
Implications of Endothelial Cell-Mediated Dysfunctions in Vasomotor Tone Regulation




1
Health Sciences Research Centre (CICS-UBI), University of Beira Interior, 6200-506 Covilhã, Portugal
2
Department of Medical Sciences, Faculty of Health Sciences (FCS-UBI), University of Beira Interior, 6200-506 Covilhã, Portugal
*
Author to whom correspondence should be addressed.
Biologics 2021, 1(2), 231-251; https://0-doi-org.brum.beds.ac.uk/10.3390/biologics1020015
Submission received: 14 July 2021
/
Revised: 3 September 2021
/
Accepted: 6 September 2021
/
Published: 8 September 2021
(This article belongs to the Section Cytokines and Allied Mediators)
Abstract
:Cardiovascular diseases (CVD) constitute the major cause of death worldwide and show a higher prevalence in the adult population. The human umbilical cord consistsof two arteries and one vein, both composed of three tunics. The tunica intima, lined with endothelial cells, regulates vascular tone through the production/release of vasoregulatory substances. These substances can be vasoactive factors released by endothelial cells (ECs) that cause vasodilation (NO, PGI2, EDHF, and Bradykinin) or vasoconstriction (ET1, TXA2, and Ang II) depending on the cell type (ECs or SMC) that reacts to the stimulus. Vascular studies using ECs are important for the analysis of cardiovascular diseases since endothelial dysfunction is an important CVD risk factor. In this paper, we will address the morphological characteristics of the human umbilical cord and its component vessels. the constitution of the vascular endothelium, and the evolution of human umbilical cord-derived endothelial cells when isolated. Moreover, the role played by the endothelium in the vasomotor tone regulation, and how it may be associated with the existence of CVD, were discussed.
1. Introduction
Cardiovascular diseases (CVD) constitute the major cause of death worldwide, with a higher prevalence in the adult population [1,2]. Examples of CVD are coronary artery disease, peripheral arterial disease, or aortic disease.
Numerous studies have been carried out that have contributed to the recognition of the role of the vascular endothelium and its involvement both in a wide variety of functions as well as in the pathophysiology of various diseases, such as atherosclerosis, diabetes, hypertension, and CVD [1,3,4]. The endothelium is involved in the regulation of blood pressure, blood flow, and coagulation [1,5].
Currently, the human umbilical cord (HUC) is a suitable source for isolation of endothelial cells (ECs) since it has no particular ethical impediments and is considered a non-tumorigenic and less immunogenic model. for this reason, HUC represents an advantageous experimental source for the isolation of endothelial cells [6]. The ECs can produce/release molecules that modulate vasoconstriction and vasorelaxation by smooth muscle cells (SMC) [7,8]. When releasing nitric oxide (NO), prostacyclin, bradykinin, and endothelium-derived hyperpolarizing factor (EDHF), the response given will be vasodilation by the SMC. If the substances released are endothelin-1 (ET-1), angiotensin II (Ang II), thromboxane A2 (TXA2), the final effect is vasoconstriction by the SMC [9].
The effect of endothelium in vivo is very difficult to study. To counteract this, several in vitro techniques were developed. Among these techniques, the use of human umbilical vein endothelial cells (HUVECs) was widely used as a source of human endothelial cells because they have some advantages. The main advantage is that it is obtained from non-pathogenic human tissues, which is not the case with other human sources, and the physiological relevance in the control of vascular pathways. Moreover, primary cultures with these cells can maintain native characteristics of endothelial cells and their intracellular signaling pathways [10].
As a source of human endothelial cells, the umbilical cord is commonly used for the isolation and culture of ECs from the umbilical vein and umbilical artery. However, most studies performed have isolated HUVECs, as artery-derived ECs are more difficult to isolate because the diameter of the artery is smaller than that of the vein [11].
In this paper, we will address the morphological characteristics of the umbilical cord and its component vessels; the constitution of the vascular endothelium, and the evolution of human umbilical cord-derived endothelial cells when isolated. Moreover, the role played by the endothelium in the physiological regulation of vascular tone and how it may be associated with the existence of CVD were analyzed.
2. Morphological Characteristics of the Human Umbilical Cord (HUC)
From a physiological point of view, the human umbilical cord (HUC) is a channel developed from the amniotic sac (which forms the epithelium), the allantois (which forms the umbilical veins and arteries), and the yolk sac [12,13].
The umbilical cord is critical for fetal development, allowing not only communication between the fetus and mother but also the exchange of gases and nutrients between them [14,15]. At an early stage of gestation, the HUC presents four umbilical vessels, two veins, and two arteries. At a later stage, one vein atrophies, leaving only two arteries and one vein [16]. Therefore, the HUC ensures gas exchange, where the two arteries carry the oxygen-poor blood, and the vein carries the oxygen-rich blood from the placenta to the fetus [17]. The placenta is responsible for gas exchange as the fetus’ lungs are not functional [17].
Additionally, the HUC is also composed of a substance of a gelatinous nature, called Wharton’s jelly [12]. This jelly consists of connective tissue cells dispersed in an amorphous surface matrix of glycosaminoglycans, such as hyaluronic acid and chondroitin sulfate, and different types of collagen [18,19]. One of the functions of Wharton’s jelly is to protect the umbilical vessels and give them flexibility. Furthermore, Warton’s jelly is responsible for giving the cord a certain firmness, due to its fibrous and porous content composed of collagen and elastin fibers [18,19].
Additionally, this fetal organ has a helical shape, due to the natural coiling that the arteries and vein exhibit [19]. It is also characterized by having a shiny, whitish appearance due to being lined by a simple epithelium from the amnion [20].
Near the end of gestation, the HUC is approximately 1–2 cm in diameter and 30–90 cm long [12]. However, HUC may be either shorter or longer. When a cord tends to be long, there is a greater likelihood of it wrapping around the fetus, while a shorter cord can lead to premature separation of the placenta from the uterine wall [12,16]. Furthermore, in approximately 0.5% of cases, the cord arises with only one artery, which can lead to vascular consequences for the newborn, such as abnormal cord length, polyhydramnios, or a decreased support of the cord blood flow which can consequently lead to intrauterine growth restriction [14]. Therefore, rigorous gestational management is necessary, not only because it can generate hypoxia or anoxia in the fetus, but also any disease that occurs in pregnancy can impact the umbilical cord [12,15]. In addition to a single artery, other changes in the morphology of the HUC (e.g., a “lean” and thicker cord, or an hypocoiled umbilical cord) may have an association with certain cardiovascular diseases. For example, when a cord is considered a thicker cord, it may be associated with some pathologies, such as diabetes mellitus, macrosomias, and hemolytic dysregulations [21].
Thus, not only the number of blood vessels but also morphological changes can contribute to the development of pathologies. We can conclude that the presence or absence of blood vessels is of extreme importance for the development of pathology.
In summary, not only the number of blood vessels but also morphological changes can contribute to the development of pathologies.
2.1. Vessels of HUC
As mentioned above, the HUC consists of two arteries and one vein. One of the functions of the HUV is the transport of oxygenated blood from the mother to the fetus [16]. In the case of HUA, it transports the poorly oxygenated blood from the fetus to the placenta [22].
Morphologically, the human umbilical artery (HUA) is a medium (1–10 mm) muscular artery composed of three tunics, morphologically different from each other (intima, media, and adventitia) [23,24].
The tunica intima, also called the endothelium, is formed by a single layer of endothelial cells having no adjacent subendothelial layer or internal elastic lamina [25]. It also plays a major role in the regulation of vascular tone since there are endothelium-dependent vasodilator and vasoconstrictor substances that are subsequently released [26].
The tunica media is composed of two different layers of smooth muscle cells (SMC). The outer media layer is less defined compared to the inner layer and has a structural appearance typically characterized by a circular arrangement, giving the outer layer this particular characteristic. This layer is of great importance because it is due to the contraction caused by this layer that the physiological closure of the umbilical arteries occurs. In the case of the SMC present in the inner media layer, these cells are arranged linearly or longitudinally concerning the axis of the artery. Furthermore, the SMC are found in a dispersed/disordered manner in an amorphous fundamental substance poor in elastic fibers, but rich in plastic fibers. Moreover, these cells have a higher plasticity, they differentiate from the contractile to the synthetic phenotypes. This phenotypic modulation allows these cells to respond to adverse conditions, such as vascular injury [12,27,28].
Finally, HUA does not have vasa vasorum or a nervi vasorum, nor a typical external tunica (adventitia). The function of this tunica is performed by Wharton’s jelly (mucous connective tissue and fibroblasts) which is surrounded by nutritional vessels and rich in glycosaminoglycans [12,16,20].
The umbilical vein has a similar structure to the umbilical arteries; however, the vein walls are a little thinner than the artery walls [20]. For this reason, the umbilical arteries are more easily identifiable as they have a smaller caliber (3 mm) compared to the veins (6 mm) [12].
Umbilical veins have a circular or triangular lumen that is permeable in cross-sections. The tunica intima of umbilical veins besides being thinner compared to arteries also has an irregular elastic tunica. The tunica media has two to three layers of muscle cells with mucopolysaccharides between them, without any type of clear cellular organization either in longitudinal or circumferential orientation [29]. Concerning the tunica adventitia, it is composed of fibrous and elastic connective tissue with variable amounts of collagen and elastic fibers. In the case of the tunica adventitia of the vein, it is composed of less dense connective tissue [30].
2.2. Vascular Endothelium
The vascular endothelium is the innermost structure that lines not only the inner walls of arteries but also the inner wall of capillaries and veins [5]. From a physiological point of view, the endothelium is characterized by being a monolayer of ECs with a slightly elongated and thin morphology with approximately 30–50 µm in length, 10–30 µm in width, and a thickness of 0.1–10 µm [5]. In vitro, the monolayer formed by these cells shows a characteristic parallelepiped-shaped pattern [5], which is in direct contact with blood components, for example, platelets. Thus, the endothelium is not only a barrier between blood and tissues but is an endocrine organ [5] and can also be a sensor of hemodynamic changes or chemical stimuli occurring in the bloodstream [25].
