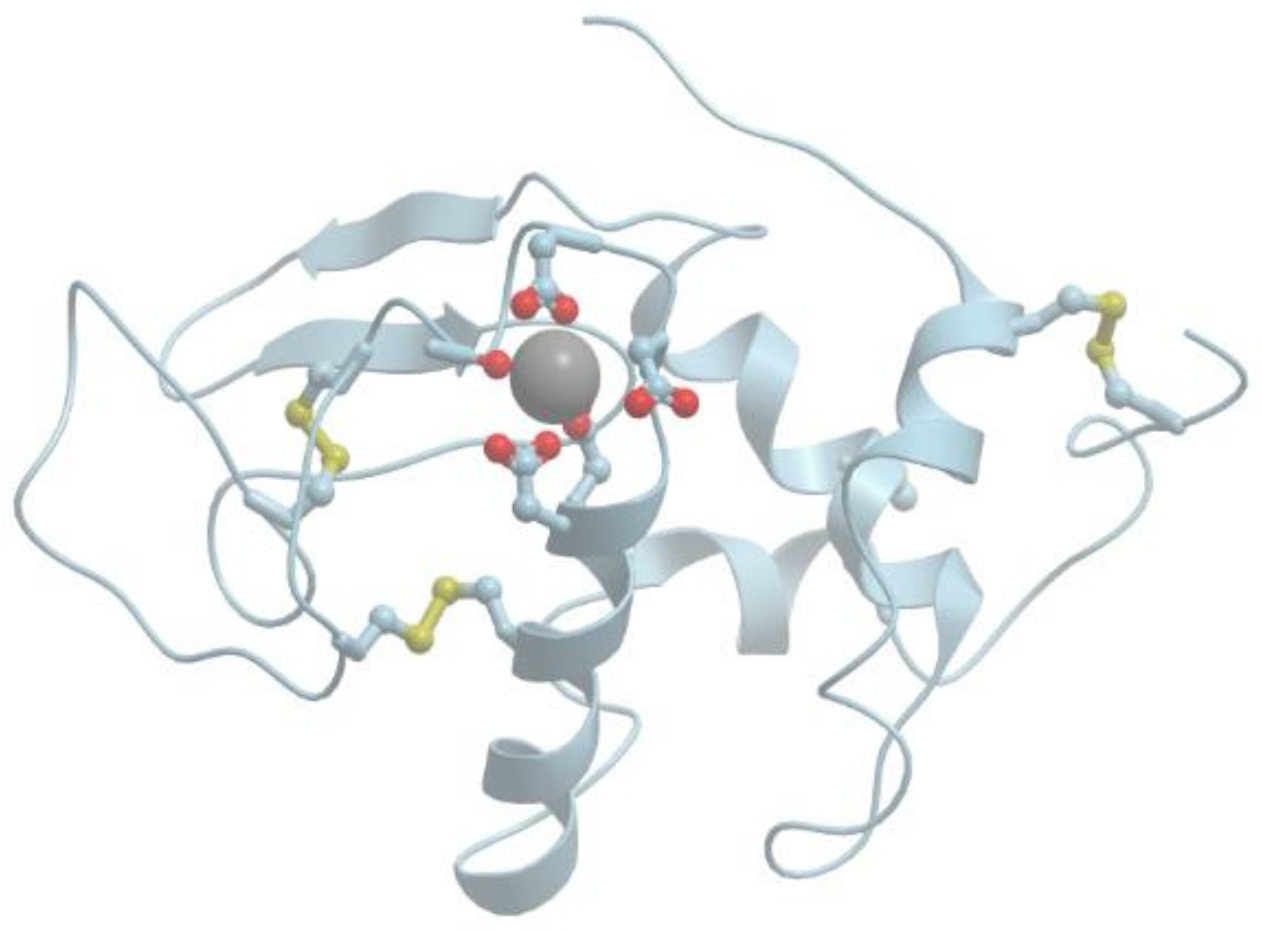{kind=link}
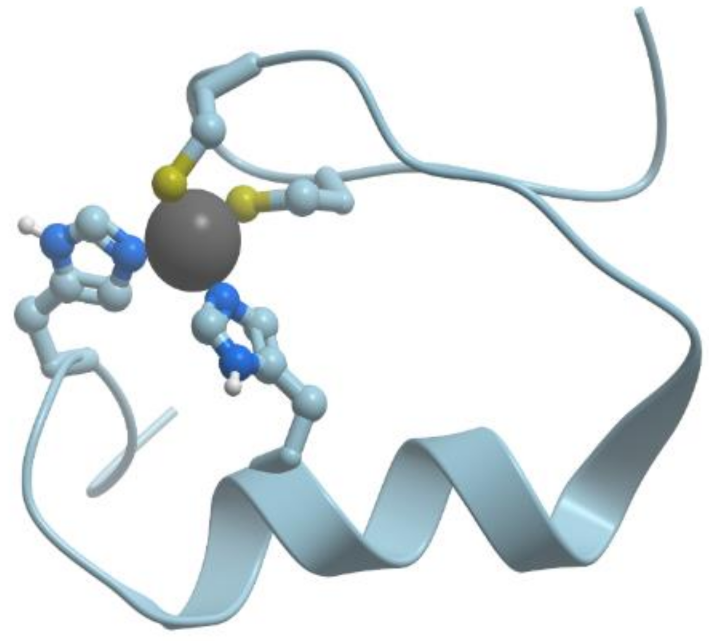{kind=link}
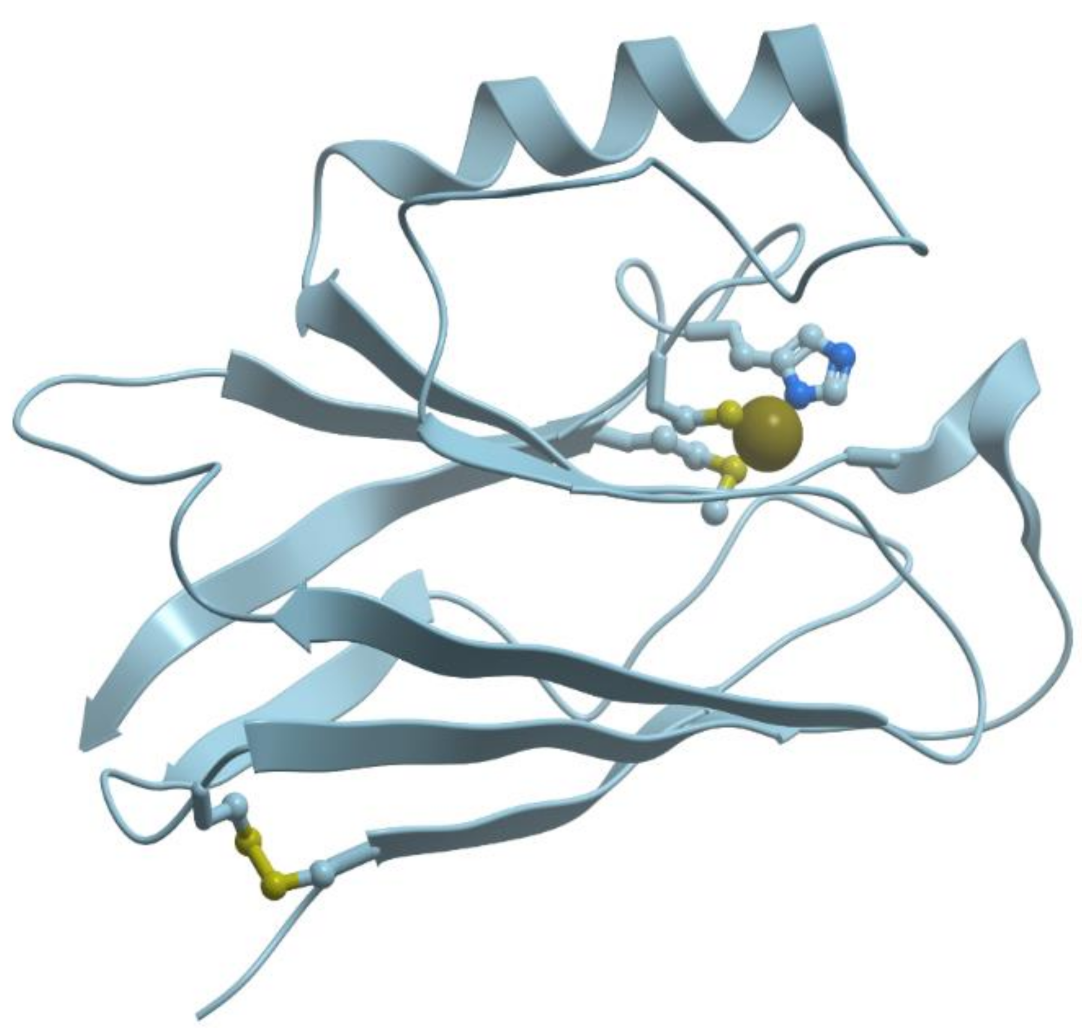{kind=link}
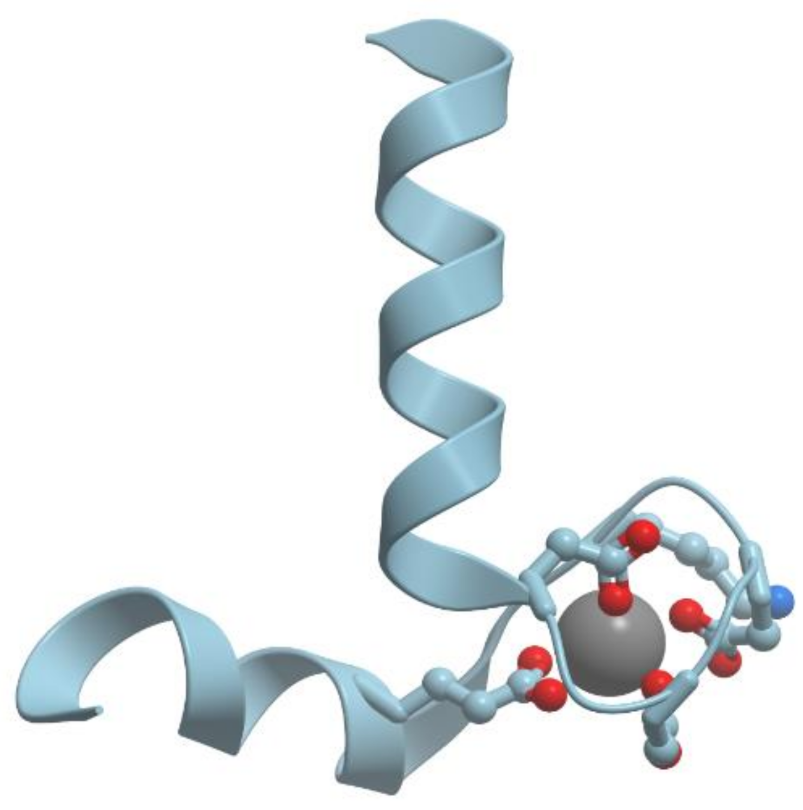{kind=link}
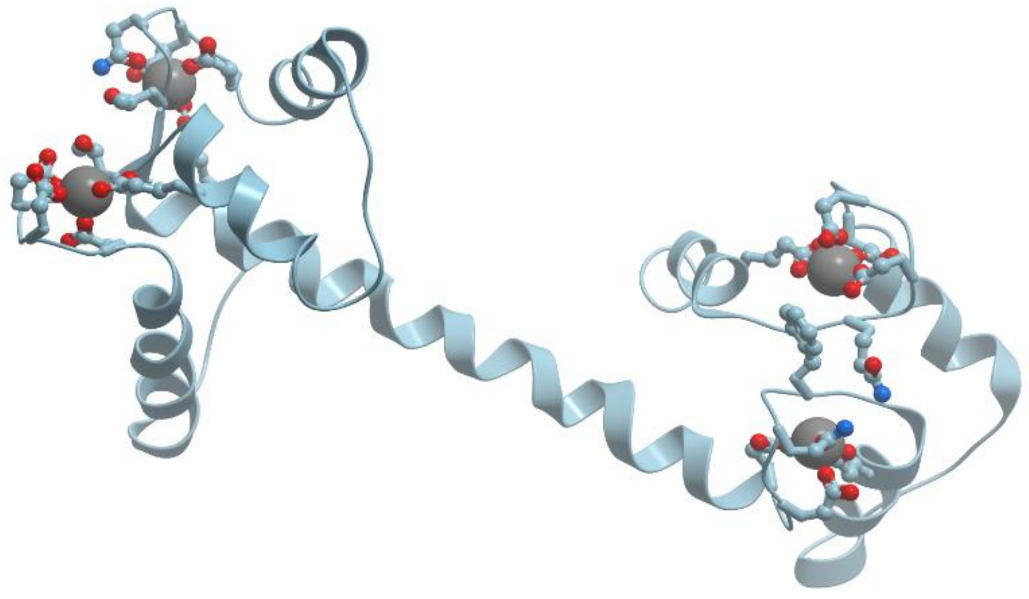{kind=link}
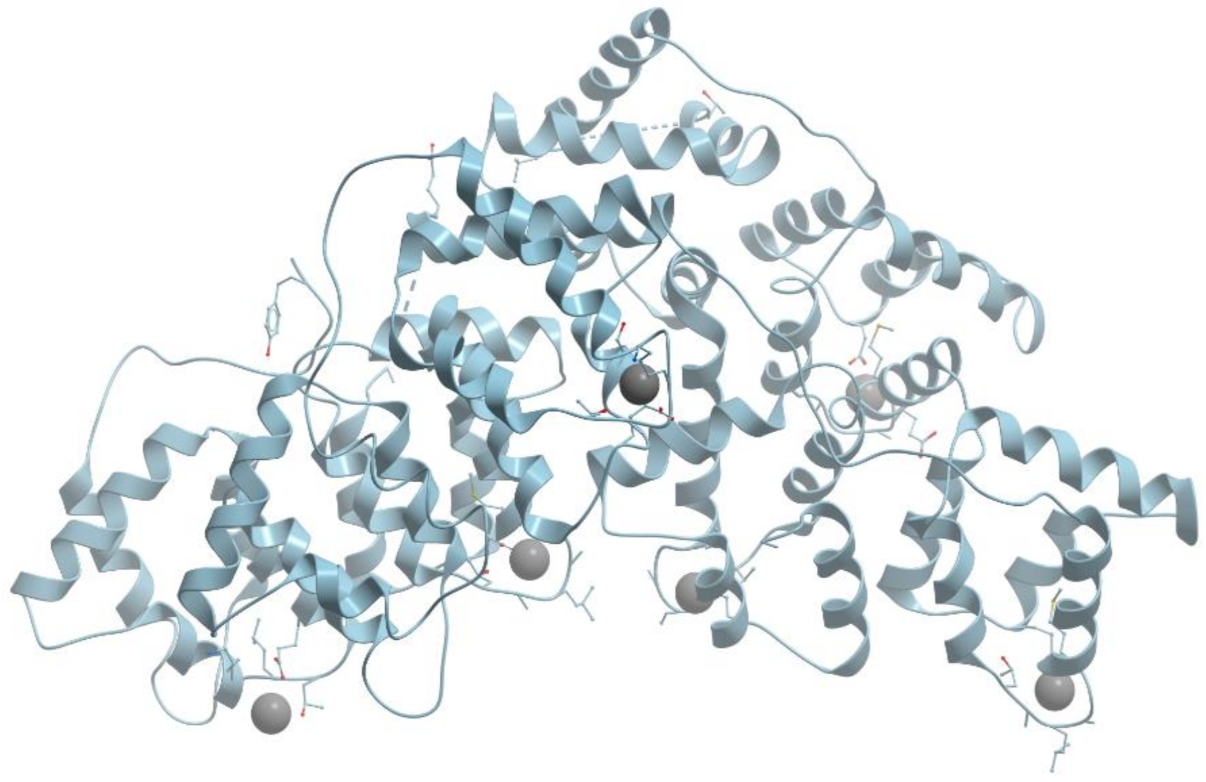{kind=link}