Additionally, ECs play an important role in regulating vascular tone [31], once they can produce different vasculoregulatory and vasculotropic molecules [1]. This coordination with SMC [5] also allows control of vascular permeability. Additionally, the vascular endothelium participates in inhibiting SMC proliferation by preventing cell migration through mechanisms, such as nitric oxide (NO) synthesis that inhibits cell proliferation, and the secretion of growth factors and other cytokines capable of exerting paracrine control on SMC [32,33]. Another function of the endothelium is to participate in the degradation and metabolic transformation of numerous endogenous drugs, such as norepinephrine, 5-hydroxytryptamine, prostaglandins E and F, leukotrienes, among others [34].
Due to its importance in the physiological regulation of SMC, several investigations have carried out the isolation of ECs from HUA and HUV, as described below.
3. Isolation of Endothelial Cells of HUC
During the last 3 decades, advances in vascular endothelial cell biology have had a great impact on the understanding of some pathophysiological conditions such as cardiovascular diseases [35,36]. Between 1922 and 1973, there was a growing interest among the scientific community in human umbilical cord vein-derived endothelial cells (HUVEC) for these investigations [1]. In 1922, studies were conducted and described the development of endothelial cells from chicken liver embryonic tissue explants [37].
In 1963, Maruyama et al. was the first to isolate HUVECs. The cells were cultured in sheets and most of the cells had a spindle shape, which demonstrates that the cultures obtained were not pure [38].
In 1973, Eric A. Jaffe successfully isolated HUVEC, which grew as a homogeneous monolayer of polygonal cells with well-defined borders. Furthermore, these authors also compared HUVECs with SMC and fibroblasts, which allowed them to conclude that HUVECs contained rod-shaped cytoplasmic organelles [39]. In this procedure, veins were perfused with collagenase and incubated for 15 min at 37 °C, and endothelial cells were collected at the end [39].
The following year, Michael A. Gimbrone et al. used the same protocol as Jaffe et al. and characterized the population growth behavior of primary cultures and subcultures of HUVECs [1,40].
Since 1974, some studies of isolation of EC have appeared intending to study the physiology of EC, most of them based on the studies and descriptions made by the following authors [11,41,42,43]. Over time, these studies have undergone some adaptations, such as the constitution of the culture medium for endothelial cells. In this sense, the constitution of the culture medium used may contain endothelial cell growth factor (ECGF), which allows long-term growth of the ECs, or an anti-PPLO agent that prevents contamination by gram-positive bacteria or mycoplasmas [41,44,45,46,47]. Culture media containing 5 ng/mL epidermal growth factor (EGF) was shown to have a higher proliferative potential compared to the anti-pleuropneumonia-like organisms’ agent (anti-PPLO agent).
Most authors use the M199 medium in their study, although they do not provide any justification for this use. There are other authors who refer to different culture media, for example TC 199 [39], MCDB 131 [48], M200 or M231 [11], Dulbecco’s modified Eagle’s medium (DMEM) [49], YLH medium [38], and EGM-2 culture medium [43,50,51].
Based on what was described by Jaffe et al. 1973 [39], Psefteli et al. 2021 also performed the isolation of the HUVECs with a similar protocol and demonstrated the effect of the endothelial glycocalyx in the HUVECs. This endothelial glycocalyx may represent a potential therapeutic target against EC dysfunction in cardiovascular disease [52]. Furthermore, the performance of cultures of HUVECs led to interesting findings, namely the identification of nitric oxide (NO) as an endothelium-derived relaxation factor [1,53].
According to Martin de Llano, the isolation of ECs from either the human umbilical vein or the human umbilical artery from the same cord allows a functional comparative study to be made between these cells [11]. Regarding the isolation of endothelial cells derived from the human umbilical cord artery, there are still few studies performed possibly due to the small diameter of the arteries which makes isolation more difficult to perform [11]. However, some studies have been described, namely Ulrich-Merzenich et al. (2022), Martin de Llan et al. (2007), and Provitera et al. (2019) [11,42,54].
The main objective of carrying out a primary culture is always to obtain pure cultures. Baudin et al., by isolating ECs from the umbilical vein vascular wall, described SMC and fibroblast contaminations in the cultures. When confluent ECs were obtained, the contaminations were visible by the more elongated morphology of SMC/fibroblasts instead of tightly packed polygonal ECs [45]. In these cases, the cultures should be discarded. However, the authors do not mention how to avoid these contaminations.
The isolation of ECs was always performed with digestive enzymes, but some of these studies also used the explant method after digestion [55] (please see Table 1). In this process, where usually no enzyme is used, an original tissue or organ is excised (“or explanted”) into smaller pieces and planted in culture flasks (i.e., “ex vivo”). Then, the cells migrate out of the tissue and adhere to the surface of the culture flasks, where they will be used for different experimental studies [56]. Mostly, the enzyme of choice is collagenase, however, the type of collagenase varies. Another procedure to perform enzymatic digestion is by using an enzyme mixture, and this is usually, the joining of collagenase with type II dispase [11,42,57]. After enzymatic digestion and subsequent centrifugation, the ECs are cultured.
In most studies, the coating is performed with gelatin at different percentages (0.2–1%). However, other studies performed the coating with fibronectin [11,42,43,57].
Meanwhile, some authors performed with the same umbilical cord the culture of HUVECs and the culture of HUAECs, and observed that cell isolation by enzymatic digestion is easy and fast when the artery and vein are used from a single umbilical cord [42]. Furthermore, other authors performed the isolation of the HUVECs and the HUCMSC (umbilical cord mesenchymal stem cells) from the same umbilical cord [57], as shown in Table 1. The culture of HUCMSC may also allow us to direct the fate towards an endothelial cell lineage, which can represent economical and commercially viable option manly for cell replacement therapy due to its noninvasive collection procedure [57].
Over the last decades, some markers that allow the identification of ECs have also been studied. The enzymes that are cellular markers are angiotensin-converting enzyme (ACE) and endothelial NO synthase (eNOS) [58]. In addition to enzymes considered markers, there are also protein markers such as CD31, CD36, CD34, CD144, ICAM-1 and VCAM-1, PECAM-1, endothelin 1 (ET-1), E-selectin and P-selectin, among others [1,59,60,61]. However, the ECs are negative for other biomarkers, such as CD45 and CD133 [62].
The structural heterogeneity of ECs includes variations in cell morphology (size, thickness, and the position of the nucleus) as well as in the gene expression profile, extracellular matrix production (basal lamina components), and finally in cell surface properties [5]. The E-selectin marker is an inducible gene restricted to the endothelium, but which is only expressed in activated endothelium and not in endothelium considered at “rest” [63]. Regarding the markers used to identify ECs present in large vessels, the most used are fibronectin, collagen 5α1, collagen 5α2, and osteonectin, which are involved in the biosynthesis and remodeling of the extracellular matrix (ECM). These differences are probably related, in part, to the relatively thick vascular wall surrounding the endothelium of large vessels. On the other hand, microvascular ECs express genes encoding basal membrane proteins, such as laminin, collagen 4α1, collagen 4α2 and collagen 4α-binding protein, and proteins that interact with ECM, such as CD36, integrin α1, integrin α4, integrin α9, and integrin β4, perhaps related to the intimate association of microvascular ECs with the basal membrane and ECM [64].
In summary, and as shown in Table 1, the most used culture medium is M199 with gelatin coating and the enzyme collagenase. Vascular studies using ECs are very important to analyze several cardiovascular pathologies since endothelial dysfunction is considered a major risk factor for cardiovascular diseases [1,32].
4. Regulation of Vascular Tone by Endothelial Cells
Endothelial cells are crucial cells to control vascular tone by producing and releasing paracrine signals for the vascular SMC and are responsible for promoting contraction or dilatation in the vessels. The equilibrium between vasodilation and vasoconstrictive signals are responsible for the regulation of the blood flow and blood pressure according to the requirements of tissues and organs. In this sense, the dysregulation of ECs is directly associated with the beginning and progression of cardiovascular diseases, including atherosclerosis and hypertension [31]. The vasoactive factors released by endothelium are nitric oxide (NO), prostacyclin (PGI 2), thromboxane (TXA2), endothelin 1 (ET-1), angiotensin II (Ang II), bradykinin, and EDHF [77]. Some factors induce vasodilatory mechanism, for example, NO, PGI2, and EDHF. On the other hand, TXA2 induce vasoconstrictor mechanism [78]. However, ET-1, bradykinin, and Ang II may induce both effects (Table 2). The main mechanisms involved in the regulation of vascular tone by the vasoactive factor release by the endothelial cells are shown in Figure 1.
4.1. Nitric Oxide
Nitric oxide (NO) is an endothelium-dependent vasodilator of vascular smooth muscle that was first identified in 1980 by Furchgott and Zawadzki [79].
In endothelial cells, NO is synthesized by eNOS through the reaction of oxygen and L-arginine [32]. In this oxidation reaction of L-arginine, calcium (Ca2+) and calmodulin (CaM) act as co-factors, since the complex Ca2+-calmodulin binds to NO synthase. Then, stoichiometric amounts of NO and L-citrulline are produced [32,80]. The eNOS can be activated by fluid shear stress and by vasoactive agents (such as ATP, acetylcholine, or bradykinin) via phosphorylating proteins, protein–protein interactions, Ca2+ signaling, and subcellular protein translocations [7,31].
Activation of eNOS leads to a release of the NO radical, which diffuses easily across the membrane of ECs.
Afterward, NO activates soluble guanylyl cyclase (GC) in smooth muscle cells. Subsequently, a conversion of GTP to cGMP occurs, leading to an increase in intracellular cGMP. This second messenger activates a protein kinase, leading to the inhibition of calcium influx into the cell and decreasing calcium–calmodulin stimulation of the myosin light chain kinase. In the final stage of this process occurs a decrease of the myosin light chains phosphorylation, decreasing the development of smooth muscle tension, causing vasodilation of the adjacent smooth muscle cells [7,32].
Thus, NO is an essential determinant of cardiovascular homeostasis by regulating vasomotor tone, thereby maintaining the integrity of the vasculature and protecting against cellular injury [31,77]. Studies have demonstrated that the endothelium of HUA expresses eNOS and can release NO that, in turn, acts on SMC. Currently, the vasodilatory role of NO in regulating vascular tone in HUA is well described [81,82,83].
Some studies used HUVECS to investigate NO production regulated by inflammatory cytokines [84], ketamines [85], and a flavonoid with cardiovascular protective effects (rutin) [86]. These studies concluded that NO synthesis is decreased when exposed to a concentration of ketamines. The authors also concluded that there is an inhibition of pre-translation of eNOS expression, a decrease in post-translation of endothelial NO synthase activity due to reduced intracellular calcium levels [85]. When HUVECs were exposed to rutin and inflammatory cytokines, an increase in NO biosynthesis occurred. When exposed to rutin, this increase was due to an increase in eNOS gene expression and consequently an increase in eNOS activity [86].
Endothelial dysfunctions in the NO synthesis contribute to CVD by favoring the vasoconstrictor state [8]. Specifically, a reduction in the biological activity of NO seems to be harmful to the normal function of the endothelium and has an important role in atherogenesis promotion [8,32]. In these pathological states, the vascular production of superoxide () increase and the reaction of both molecules may form the strong oxidant peroxynitrite (). Finally, DNA is damaged and the cell dies, which leads ultimately to atherosclerosis [32,87].
4.2. Endothelin 1
Endothelin (ET) was identified by Yanagisawa and his colleagues as an endothelium-derived constriction factor (EDCF) in 1988 [58].
ET is a vasoconstrictor agent that has three isoforms, ET-1, ET-2, and ET-3. However, ECs can express and produce only one of these isoforms, ET-1 [77,88]. Prepo-endothelin-1 is converted to big ET-1 and then to ET-1 [88].
The biological effect of ET-1 is mediated by the activation of two receptors, ETA and ETB, both G-protein coupled receptors, thus sharing the same signaling pathway [89,90]. Both receptors (ETA and ETB) are expressed in SMC mediating the vasoconstrictor effect of ET-1. In the case of endothelial cells, only ETB receptors are expressed, where activation of this receptor leads to vasodilation mediated by prostacyclins and NO release [32]. Depending on where the ETB receptor is expressed, it leads to different end effects. Thus, when ETB receptors are expressed on SMC an activation of Gq protein occurs, activation of PKC, releasing IP3, and a subsequent release of Ca2+, leading to vasoconstriction [91]. On the other hand, when ET-1 binds to the ETB receptor present on endothelial cells it causes a vasorelaxation of SMC cells. This vasorelaxation is due to the release of Ca2+, which will bind to calmodulin to activate calmodulin kinase, responsible for the phosphorylation of eNOS, initiating NO synthesis in the endothelial cell [90,92]. However, this binding can also trigger prostacyclin production, such as PGI2, which leads to an increase in cAMP and subsequently vasodilation [92].
ET-1 and NO are also known to be interdependent, as there is a strong inhibitory effect of ET-1 on NO-mediated dilation [93]. ET-1, when it binds to the ETB receptor present on the endothelial cell, will stimulate the production of eNOS, and this occurs through the activation of PI3-K/AKT by ET-1. Then, a stimulation of eNOS phosphorylation occurs with an increase in NO production [94]. However, the NO produced can inhibit ET-1 production, at the level of transcription [9].
Increased production of ET-1 and its receptors can mediate many pathophysiological events, contributing to the development of atherosclerosis and vascular complications in diabetes mellitus [95].
4.3. Prostacyclin
The endothelium also can generate an eicosanoid, PGI2 (prostacyclin/prostaglandin) which mediates relaxant effects on vascular SMC [5].
Phospholipids from the plasma membrane are hydrolyzed by phospholipase A2 (PLA2) producing arachidonic acid (AA). Then, released AA is metabolized by cyclooxygenases (COXs) to form prostaglandin G2 (PGG2) which, subsequently, via a peroxidase, gives rise to the formation of prostaglandin H2 (PGH2). Through PG synthetases, PGI2 is formed [96,97]. PGI2 release is stimulated by shear and in response to acetylcholine [32]. This release activates surface prostacyclin receptors (IP), which are G-protein coupled receptors, more specifically a Gs protein, and their activation induces smooth muscle cell relaxation due to increased intracellular cAMP concentration [98,99].
PGI 2 also appears to be involved in the regulation of vascular tone when resting vascular tone [5]. Like NO, PGI2 also can inhibit leukocyte adhesion and smooth muscle cell proliferation, in addition to also facilitating NO release by EC [32].
Prostacyclin is the preferred ligand of prostacyclin (IP) receptors, when it binds to the PI receptors present on SMC it activates adenylyl cyclase-producing cAMP, stimulating PKA, and consequently leads to relaxation [32,100,101]. However, prostacyclin does not always activate adenylyl cyclase [100]. In addition to the classic IP-cAMP receptor pathway, there is evidence to suggest that PGI 2 may act through the peroxisome proliferator-activated receptor δ (PPAR) [102].
If PGI2 binds to the IP receptor present on the endothelial cell membrane, it leads to NO production and consequently diffuses into the SMC membrane and activates soluble guanylyl cyclase. This activation leads to an increase in cGMP and activation of PKG, causing relaxation [101].
Several studies were performed to understand the effects that PGI2 exerts in the HUC artery [82,103,104]. These studies show that PGI2 induce vasorelaxation in the HUA. Moreover, other studies were also performed in HUVECs that allowed assessing PGI2 production in endothelial cells under the influence of some factors, such as oxidative stress and some agents such as antioxidant enzymes (superoxide dismutase and cata-lase), oxygen-free radical scavengers (vitamin E), and eicosanoids (arachidonic acid, eicosapentanoic acid). The authors concluded that when ECs are subjected to oxidative stress and antioxidant enzymes, an increase in PGI2 production occurs [105].
PGI2 appears to be involved in endothelial dysfunction in patients with hypertension, mainly in preeclampsia. PGI2 deficiency and PGI2/TXA2 imbalance might be critical in the pathogenesis of preeclampsia and could partially explain the increased vascular reactivity in gestational hypertension [106]. During preeclampsia a decrease of 6-keto-PGF1α, a hydration product of PGI2 has already been observed [107]. However, the mechanism that led to the decrease of PGI2 production remains unknown.
4.4. Endothelium-Derived Hyperpolarization Factor
All endothelial cells produce a hyperpolarization factor (EDHF) that is produced by CYP epoxygenases of cytochrome P450 [5]. While NO mostly regulates the vascular tone of conduction vessels (e.g., aorta and epicardial coronary arteries), EDHF regulates smaller resistance vessels (e.g., small mesenteric arteries and coronary microvessels) [108,109]. On the other hand, if the vasodilatory response persists after combined inhibition of NO (by L-NAME) and PGI2 (by aspirin), this vasodilator response is thought to be attributed to substances that cause hyperpolarization of vascular smooth muscle via a mechanism involving potassium (K+) conductance. One of the substances that act by this mechanism is EDHF [32]. Regarding aspirin’s mode of action, it is an inhibitor of the cyclooxygenase type 2 enzyme, present in endothelial cells and consequently inhibits the conversion of AA into PGG2 and PGH2, which results in an inhibition of PGI2. This inhibition has the consequence of PGI2 not binding to the IP receptor, present in SMC [110]. Regarding L-NAME, this is considered a non-selective inhibitor of eNOS widely used as a pharmacological tool [111]. In this case, inhibition by L-NAME causes a reduction in eNOS activity, which leads to a reduction in NO production [8,112].
It is described that EDHF opens Ca2+-activated K+ channels in vascular smooth cells producing vasorelaxation [80]. This vasoactive agent acts through two different phases: in the first phase, it promotes an increase in intracellular Ca2+ concentration, which leads to an activation of Ca2+-dependent K+ channels and increases K+ efflux followed by hyperpolarization [113]; and in the second phase, it reflects the mechanism by which hyperpolarization in the endothelial cell is transferred to SMC [113]. After this transfer, SMC activates K+ channels thus causing hyperpolarization of SMC which is accompanied by the closure of voltage sensitive Ca2+ channels, ultimately resulting in a relaxation of SMC [113].
Some molecules and mediators can act as the EDHF, for example, K+, cytochrome P450 metabolites, lipoxygenase products, reactive oxygen species, C-type natriuretic peptide, and electrical coupling via myoendothelial communicating junctions. [32] Currently, there are still no studies performed on the human umbilical cord or HUC-derived endothelial cells.
EDHF appears to be involved in endothelial dysfunction in patients with diabetes and hypertension. In various animal models of type I and type II diabetes, EDHF-mediated responses are depressed. Regarding diabetes in humans, it seems there exists a difference concerning the type of diabetes. Controlled patients (good glycemic control and without albuminuria) with type I diabetes show a normal endothelial function and both the NO- and the EDHF-mediated responses are conserved. However, in uncontrolled patients (with microalbuminuria) a decrease in the endothelium-dependent vasodilatation is observed. However, it is not known if NO or EDHF are modified by uncontrolled type I patients and patients with type II diabetes [114]. EDHF may act as a compensatory vasodilator mechanism despite the reduced bioavailability of NO [115]. Regarding hypertension patients, it seems that hypertension per se does not produce a consistent depression of the EDHF-mediated responses (please see the review [114]).
4.5. Thromboxane A2
Thromboxane A2 plays an important role in cardiovascular diseases through vasoconstriction, platelet aggregation, and proliferation of SMC, but it is also involved in allergies, modulation of acquired immunity, and cancer cell metastasis [100,116].