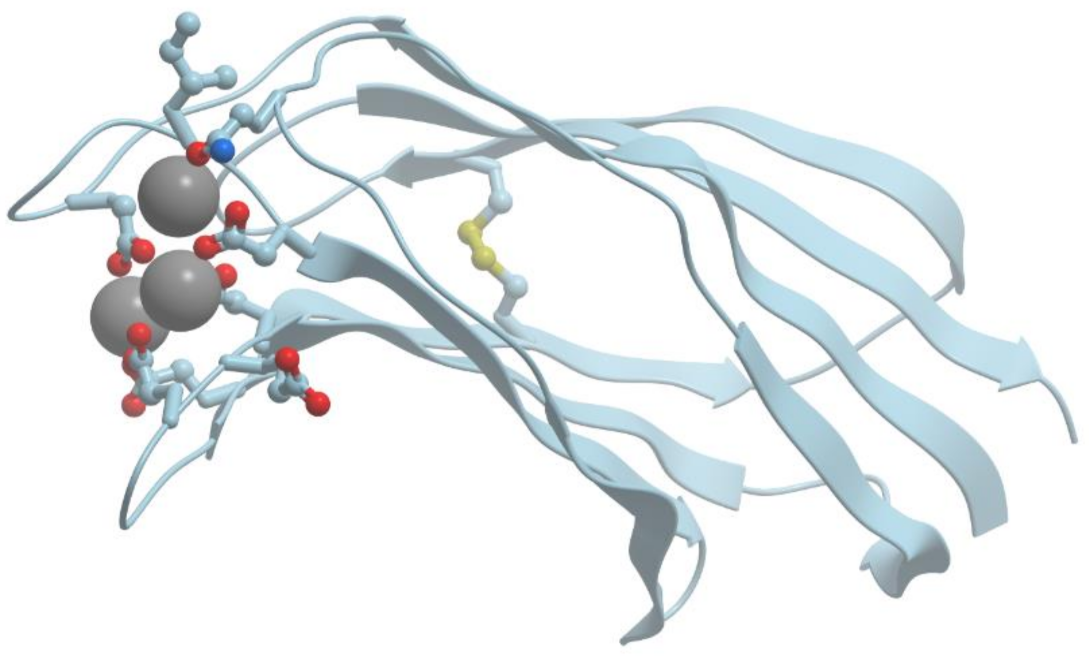{kind=link}
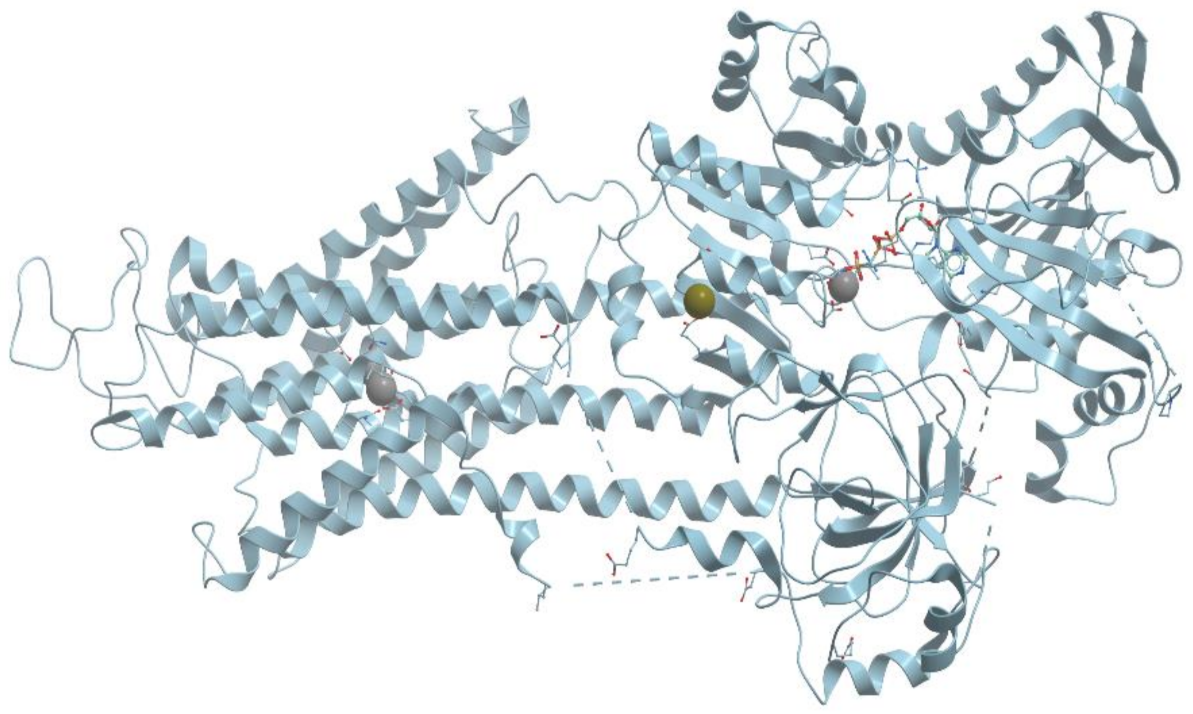{kind=link}
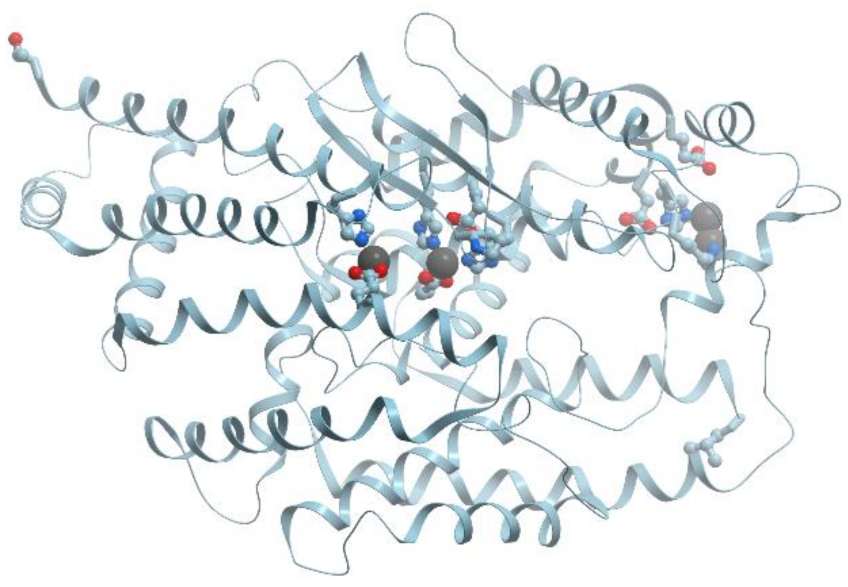{kind=link}
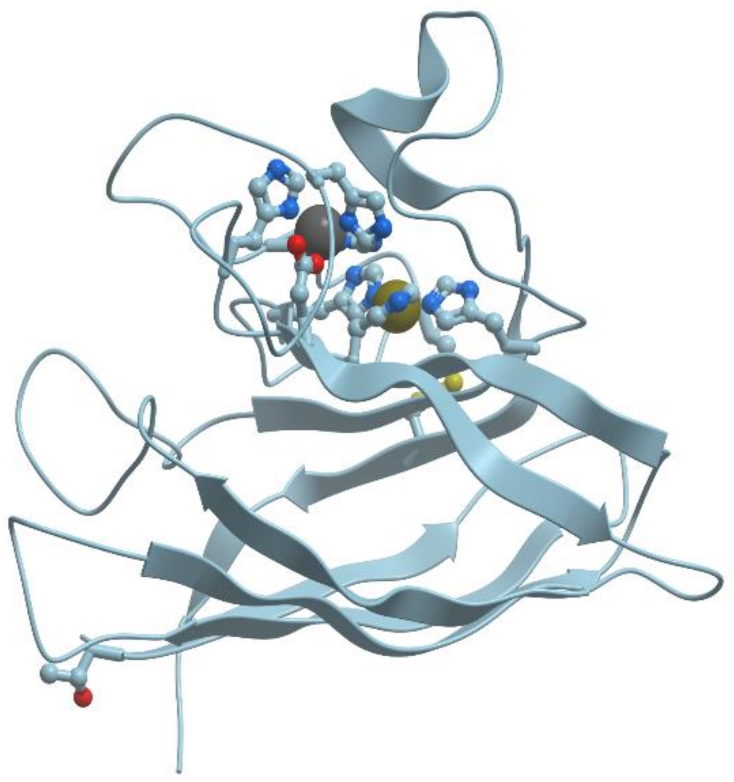{kind=link}
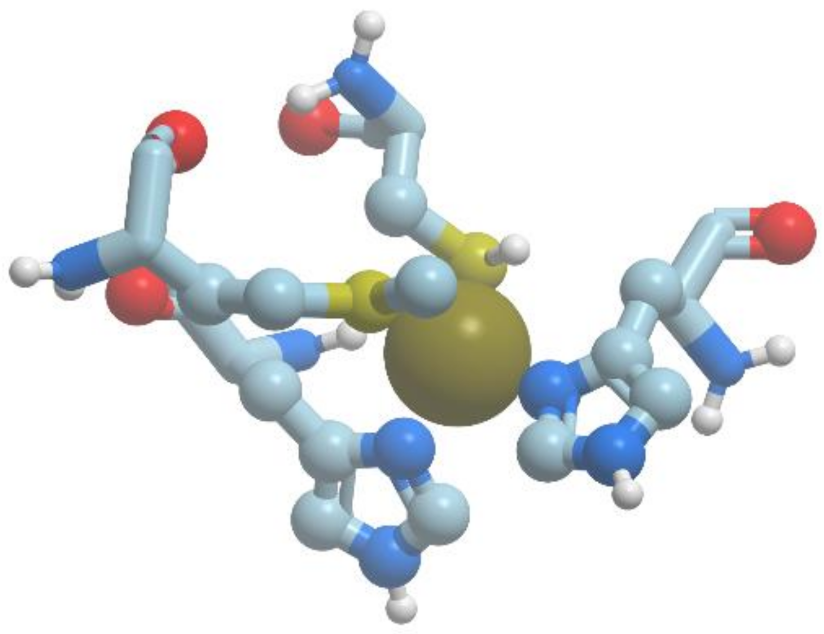{kind=link}
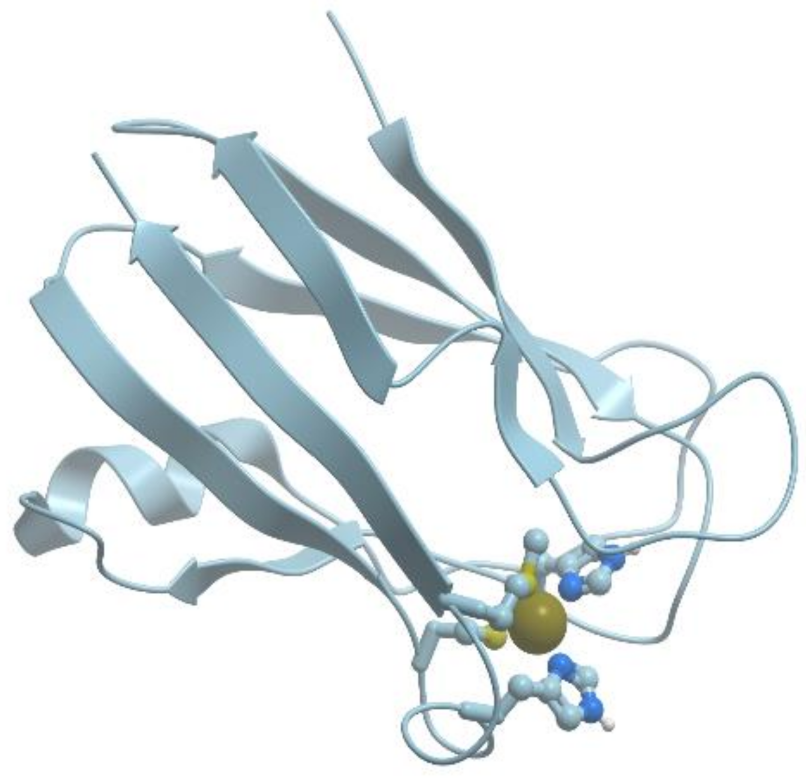{kind=link}