As mentioned above, TXA2 formation occurs in three steps similarly to PGI2 formation. The first is considered the limiting step, where mobilization induced by arachidonic acid (AA) derived from cell membrane phosphoglycerides through diaglycerol (DAG) and phospholipase A2 (PLA2) occurs. In the second step, oxidation of AA into endoperoxides (PG) occurs via COX. In this second step, AA is sequentially converted to PGG2, which is then converted back to PGH2. Finally, in the third step, isomerization of PGH2 occurs to form TXA2 by TXA2 synthetase [117]. TXA2 is considered the preferred ligand of thromboxane receptors (TP receptors) [100].
When TXA2 binds to the TP receptor, present on the membrane of smooth muscle cells, a coupling of Gq protein occurs, stimulating phospholipase C and increased production of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). The formed IP3 binds to the endoplasmic reticulum and releases Ca2+. Then, the DAG will activate protein kinase C (PKC) along with the Ca2+ released from the endoplasmic reticulum which leads to vasoconstriction [23,116].
Reactive oxygen species not only increase the stability but also increase the density of functional TP receptors present in the cell membrane. Activation of TP receptors inhibits NO production in endothelial cells [100].
Concerning the effects of TXA2 in HUA, only Bodelsson et al. proved that this agent also induced vasoconstriction in the umbilical artery [118]. In addition, studies were performed in HUVECs and demonstrated that TXA2 are also produced by these cells under various conditions. The results suggest that when HUVECs are under oxidative stress, vitamin E, and arachidonic acid, there is an increase in TXA2 levels [105].
Studies have shown that TXA2 production is elevated in various states of CVD, promoting endothelial dysfunction [119] by supporting ROS formation. In diabetes mellitus, typically associated with endothelial dysfunction, it has been shown that the increased production of this vasoconstrictor agent is accompanied by a decrease in responses to NO and EDH, contributing to an altered vascular reactivity [120,121]. Furthermore, the activity of SKCa channels, important in NO biosynthesis, may also be another mechanism by which TXA2 promotes endothelial dysfunction.
4.6. Angiotensin II
Endothelial cells express not only the angiotensin-converting enzyme (ACE) but also the angiotensin receptors, AT1 and AT2 [32,122].
In endothelial cells, dipeptidyl carboxypeptidase converts angiotensin I to angiotensin II, which is physiologically active (Ang II) [32,123].
Angiotensin II is diffused across the membrane and binds to the AT1 receptor present on SMC. If there is a binding of angiotensin II to AT1 receptors then vasoconstriction occurs, but the same does not happen when it binds to AT2 receptors, which gives rise to a vasorelaxation effect, and both receptors are coupled to G proteins [122,124].
When Ang II binding to the AT1 receptor occurs, it results in the coupling of a Gq protein where, subsequently, stimulation of phospholipase C (PLC) occurs. The PLC activation induces the formation of diacylglycerol (DAG) and inositol triphosphate (IP3), which, via protein kinase C (PKC), leads to the activation of calcium channels. The Ca2+ binds to calmodulin and activates myosin light chain kinase (MLCK) which phosphorylates the myosin light chain and increases the interaction between actin and myosin as a consequence of SMC vasoconstriction [125,126].
The AT1 receptor mediates most of the pathophysiological effects of Ang II, including vasoconstriction, inflammation growth, and fibrosis, while the AT2 receptor can counteract many of the actions mediated by the AT1 receptor [125].
The AT2 receptor expression decreases after birth, suggesting that it may play an important role in fetal development [126].
After the AT2 receptor is activated, it will stimulate the B2 receptor, which in turn induces phosphorylation of endothelial nitric oxide synthase (eNOS). Thus, NO production is increased, leading to the activation of GC, which synthesizes cGMP, promoting a vasodilation of SMC [127].
The actions of angiotensin II are associated with dysfunction/uncoupling of endothelial NOS (eNOS), which leads to decreased NO levels and increased superoxide production [118]. Moreover, Ang II may induce vascular remodeling by the generation of reactive oxygen species (ROS) and by the activation of the sympathetic nervous system activity [128] that also leads to an increase in blood pressure. This happens mainly through the angiotensin II type 1 receptor (AT1R) [129].
In summary, the main targets of Ang II are SMC, but also has effects on ECs. In the SMC, the Ang II promote ROS production, activation of apoptotic signaling pathways, and promotion of thrombosis. In endothelial cells, Ang II regulates NO production by increasing eNOS production and, therefore, an increase in NO [130].
4.7. Bradykinin
Bradykinin is considered one of the vasoactive substances that contribute to the physiological preservation of cardiovascular system function. In addition to this, it also contributes to the progression of labor by inducing vasoconstriction of the umbilical blood vessels [131]. This molecule is released from the quininogen substrate through the action of kallikrein and is considered a potent vasodilator peptide that acts through stimulation of specific endothelial bradykinin (B2) receptors [132].
However, bradykinin is rapidly degradated (half-life 27 ± 10 s) by several metallopeptidases [133].
Bradykinin can act by binding to the B1 receptor or by binding to the B2 receptor, both of which are coupled to G proteins, translating signals through the activation of these proteins. B2 receptors are constitutively expressed in endothelial cells, smooth muscle cells, and cardiomyocytes. Contrarily, B1 receptors are weakly expressed in endothelial cells and smooth muscle cells under normal physiological conditions but are highly ex-pressed under pathophysiological conditions, such as in inflammation [134,135,136,137,138,139]. Moreover, this receptor can be activated synergistically in HUVECs by cotreatment with tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ)) [134].
When bradykinin binds to the B2 receptor present on the endothelial cell, it can activate the Gq protein, and consequently activates PLC and releases IP3 increasing Ca2+ production, leading to the production of NOS and consequently NO. This molecule activates guanylyl cyclase in SMC and increases cGMP production, contributing to the relaxation of these cells. On the other hand, after activation of the B2 receptor of the endothelial cell, there occurs an activation of a Gi protein and consequently produces EDHF and PGI2 [140]. In both cases, the final effect is a vasorelaxation of SMC.
If, instead of Bradikinin binding to the B2 receptor of endothelial cells, it binds to the B2 receptor of SMC, it activates the Gq protein and subsequently directly activates PLC, leading to an increase in Ca2+ concentration and to a constriction of vascular smooth muscle cells [141].
When B1 receptors are activated by inflammatory processes and tissue damage, they acquire hemodynamic properties of B2 receptors, for example, the final effect would be a vasorelaxation [136].
On the other hand, Nowak et al. observed in HUVECs the possible contribution of angiotensin-converting enzyme, neutral endopeptidase, and aminopeptidase P in the bradykinin inactivation pathway, and assessed how the endothelial layer is involved in this pathway. The authors observed that angiotensin-converting enzyme and neutral endopeptidase represents a relevant inactivation pathway in the human umbilical vein and that the endothelial layer plays an extremely important role in the inactivation of bradykinin. Thus, the authors were able to conclude that the use of bradykinin and angiotensin-converting enzyme and neutral endopeptidase may represent an important therapeutic target for hypertension [142].
Overall, bradykinin exerts a cardioprotective action by inducing a vasodilation, decreasing ROS production, and having an anti-inflammatory, anti-fibrinolytic, and antithrombotic effects [143]. The anti-inflammatory effects and reduction in ROS production induced by bradykinin are mediated by NO. On the other hand, the release of NO also results in an anti-apoptotic effect of bradykinin [143]. Therefore, a decrease in levels of bradykinin is harmful to the CV system, inducing endothelial dysfunction [129].
In summary, the vascular tone is regulated by the release of vasoactive substances by ECs. The vasoactive factors that are released by ECs are nitric oxide (NO), prostacyclin (PGI2), thromboxane (TXA2), endothelin 1 (ET-1), EDHF, angiotensin II (Ang II), and bradykinin. In the case of NO, PGI2, and EDHF act only by a vasodilator mechanism. Ang II, ET-1, and TXA2 cause vasoconstriction, as shown in Figure 1. However, Ang II and ET-1 also can cause a vasorelaxant effect. Bradykinin can cause a vasoconstrictor or vasodilator effect, depending on where it acts, as shown in Figure 1.
5. Endothelial Dysfunction
Increasing evidence has shown that endothelial cell dysfunction is characterized by a dysregulation of vascular tone, redox balance, and increased inflammatory reactions in the blood vessel wall. For this reason, endothelial dysfunction is associated with several disorders, including diabetes mellitus, hypertension, atherosclerosis, aging, and heart failure [8,144].
More recently, endothelial activation has also been characterized as a prominent alteration in endothelial dysfunction, which refers to the up-regulation of chemokines and adhesion molecules and other proteins involved in cell–cell interactions, thus leading to the pro-thrombotic and pro-inflammatory circumstance in blood vessels [145,146].
In CVD, and specifically in atherosclerosis, abnormal vasoconstriction occurs which is observed where plaque formation occurs [147]. This pathological state creates conditions that become favorable for platelet aggregates and leukocyte adhesion, as well as activation of cytokines that increase the permeability of the vessel wall to oxidized lipoproteins and mediators of inflammation, resulting in structural damage of the arterial wall with a proliferation of smooth muscle cells and atherosclerotic plaque formation [148,149]. Endothelial dysfunction disrupts the balance between vasoconstriction and vasodilation, which is characterized by increased endothelium-derived contracting factors (EDCFs), especially ET-1, and reduced endothelium-derived relaxing factor (EDRFs), mainly NO [149]. In addition to a significant decrease in NO bioavailability as a consequence of endothelium dysfunction, a dysfunctional endothelium also contributes to the production of various mediators, which are even more damaging to the vascular wall [8,148]. Furthermore, one of the factors that can influence the dysregulation of the endothelium is oxidative stress. Increased oxidative stress is characterized by a measurable increase in reactive oxygen species (ROS) which can result in impaired NO synthase and subsequently a decrease in L-arginine uptake [148].