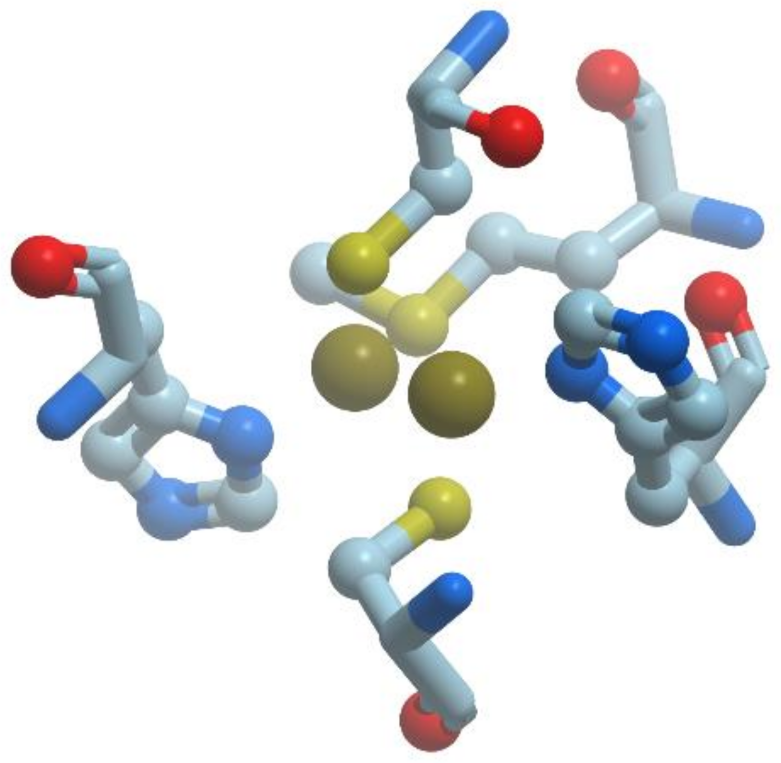{kind=link}
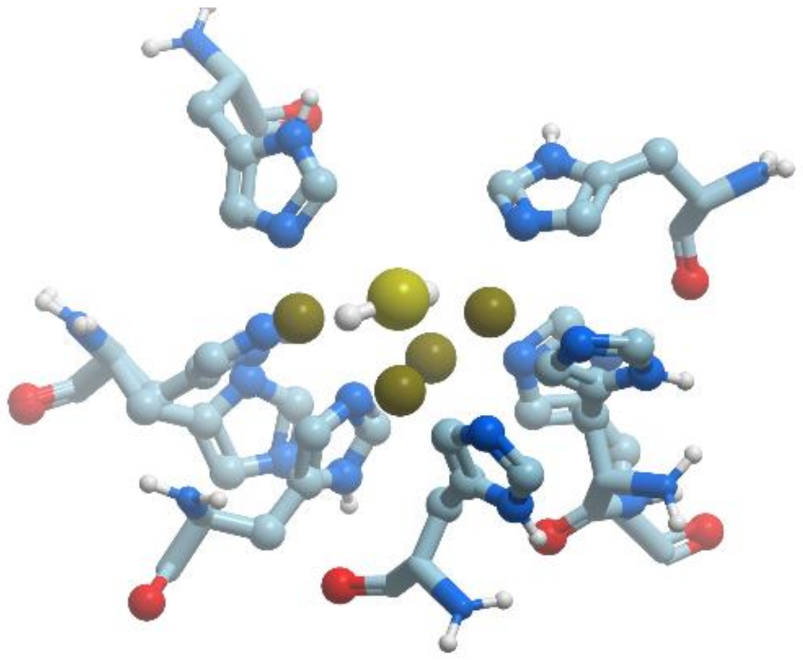{kind=link}
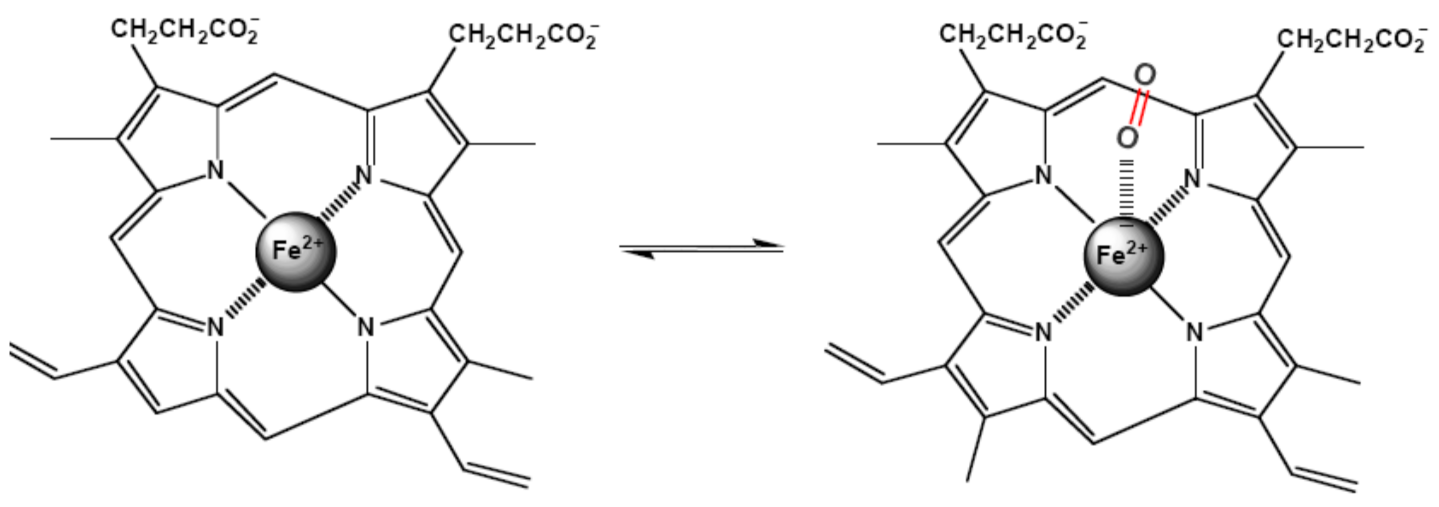{kind=link}
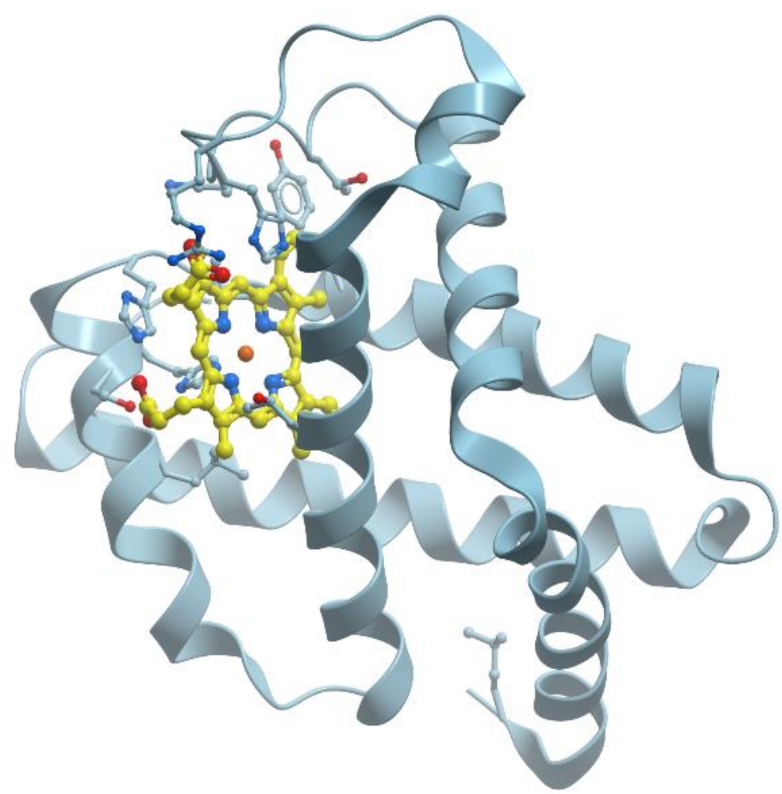{kind=link}
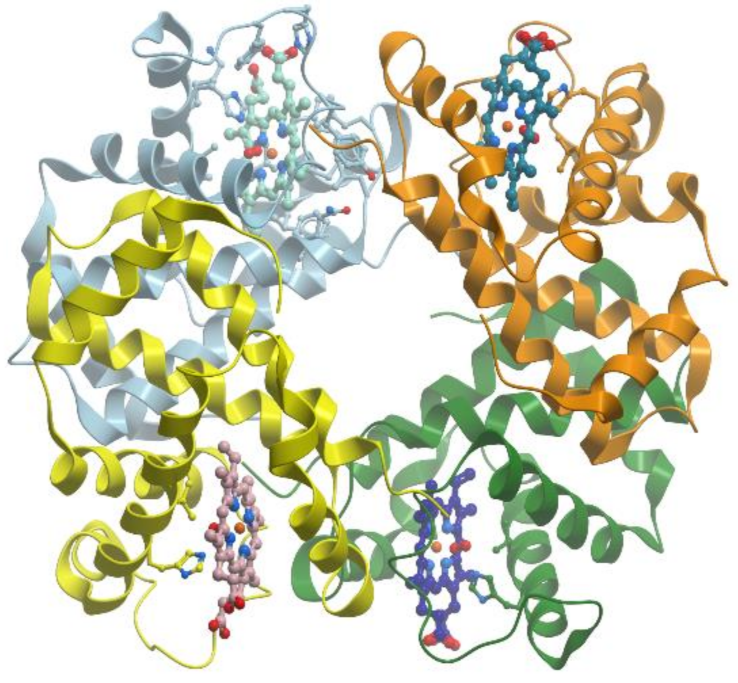{kind=link}
{kind=link}
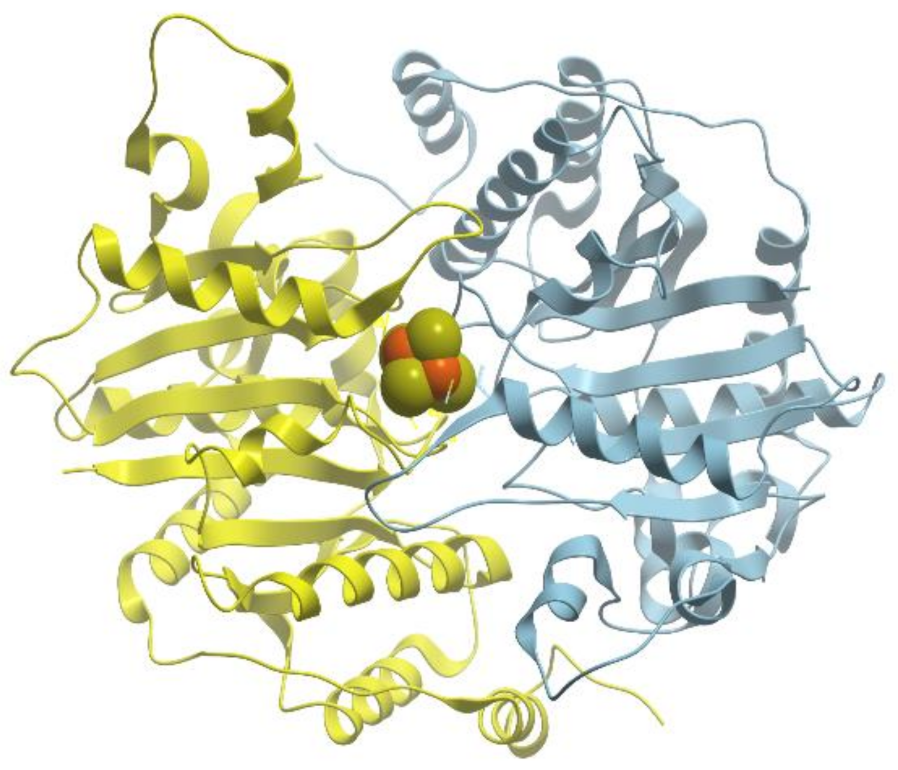{kind=link}