The vasoconstrictor effect generated by ET-1 is increased in patients suffering from type 2 diabetes [95]. If the patient is diabetic, this will induce endothelial dysfunction, leading to a decrease in NO and PGI2 and, consequently, less vasodilation of the SMC. However, it also leads to an increase in ET-1, Ang II, ROS, and COX leading to an increase in vasoconstriction [150]. When an increase in Ang II occurs, it leads to a decrease in eNOS and an increase in endothelin converting enzyme (ECE), thus increasing vasoconstriction compared to vasorelaxation [151].
Endothelial dysfunction is prevalent in CVD. However, it can also be found in patients diagnosed with neurological, gastrointestinal, rheumatological, hematological, and renal diseases [148].
Some functions must be adapted to the conditions of pregnancy, such as endocrine functions and functions of some systems including the circulatory system. These adaptations lead to metabolic changes that directly affect the function and metabolism of the endothelium. The endothelium thus plays a key role in the pathogenesis of metabolic dis-orders associated with pregnancy. It is affected by metabolic disturbances by losing its ability to control permeability [152].
Furthermore, endothelial dysfunction is associated with the development of diseases in pregnancy. One of the pathologies associated with pregnancy is gestational diabetes mellitus. Maternal hyperglycemia increases fetal circulating glucose levels, triggering the development of fetoplacental endothelium alterations, which result in NO synthesis de-regulation [152].
Other pathologies associated with endothelial dysfunction are hypertensive pathologies, such as pre-eclampsia. These individuals with PE fail to develop insensitivity to vasoconstrictors, Ca2+ signaling and increased vasodilator production are reduced or absent [153]. The reduced placental perfusion seen in these circumstances also creates changes in the placental environment, where it leads to ROS production and the activation of endothelial cells by different mechanisms that result in endothelial dysfunction [154]. This maternal vascular endothelium dysfunction leads to an increase of the formation of vasoactive factors like ET-1 and TXA2, increases of vascular sensitivity to angiotensin II, and decreases the formation of vasodilators, such as NO and PGI2. Furthermore, it enhances the synthesis of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin (IL). This TNF-α may induce structural and functional alterations in placental endothelial cells during trophoblast development, which is associated with increased blood pressure, mainly preeclampsia [155].
In summary, dysregulation of vascular tone has caused endothelial cell dysfunction and is consequently associated with various diseases. One of the consequences of endothelial dysfunction is a decrease in NO bioavailability. However, an increase in ET-1, Ang II, ROS, and COX production may also occur, contributing to vasoconstriction. Endothelial dysfunction is then associated with several diseases.
6. Conclusions
Cardiovascular diseases (CVD), such as coronary artery disease, peripheral arterial disease, or aortic disease, are a major cause of death worldwide and affect mainly the arteries. The vascular endothelium, formed by a monolayer of endothelial cells (ECs), is the innermost structure that lines not only the inner walls of arteries but also the inner walls of capillaries and veins. Human umbilical vein endothelial cells (HUVECs) are currently the most used ECs in in vitro studies, since their first isolation in 1973, due to being an easy, fast, and economical process. However, increasing evidence has shown that the endothelium is heterogeneous and depends on the vascular bed and that, therefore, arterial ECs are different from venous ECs. Furthermore, the circulation in the HUC is special in that the vein transports oxygen-rich blood and the arteries transport oxygen-poor blood, contrary to the systemic circulation, in which the arteries transport oxygen-rich blood and veins transport oxygen-poor blood. This raises the question as to whether umbilical cord-derived endothelium is really representative for the endothelium of other locations in the human body. Currently, there is confidence in saying that the results obtained with these cells can be transferred to other types of ECs, in different parts of the body. Therefore, both HUVECs and HUAECs are potential cell sources for cardiovascular research. In this sense, the vascular endothelium may be used as a target in the therapy of many of these diseases, namely CVD, since the endothelial cell-mediated dysfunctions have implications in vasomotor tone regulation and are associated with most CVD. However, more studies are needed to improve the clinical applications of endothelium in this field.
Author Contributions
Conceptualization, E.C.; writing—original draft preparation, C.M., M.L. and E.C.; writing—review and editing, C.M., M.L. and E.C.; supervision, E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by the Foundation for Science and Technology (FCT), through funds from the State Budget, and by the European Regional Development Fund (ERDF), under the Portugal 2020 Program, through the Regional Operational Program of the Center (Centro2020), through the Project with the reference UIDB/00709/2020. M.L. acknowledges the PhD fellowship from FCT (Reference: 2020.06616.BD).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Medina-Leyte, D.J.; Domínguez-Pérez, M.; Mercado, I.; Villarreal-Molina, M.T.; Jacobo-Albavera, L. Use of Human Umbilical Vein Endothelial Cells (HUVEC) as a Model to Study Cardiovascular Disease: A Review. Appl. Sci. 2020, 10, 938. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.; Gossen, M.; Lendlein, A.; Jung, F. Venous and Arterial Endothelial Cells from Human Umbilical Cords: Potential Cell Sources for Cardiovascular Research. Int. J. Mol. Sci. 2021, 22, 978. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxidative Med. Cell. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef] [Green Version]
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campesi, I.; Franconi, F.; Montella, A.; Dessole, S.; Capobianco, G. Human Umbilical Cord: Information Mine in Sex-Specific Medicine. Life 2021, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Dey, S.K.; Kundu, S. Functional implications of vascular endothelium in regulation of endothelial nitric oxide synthesis to control blood pressure and cardiac functions. Life Sci. 2020, 259, 118377. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.; Radomski, M.W. The nitric oxide-endothelin-1 connection. Heart Fail. Rev. 2003, 8, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Storch, A.S.; de Mattos, J.D.; Alves, R.; Galdino, I.d.S.; Rocha, H.N.M. Methods of Endothelial Function Assessment: Description and Applications. Int. J. Cardiovasc. Sci. 2017, 30, 262–273. [Google Scholar] [CrossRef]
- Martin de Llano, J.J.; Fuertes, G.; Garcia-Vicent, C.; Torro, I.; Fayos, J.L.; Lurbe, E. Procedure to consistently obtain endothelial and smooth muscle cell cultures from umbilical cord vessels. Transl. Res. 2007, 149, 1–9. [Google Scholar] [CrossRef]
- Lorigo, M.; Mariana, M.; Feiteiro, J.; Cairrao, E. Human Umbilical Artery Smooth Muscle Cells: Vascular Function and Clinical Importance. In Horizons in World Cardiovascular Research; Nova Science Publishers: New York, NY, USA, 2019; Volume 16, pp. 81–137. [Google Scholar]
- Yampolsky, M.; Salafia, C.M.; Shlakhter, O.; Haas, D.; Eucker, B.; Thorp, J. Modeling the variability of shapes of a human placenta. Placenta 2008, 29, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshiri, M.; Zaidi, S.F.; Robinson, T.J.; Bhargava, P.; Siebert, J.R.; Dubinsky, T.J.; Katz, D.S. Comprehensive imaging review of abnormalities of the umbilical cord. Radiographics 2014, 34, 179–196. [Google Scholar] [CrossRef]
- Chillakuru, S.; Velichety, S.D.; Rajagopalan, V. Human umbilical cord and its vessels: A histomorphometric study in difference severity of hypertensive disorders of pregnancy. Anat. Cell Biol. 2020, 53, 68–75. [Google Scholar] [CrossRef]
- Bosselmann, S.; Mielke, G. Sonographic Assessment of the Umbilical Cord. Geburtshilfe Frauenheilkd. 2015, 75, 808–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filiz, A.A.; Rahime, B.; Keskin, H.L.; Esra, A.K. Positive correlation between the quantity of Wharton’s jelly in the umbilical cord and birth weight. Taiwan. J. Obstet. Gynecol. 2011, 50, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arutyunyan, I.; Elchaninov, A.; Makarov, A.; Fatkhudinov, T. Umbilical Cord as Prospective Source for Mesenchymal Stem Cell-Based Therapy. Stem Cells Int. 2016, 2016, 6901286. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, V.L.; Dodson, R.B. Bioengineering aspects of the umbilical cord. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 144, S108–S113. [Google Scholar] [CrossRef]