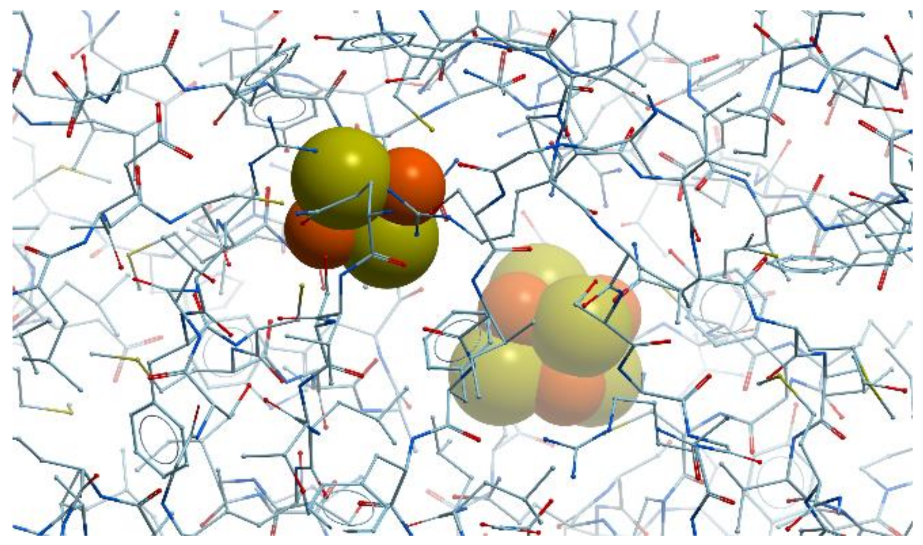{kind=link}
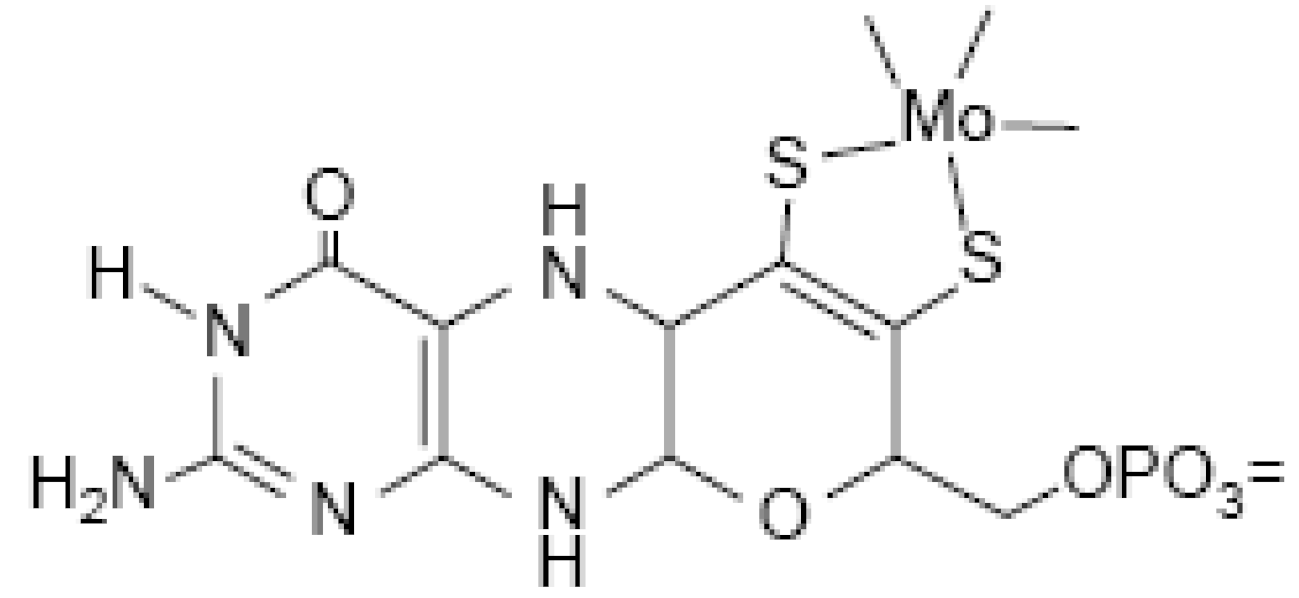{kind=link}
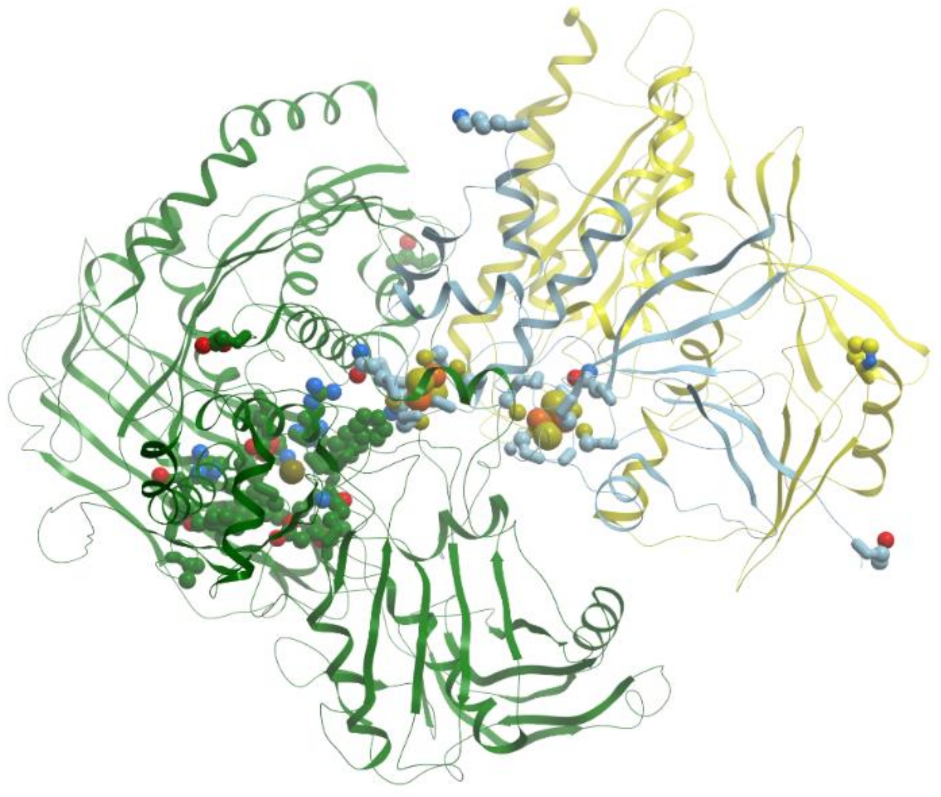{kind=link}
{kind=link}
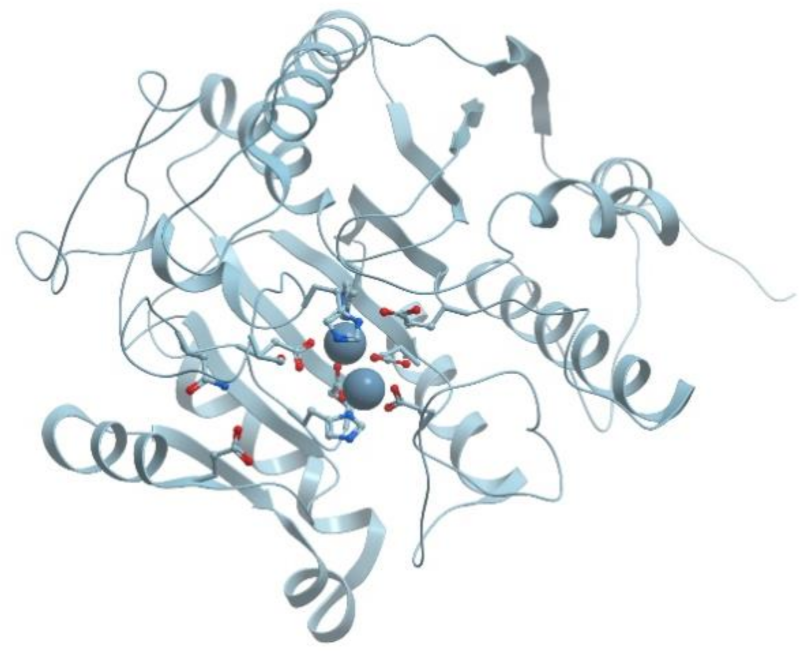{kind=link}
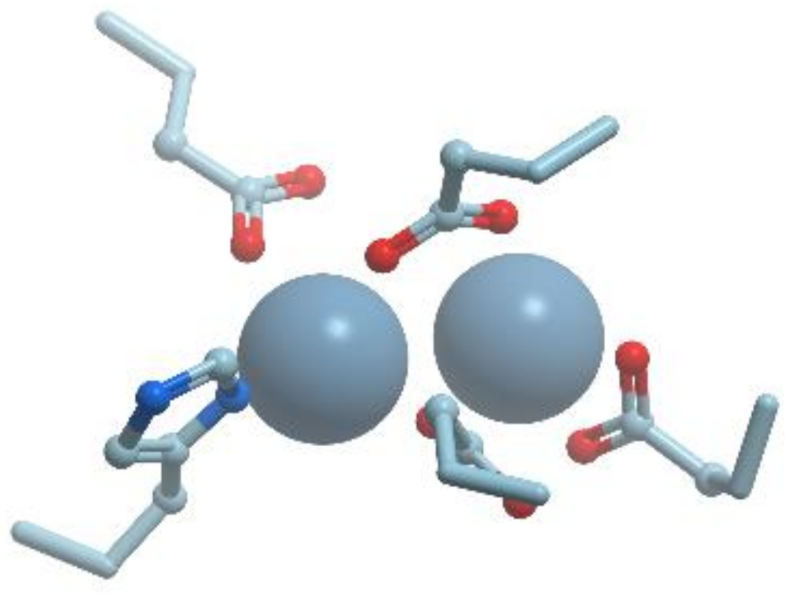{kind=link}
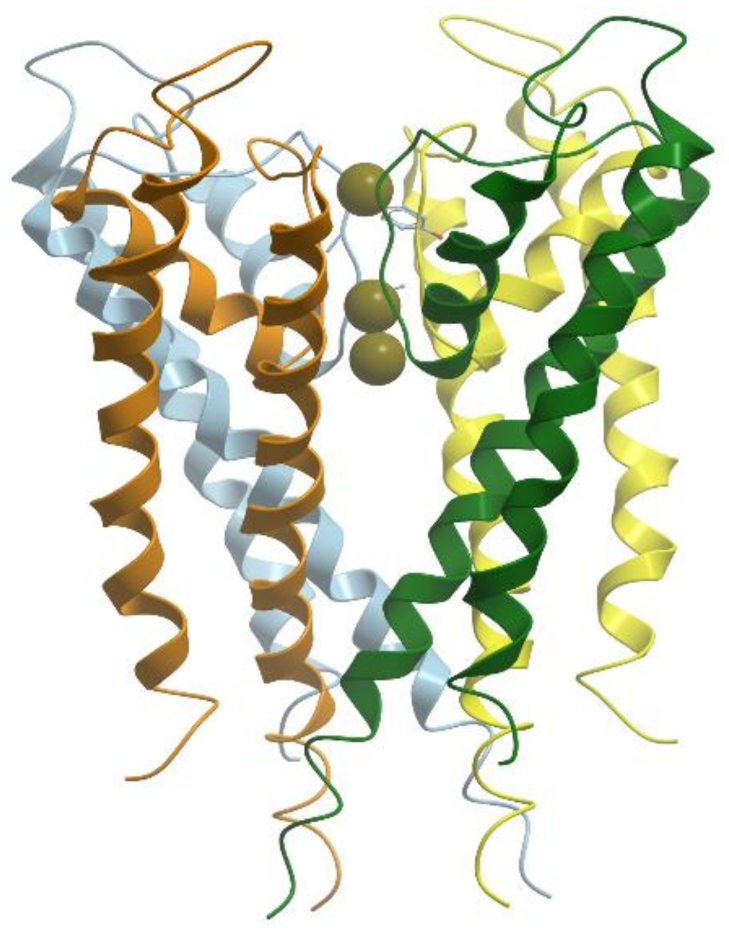{kind=link}
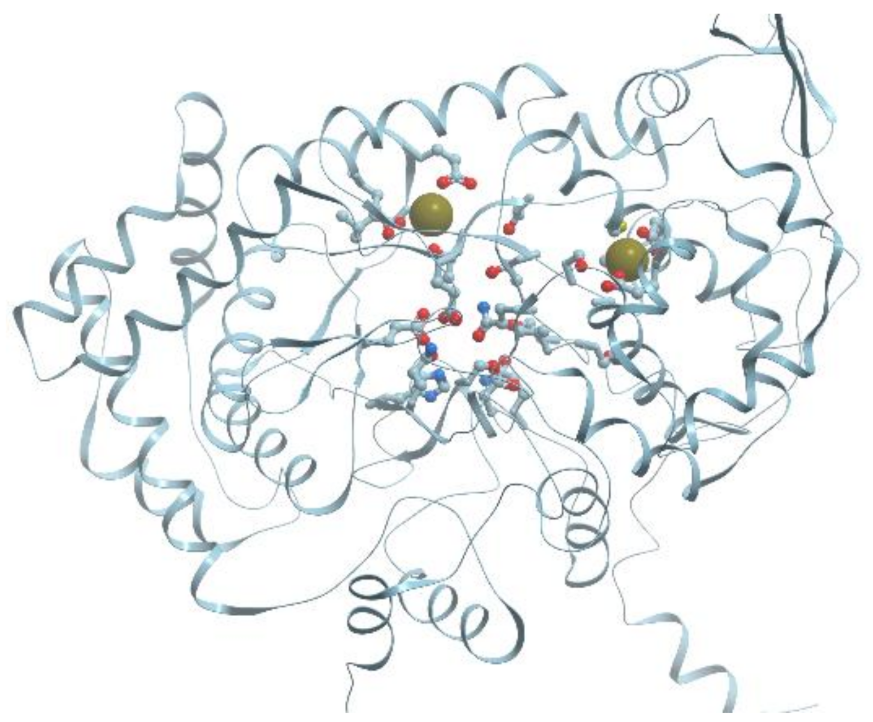{kind=link}
Table 1.
A fragment of Mendelleev periodic table of elements.
| Periods | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| III | Na | Mg | ||||||||||
| IV | K | Ca | Mn | Fe | Co | Ni | Cu | Zn | ||||
| V | Mo | |||||||||||
| VI | W |
Table 2.
Ionic radii of biologically significant metals.