- Kellow, Z.S.; Feldstein, V.A. Ultrasound of the placenta and umbilical cord: A review. Ultrasound Q. 2011, 27, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, W. Time for a more detailed prenatal examination of the umbilical cord? Ultrasound Obs. Gynecol. 1999, 13, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Spurway, J.; Logan, P.; Pak, S. The development, structure and blood flow within the umbilical cord with particular reference to the venous system. Australas. J. Ultrasound Med. 2012, 15, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; McGrath, B.C.; Bai, Y.; Tang, X.; Cavener, D.R. PERK regulates Gq protein-coupled intracellular Ca2+ dynamics in primary cortical neurons. Mol. Brain 2016, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Lorigo, M.; Mariana, M.; Feiteiro, J.; Cairrao, E. How is the human umbilical artery regulated? J. Obstet. Gynaecol. Res. 2018, 44, 1193–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoutte, P.M. Endothelium and control of vascular function. State of the Art lecture. Hypertension 1989, 13, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cetin, A.; Kukner, A.; Ozturk, F. Ultrastructure of human umbilical vessels in pre-eclampsia. J. Matern.-Fetal Neonatal Med. 2002, 12, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Rockelein, G.; Schneider, R. Three-dimensional analysis of the tunica media of umbilical arteries. Scanning electron microscopy study. Z. Geburtshilfe Perinatol. 1992, 196, 266–272. [Google Scholar] [PubMed]
- Meyer, W.W.; Rumpelt, H.J.; Yao, A.C.; Lind, J. Structure and closure mechanism of the human umbilical artery. Eur. J. Pediatr. 1978, 128, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, V.E.; Martinez-Gonzalez, B.; Quiroga-Garza, A.; Reyes-Hernandez, C.G.; de la Fuente-Villarreal, D.; de la Garza-Castro, O.; Guzman-Lopez, S.; Elizondo-Omana, R.E. Human Umbilical Vessels: Choosing the Optimal Decellularization Method. ASAIO J. 2018, 64, 575–580. [Google Scholar] [CrossRef] [PubMed]
- DeFreitas, M.J.; Mathur, D.; Seeherunvong, W.; Cano, T.; Katsoufis, C.P.; Duara, S.; Yasin, S.; Zilleruelo, G.; Rodriguez, M.M.; Abitbol, C.L. Umbilical artery histomorphometry: A link between the intrauterine environment and kidney development. J. Dev. Orig. Health Dis. 2017, 8, 349–356. [Google Scholar] [CrossRef]
- Pi, X.; Xie, L.; Patterson, C. Emerging Roles of Vascular Endothelium in Metabolic Homeostasis. Circ. Res. 2018, 123, 477–494. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. [Google Scholar] [PubMed]
- Vanhoutte, P.M. COX-1 and vascular disease. Clin. Pharmacol. Ther. 2009, 86, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, P.S.; Joseph, L.B.; Titterington, L.; Stephens, R.E. Methods for the Initiation and Maintenance of Human Endothelial Cell Culture. Vasc. Surg. 1987, 21, 391–400. [Google Scholar] [CrossRef]
- Siow, R.C. Culture of human endothelial cells from umbilical veins. Methods Mol. Biol. 2012, 806, 265–274. [Google Scholar]
- Lewis, W.H. Endothelium in tissue cultures. Am. J. Anat. 1922, 30, 39–59. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, Y. The human endothelial cell in tissue culture. Z. Zellforsch. Mikrosk. Anat. 1963, 60, 69–79. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; Cotran, R.S.; Folkman, J. Human vascular endothelial cells in culture. Growth and DNA synthesis. J. Cell Biol. 1974, 60, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Marin, V.; Kaplanski, G.; Grès, S.; Farnarier, C.; Bongrand, P. Endothelial cell culture: Protocol to obtain and cultivate human umbilical endothelial cells. J. Immunol. Methods 2001, 254, 183–190. [Google Scholar] [CrossRef]
- Ulrich-Merzenich, G.; Metzner, C.; Bhonde, R.R.; Malsch, G.; Schiermeyer, B.; Vetter, H. Simultaneous Isolation of Endothelial and Smooth Muscle Cells from Human Umbilical Artery or Vein and Their Growth Response to Low-Density Lipoproteins. In Vitr. Cell. Dev. Biol.-Anim. 2002, 38, 265–272. [Google Scholar] [CrossRef]
- Yang, S.J.; Son, J.K.; Hong, S.J.; Lee, N.E.; Shin, D.Y.; Park, S.H.; An, S.B.; Sung, Y.C.; Park, J.B.; Yang, H.M.; et al. Ectopic vascularized bone formation by human umbilical cord-derived mesenchymal stromal cells expressing bone morphogenetic factor-2 and endothelial cells. Biochem. Biophys. Res. Commun. 2018, 504, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, T.; Evensen, S.A.; Elgjo, R.F.; Vefling, A. Human fetal endothelial cells in cluture. Scand. J. Haematol. 1975, 14, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Baudin, B.; Bruneel, A.; Bosselut, N.; Vaubourdolle, M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nat. Protoc. 2007, 2, 481–485. [Google Scholar] [CrossRef]
- Cheung, A.L. Isolation and culture of human umbilical vein endothelial cells (HUVEC). Curr. Protoc. Microbiol. 2007, 4. [Google Scholar] [CrossRef]
- Kunkanjanawan, H.; Kunkanjanawan, T.; Khemarangsan, V.; Yodsheewan, R.; Theerakittayakorn, K.; Parnpai, R. A Xeno-Free Strategy for Derivation of Human Umbilical Vein Endothelial Cells and Wharton’s Jelly Derived Mesenchymal Stromal Cells: A Feasibility Study toward Personal Cell and Vascular Based Therapy. Stem Cells Int. 2020, 2020, 8832052. [Google Scholar] [CrossRef]
- Larrivee, B.; Karsan, A. Isolation and culture of primary endothelial cells. Methods Mol. Biol. 2005, 290, 315–329. [Google Scholar] [PubMed]
- Pipino, C.; Shah, H.; Prudente, S.; Di Pietro, N.; Zeng, L.; Park, K.; Trischitta, V.; Pennathur, S.; Pandolfi, A.; Doria, A. Association of the 1q25 Diabetes-Specific Coronary Heart Disease Locus With Alterations of the gamma-Glutamyl Cycle and Increased Methylglyoxal Levels in Endothelial Cells. Diabetes 2020, 69, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Brodowski, L.; Burlakov, J.; Hass, S.; von Kaisenberg, C.; von Versen-Hoynck, F. Impaired functional capacity of fetal endothelial cells in preeclampsia. PLoS ONE 2017, 12, e0178340. [Google Scholar] [CrossRef]
- Amrithraj, A.I.; Kodali, A.; Nguyen, L.; Teo AK, K.; Chang, C.W.; Karnani, N.; Ng, K.L.; Gluckman, P.D.; Chong, Y.S.; Stunkel, W. Gestational Diabetes Alters Functions in Offspring’s Umbilical Cord Cells with Implications for Cardiovascular Health. Endocrinology 2017, 158, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Psefteli, P.M.; Kitscha, P.; Vizcay, G.; Fleck, R.; Chapple, S.J.; Mann, G.E.; Fowler, M.; Siow, R.C. Glycocalyx sialic acids regulate Nrf2-mediated signaling by fluid shear stress in human endothelial cells. Redox Biol. 2021, 38, 101816. [Google Scholar] [CrossRef]
- Bachetti, T.; Morbidelli, L. Endothelial cells in culture: A model for studying vascular functions. Pharmacol. Res. 2000, 42, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Provitera, L.; Cavallaro, G.; Griggio, A.; Raffaeli, G.; Amodeo, I.; Gulden, S.; Lattuada, D.; Ercoli, G.; Lonati, C.; Tomaselli, A.; et al. Cyclic nucleotide-dependent relaxation in human umbilical vessels. J. Physiol. Pharmacol. 2019, 70, 619–630. [Google Scholar]
- Di Tomo, P.; Lanuti, P.; Di Pietro, N.; Baldassarre MP, A.; Marchisio, M.; Pandolfi, A.; Consoli, A.; Formoso, G. Liraglutide mitigates TNF-alpha induced pro-atherogenic changes and microvesicle release in HUVEC from diabetic women. Diabetes Metab. Res. Rev. 2017, 33, e2925. [Google Scholar] [CrossRef] [PubMed]
- Hendijani, F. Explant culture: An advantageous method for isolation of mesenchymal stem cells from human tissues. Cell Prolif. 2017, 50, e12334. [Google Scholar] [CrossRef] [Green Version]
- Kadam, S.S.; Tiwari, S.; Bhonde, R.R. Simultaneous isolation of vascular endothelial cells and mesenchymal stem cells from the human umbilical cord. In Vitr. Cell. Dev. Biol.-Anim. 2009, 45, 23–27. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fina, L.; Molgaard, H.V.; Robertson, D.; Bradley, N.J.; Monaghan, P.; Delia, D.; Sutherland, D.R.; Baker, M.A.; Greaves, M.F. Expression of the CD34 gene in vascular endothelial cells. Blood 1990, 75, 2417–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutin, M.; Dignat-George, F.; Sampol, J. Immunologic phenotype of cultured endothelial cells: Quantitative analysis of cell surface molecules. Tissue Antigens 1997, 50, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; Ward, P.A. C5a-induced expression of P-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Pei, H.; Wang, S.; Zhang, B.; Fan, Z.; Liu, Y.; Xie, X.; Yang, Z.; Xu, L.; Jia, Y.; et al. Arterial endothelium creates a permissive niche for expansion of human cord blood hematopoietic stem and progenitor cells. Stem Cell Res. Ther. 2020, 11, 358. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA. 2003, 100, 10623–10628. [Google Scholar] [CrossRef] [Green Version]
- Mann, G.E.; Pearson, J.D.; Sheriff, C.J.; Toothill, V.J. Expression of amino acid transport systems in cultured human umbilical vein endothelial cells. J. Physiol. 1989, 410, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Sobrevia, L.; Cesare, P.; Yudilevich, D.L.; Mann, G.E. Diabetes-induced activation of system y+ and nitric oxide synthase in human endothelial cells: Association with membrane hyperpolarization. J. Physiol. 1995, 489, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahabeleshwar, G.H.; Somanath, P.R.; Byzova, T.V. Methods for isolation of endothelial and smooth muscle cells and in vitro proliferation assays. Methods Mol. Med. 2006, 129, 197–208. [Google Scholar] [PubMed]
- Crampton, S.P.; Davis, J.; Hughes, C.C. Isolation of human umbilical vein endothelial cells (HUVEC). J. Vis. Exp. 2007, 3, e183. [Google Scholar]
- Casanello, P.; Krause, B.; Torres, E.; Gallardo, V.; González, M.; Prieto, C.; Escudero, C.; Farías, M.; Sobrevia, L. Reduced l-arginine transport and nitric oxide synthesis in human umbilical vein endothelial cells from intrauterine growth restriction pregnancies is not further altered by hypoxia. Placenta 2009, 30, 625–633. [Google Scholar] [CrossRef]
- Krause, B.J.; Prieto, C.P.; Munoz-Urrutia, E.; San Martin, S.; Sobrevia, L.; Casanello, P. Role of arginase-2 and eNOS in the differential vascular reactivity and hypoxia-induced endothelial response in umbilical arteries and veins. Placenta 2012, 33, 360–366. [Google Scholar] [CrossRef]
- Pang, H. Make Human Umbilical Vein Endothelial Cells from Cords. Bio-Protocol 2012, 2, e204. [Google Scholar] [CrossRef]
- Lattuada, D.; Roda, B.; Pignatari, C.; Magni, R.; Colombo, F.; Cattaneo, A.; Zattoni, A.; Cetin, I.; Reschiglian, P.; Bolis, G. A tag-less method for direct isolation of human umbilical vein endothelial cells by gravitational field-flow fractionation. Anal. Bioanal. Chem. 2013, 405, 977–984. [Google Scholar] [CrossRef]
- Lei, J.; Peng, S.; Samuel, S.B.; Zhang, S.; Wu, Y.; Wang, P.; Li, Y.F.; Liu, H. A simple and biosafe method for isolation of human umbilical vein endothelial cells. Anal. Biochem. 2016, 508, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Krause, B.J.; Hernandez, C.; Caniuguir, A.; Vasquez-Devaud, P.; Carrasco-Wong, I.; Uauy, R.; Casanello, P. Arginase-2 is cooperatively up-regulated by nitric oxide and histone deacetylase inhibition in human umbilical artery endothelial cells. Biochem. Pharmacol. 2016, 99, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Suhaila, R.N.; Safuan, S. Isolation Methods and Culture Conditions of Human Umbilical Vein Endothelial Cells from Malaysian Women. Sains Malays. 2017, 46, 463–468. [Google Scholar] [CrossRef]
- Thormodsson, F.R.; Olafsson, I.H.; Vilhjalmsson, D.T. Preparation and Culturing of Human Primary Vascular Cells. Methods Mol. Biol. 2018, 1779, 355–369. [Google Scholar]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Vanhoutte, P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989, 3, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405. [Google Scholar] [CrossRef]
- Izumi, H.; Makino, Y.; Shirakawa, K.; Garfield, R.E. Role of nitric oxide on vasorelaxation in human umbilical artery. Am. J. Obstet. Gynecol. 1995, 172, 1477–1484. [Google Scholar] [CrossRef]
- Izumi, H.; Makino, Y.; Mohtai, H.; Shirakawa, K.; Garfield, R.E. Comparison of nitric oxide and prostacyclin in endothelium-dependent vasorelaxation of human umbilical artery at midgestation. Am. J. Obstet. Gynecol. 1996, 175, 375–381. [Google Scholar] [CrossRef]
- Lovren, F.; Triggle, C. Nitric oxide and sodium nitroprusside-induced relaxation of the human umbilical artery. Br. J. Pharmacol. 2000, 131, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenkranz-Weiss, P.; Sessa, W.C.; Milstien, S.; Kaufman, S.; Watson, C.A.; Pober, J.S. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J. Clin. Investig. J. 1994, 93, 2236–2243. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.M.; Chen, T.L.; Lin, Y.L.; Chen, T.G.; Tai, Y.T. Ketamine reduces nitric oxide biosynthesis in human umbilical vein endothelial cells by down-regulating endothelial nitric oxide synthase expression and intracellular calcium levels. Crit. Care Med. 2005, 33, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Ugusman, A.; Zakaria, Z.; Chua, K.H.; Nordin, N.A.; Abdullah Mahdy, Z. Role of rutin on nitric oxide synthesis in human umbilical vein endothelial cells. Sci. World J. 2014, 2014, 169370. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Brunner, F.; Bras-Silva, C.; Cerdeira, A.S.; Leite-Moreira, A.F. Cardiovascular endothelins: Essential regulators of cardiovascular homeostasis. Pharmacol. Ther. 2006, 111, 508–531. [Google Scholar] [CrossRef]
- Sanchez, A.; Martinez, P.; Munoz, M.; Benedito, S.; Garcia-Sacristan, A.; Hernandez, M.; Prieto, D. Endothelin-1 contributes to endothelial dysfunction and enhanced vasoconstriction through augmented superoxide production in penile arteries from insulin-resistant obese rats: Role of ET(A) and ET(B) receptors. Br. J. Pharmacol. 2014, 171, 5682–5695. [Google Scholar] [CrossRef] [Green Version]
- Enevoldsen, F.C.; Sahana, J.; Wehland, M.; Grimm, D.; Infanger, M.; Kruger, M. Endothelin Receptor Antagonists: Status Quo and Future Perspectives for Targeted Therapy. J. Clin. Med. 2020, 9, 824. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; VanBeusecum, J.P.; Inscho, E.W. Endothelin and the renal microcirculation. Semin. Nephrol. 2015, 35, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasser, S.A.; El-Mas, M.M. Endothelin ETA receptor antagonism in cardiovascular disease. Eur. J. Pharmacol. 2014, 737, 210–213. [Google Scholar] [CrossRef]
- Thorin, E.; Webb, D.J. Endothelium-derived endothelin-1. Pflug. Arch. 2010, 459, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Khimji, A.K.; Rockey, D.C. Endothelin--biology and disease. Cell. Signal. 2010, 22, 1615–1625. [Google Scholar] [CrossRef]
- Pernow, J.; Shemyakin, A.; Bohm, F. New perspectives on endothelin-1 in atherosclerosis and diabetes mellitus. Life Sci. 2012, 91, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Majed, B.H.; Khalil, R.A. Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol. Rev. 2012, 64, 540–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabito Colafella, K.M.; Neuman, R.I.; Visser, W.; Danser AH, J.; Versmissen, J. Aspirin for the prevention and treatment of pre-eclampsia: A matter of COX-1 and/or COX-2 inhibition? Basic Clin. Pharmacol. Toxicol. 2020, 127, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Villar, I.C.; Francis, S.; Webb, A.; Hobbs, A.J.; Ahluwalia, A. Novel aspects of endothelium-dependent regulation of vascular tone. Kidney Int. 2006, 70, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eivers, S.B.; Kinsella, B.T. Regulated expression of the prostacyclin receptor (IP) gene by androgens within the vasculature: Combined role for androgens and serum cholesterol. Biochim. Biophys. Acta 2016, 1859, 1333–1351. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M.; Huang, Y.; Vanhoutte, P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br. J. Pharmacol. 2011, 164, 894–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, S.W.S. The vascular impact of IP-TP receptor interactions. Acta Physiol. 2020, 231, e13577. [Google Scholar]
- Lim, H.; Dey, S.K. A novel pathway of prostacyclin signaling-hanging out with nuclear receptors. Endocrinology 2002, 143, 3207–3210. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, K.; Sintetos, A.; Ramwell, P. The effect of prostacyclin on the human umbilical artery. Prostaglandins 1978, 15, 1035–1044. [Google Scholar] [CrossRef]
- Klockenbusch, W.; Braun, M.S.; Schröder, H.; Heckenberger, R.E.; Strobach, H.; Schrör, K. Prostacyclin rather than nitric oxide lowers human umbilical artery tone in vitro. Eur. J. Obstet. Gynecol. Reprod. Biol. 1992, 47, 109–115. [Google Scholar] [CrossRef]
- Estrada-García, L.; Carrera-Rotllan, J.; Puig-Parellada, P. Effects of oxidative stress and antioxidant treatments on eicosanoid synthesis and lipid peroxidation in long term human umbilical vein endothelial cells culture. Prostaglandins Other Lipid Mediat. 2002, 67, 13–25. [Google Scholar] [CrossRef]
- Walsh, S.W. Eicosanoids in preeclampsia. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liu, Y.; Zhang, Y.; Zhang, Y.; Li, H.; Zheng, Q.; Li, N.; Tang, J.; Xu, Z. New views on endothelial dysfunction in gestational hypertension and potential therapy targets. Drug Discov. Today 2021, 26, 1420–1436. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Durante, W.; Sowers, J.R. Endothelium-Derived Hyperpolarizing Factors: A Potential Therapeutic Target for Vascular Dysfunction in Obesity and Insulin Resistance. Diabetes 2016, 65, 2118–2120. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, H.; Godo, S. Nitric oxide and endothelium-dependent hyperpolarization mediated by hydrogen peroxide in health and disease. Basic Clin. Pharmacol. Toxicol. 2020, 127, 92–101. [Google Scholar] [CrossRef]
- Heptinstall, S. Antiplatelet Agents: Current and Novel. In Antiplatelet and Anticoagulation Therapy; Ferro, A., Garcia, D.A., Eds.; Springer: London, UK, 2013; pp. 1–44. [Google Scholar]
- Wu, Z.; Yao, H.; Xu, H.; Wang, Y.; Hu, W.; Lou, G.; Zhang, L.; Huang, C.; Jiang, C.; Zhou, S.; et al. Inhibition of eNOS by L-NAME resulting in rat hind limb developmental defects through PFKFB3 mediated angiogenetic pathway. Sci. Rep. 2020, 10, 16754. [Google Scholar] [CrossRef]
- Suksawat, M.; Techasen, A.; Namwat, N.; Boonsong, T.; Titapun, A.; Ungarreevittaya, P.; Yongvanit, P.; Loilome, W. Inhibition of endothelial nitric oxide synthase in cholangiocarcinoma cell lines—A new strategy for therapy. FEBS Open Bio 2018, 8, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Luksha, L.; Agewall, S.; Kublickiene, K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis 2009, 202, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M. Calcium-activated potassium channels and endothelial dysfunction: Therapeutic options? Br. J. Pharmacol. 2009, 156, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Martinez-Lemus, L.A.; Zhang, C. Endothelium-derived hyperpolarizing factor and diabetes. World J. Cardiol. 2011, 3, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, H. Role of thromboxane A2 signaling in endothelium-dependent contractions of arteries. Prostaglandins Other Lipid Mediat. 2018, 134, 32–37. [Google Scholar] [CrossRef]
- Sellers, M.M.; Stallone, J.N. Sympathy for the devil: The role of thromboxane in the regulation of vascular tone and blood pressure. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1978–H1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodelsson, G.; Maršál, K.; Stjernquist, M. Reduced contractile effect of endothelin-1 and noradrenalin in human umbilical artery from pregnancies with abnormal umbilical artery flow velocity waveforms. Early Hum. Dev. 1995, 42, 15–28. [Google Scholar] [CrossRef]
- Ellinsworth, D.C.; Shukla, N.; Fleming, I.; Jeremy, J.Y. Interactions between thromboxane A(2), thromboxane/prostaglandin (TP) receptors, and endothelium-derived hyperpolarization. Cardiovasc. Res. 2014, 102, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Kakami, M.; Noguchi, E.; Kobayashi, T.; Kamata, K. Imbalance between endothelium-derived relaxing and contracting factors in mesenteric arteries from aged OLETF rats, a model of Type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1480–H1490. [Google Scholar] [CrossRef]