| Metal | Ion | Ionic Radius, Å |
|---|---|---|
| Na | Na+ | 0.95 |
| K | K+ | 1.33 |
| Mg | Mg2+ | 0.66 |
| Ca | Ca2+ | 0.99 |
| Mn | Mn2+ | 0.80 |
| Mn3+ | 0.66 | |
| Mn4+ | 0.52 | |
| Fe | Fe2+ | 0.76 |
| Fe3+ | 0.63 | |
| Co | Co2+ | 0.76 |
| Co3+ | 0.63 | |
| Ni | Ni2+ | 0.83 |
| Ni3+ | 0.74 | |
| Cu | Cu+ | 0.91 |
| Cu2+ | 0.69 | |
| Zn | Zn2+ | 0.74 |
| Mo | Mo+3 | 0.83 |
| Mo+4 | 0.79 | |
| Mo+6 | 0.73 | |
| W | W+4 | 0.80 |
| W+5 | 0.76 | |
| W+6 | 0.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Permyakov, E.A. Metal Binding Proteins. Encyclopedia 2021, 1, 261-292. https://0-doi-org.brum.beds.ac.uk/10.3390/encyclopedia1010024
AMA Style
Permyakov EA. Metal Binding Proteins. Encyclopedia. 2021; 1(1):261-292. https://0-doi-org.brum.beds.ac.uk/10.3390/encyclopedia1010024
Chicago/Turabian StylePermyakov, Eugene A. 2021. "Metal Binding Proteins" Encyclopedia 1, no. 1: 261-292. https://0-doi-org.brum.beds.ac.uk/10.3390/encyclopedia1010024