- Matsumoto, T.; Noguchi, E.; Ishida, K.; Kobayashi, T.; Yamada, N.; Kamata, K. Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1165–H1176. [Google Scholar] [CrossRef] [Green Version]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719–741. [Google Scholar] [CrossRef]
- Tirapelli, C.R.; Bonaventura, D.; Tirapelli, L.F.; de Oliveira, A.M. Mechanisms underlying the vascular actions of endothelin 1, angiotensin II and bradykinin in the rat carotid. Pharmacology 2009, 84, 111–126. [Google Scholar] [CrossRef]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar] [PubMed]
- Montezano, A.C.; Nguyen Dinh Cat, A.; Rios, F.J.; Touyz, R.M. Angiotensin II and vascular injury. Curr. Hypertens. Rep. 2014, 16, 431. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Faria-Costa, G.; Leite-Moreira, A.; Henriques-Coelho, T. Cardiovascular effects of the angiotensin type 2 receptor. Rev. Port. Cardiol. 2014, 33, 439–449. [Google Scholar] [CrossRef]
- Masi, S.; Uliana, M.; Virdis, A. Angiotensin II and vascular damage in hypertension: Role of oxidative stress and sympathetic activation. Vasc. Pharm. 2019, 115, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Ancion, A.; Tridetti, J.; Nguyen Trung, M.L.; Oury, C.; Lancellotti, P. A Review of the Role of Bradykinin and Nitric Oxide in the Cardioprotective Action of Angiotensin-Converting Enzyme Inhibitors: Focus on Perindopril. Cardiol. Ther. 2019, 8, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Schena, M.; Mulatero, P.; Schiavone, D.; Mengozzi, G.; Tesio, L.; Chiandussi, L.; Veglio, F. Vasoactive hormones induce nitric oxide synthase mRNA expression and nitric oxide production in human endothelial cells and monocytes. Am. J. Hypertens. 1999, 12, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Radenkovic, M.; Grbovic, L.; Radunovic, N.; Momcilov, P. Pharmacological evaluation of bradykinin effect on human umbilical artery in normal, hypertensive and diabetic pregnancy. Pharmacol. Rep. 2007, 59, 64–73. [Google Scholar]
- Hornig, B.; Drexler, H. Endothelial function and bradykinin in humans. Drugs 1997, 54, 42–47. [Google Scholar] [CrossRef]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The kallikrein-kinin system: Current and future pharmacological targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumbadinga, G.A.; Desormeaux, A.; Adam, A.; Marceau, F. Effect of interferon-gamma on inflammatory cytokine-induced bradykinin B1 receptor expression in human vascular cells. Eur. J. Pharmacol. 2010, 647, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marceau, F.; Bachelard, H.; Bouthillier, J.; Fortin, J.P.; Morissette, G.; Bawolak, M.T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 106305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tom, B.; de Vries, R.; Saxena, P.R.; Danser, A.H. Bradykinin potentiation by angiotensin-(1-7) and ACE inhibitors correlates with ACE C- and N-domain blockade. Hypertension 2001, 38, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Takano, M.; Matsuyama, S. Intracellular and nuclear bradykinin B2 receptors. Eur. J. Pharmacol. 2014, 732, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.; Rhaleb, I.A.; Kassem, K.M.; Rhaleb, N.E. Role of Kinins in Hypertension and Heart Failure. Pharmaceuticals 2020, 13, 347. [Google Scholar] [CrossRef]
- Bai, B.; Liu, L.; Zhang, N.; Wang, C.; Jiang, Y.; Chen, J. Heterodimerization of human apelin and bradykinin 1 receptors: Novel signal transduction characteristics. Cell. Signal. 2014, 26, 1549–1559. [Google Scholar] [CrossRef]
- Murphey, L. Contribution of bradykinin to the cardioprotective effects of ACE inhibitors. Eur. Heart J. Suppl. 2003, 5, A37–A41. [Google Scholar] [CrossRef] [Green Version]
- Conlon, J.M. The kallikrein-kinin system: Evolution of form and function. Ann. N. Y. Acad. Sci. 1998, 839, 1–8. [Google Scholar] [CrossRef]
- Nowak, W.; Errasti, A.E.; Armesto, A.R.; Santin Velazque, N.L.; Rothlin, R.P. Endothelial angiotensin-converting enzyme and neutral endopeptidase in isolated human umbilical vein: An effective bradykinin inactivation pathway. Eur. J. Pharmacol. 2011, 667, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Bortolotto, L. Unraveling the Pivotal Role of Bradykinin in ACE Inhibitor Activity. Am. J. Cardiovasc. Drugs 2016, 16, 309–321. [Google Scholar] [CrossRef]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharm. 2019, 10, 1568. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Yuyun, M.F.; Ng, L.L.; Ng, G.A. Endothelial dysfunction, endothelial nitric oxide bioavailability, tetrahydrobiopterin, and 5-methyltetrahydrofolate in cardiovascular disease. Where are we with therapy? Microvasc. Res. 2018, 119, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Grover-Paez, F.; Zavalza-Gomez, A.B. Endothelial dysfunction and cardiovascular risk factors. Diabetes Res. Clin. Pract. 2009, 84, 1–10. [Google Scholar] [CrossRef]
- Widmer, R.J.; Lerman, A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014, 2014, 291–308. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Z.; Zhang, L.; Wang, Y. Roles of Cells from the Arterial Vessel Wall in Atherosclerosis. Mediat. Inflamm. 2017, 2017, 8135934. [Google Scholar] [CrossRef]
- Knapp, M.; Tu, X.; Wu, R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharm. Sin. 2019, 40, 1–8. [Google Scholar] [CrossRef]
- Radenkovic, M.; Stojanovic, M.; Nesic, I.M.; Prostran, M. Angiotensin receptor blockers & endothelial dysfunction: Possible correlation & therapeutic implications. Indian J. Med. Res. 2016, 144, 154–168. [Google Scholar] [PubMed]
- Echeverria, C.; Eltit, F.; Santibanez, J.F.; Gatica, S.; Cabello-Verrugio, C.; Simon, F. Endothelial dysfunction in pregnancy metabolic disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165414. [Google Scholar] [CrossRef]
- Boeldt, D.S.; Bird, I.M. Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia. J. Endocrinol. 2017, 232, R27–R44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Aranguren, L.C.; Prada, C.E.; Riano-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Nakhla, S.; Makris, A.; Hennessy, A. TNF-α inhibits trophoblast integration into endothelial cellular networks. Placenta 2011, 32, 241–246. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the main mechanisms involved in the regulation of vascular tone by endothelial cells. The EDHF pathway (represented in orange) promotes an increase in intracellular Ca2+, activating K+ channels, and increasing K+ efflux. In SMC, it activates K+ channels causing a hyperpolarization of SMC, leading to the closure of voltage-sensitive Ca2+ channels (kCa), and a relaxation of SMC. In ECs, AA is converted to PGG2, which is converted back to PGH2. PGH2 will form PGI2. PGI2 is released and activates G-protein coupled IP receptors, increasing cAMP and activating PKA (red). Subsequently, this decreases Ca2+ concentration, decreases Ca2+-Cam binding, and leads to SMC relaxation (black). PGI2 can also bind to the IP receptor present on the endothelial cell, leading to an activation of eNOS, increases cGMP, and activates PKG, causing vasorelaxation. In ECs, L-arginine (L-arg) is converted to NO and L-citrulline via eNOS (green mechanism). After diffusing into the membrane, it activates guanylyl cyclase (GC), which in turn activates PKG and increases cGMP. This increase leads to a decrease in Ca2+, a decrease in Ca2+-CaM binding and decreases the phosphorylation of myosin light chains (MLCK) (common black mechanism). When bradykinin binds to the B2 receptor of the endothelial cell, it activates a Gq protein, which activates PLC and releases IP3, increasing Ca2+ production and leading to activation of eNOS, producing NO (blue pathway). If it binds to the B2 receptor of SMC, it will activate the Gq protein, activating PLC, releasing IP3 and DAG. This release will activate PKC which increases Ca2+ concentration and causes vasocontraction (common black pathway). Thus, similarly to PGI2, the TXA2 formation pathway is similar in ECs. After PGH2 is formed, it is converted into TXA2 (pink pathway). In SMC, TXA2 binds to the TP receptor, activating a Gq protein activating PLC, releasing IP3 and DAG. This release will activate PKC which increases Ca2+ concentration and causes vasocontraction (common black pathway). In ECs, Ang I is converted to Ang II, by ACE. Ang II will bind to the AT1 receptor (gray pathway) present in SMC, activating a Gq protein activating PLC, releasing IP3 and DAG. This release will activate PKC which increases Ca2+ concentration and causes vasocontraction (common black pathway). Ang II can also bind to the AT2 receptor present in ECs, thus activating the B2 receptor, and triggering the same mechanism that leads to vasorelaxation (gray pathway). Prepro-ET1 is converted to Big ET1 which, via the ECE, is converted back to ET1 (purple pathway) in the ECs. When ETB receptors are expressed on SMC, an activation of Gq protein and an activation of PKC occur, releasing IP3, and a subsequent release of Ca2+, leading to vasoconstriction (common black pathway). On the other hand, when ET-1, binds to the ETB receptor present on endothelial cells the release of Ca2+ occurs, which will bind to calmodulin to activate calmodulin kinase, responsible for the phosphorylation of eNOS, initiating NO synthesis in the endothelial cell. However, this binding can also trigger prostacyclin production, such as PGI2, which leads to an increase in cAMP and subsequently vasodilation.
Figure 1.
Schematic representation of the main mechanisms involved in the regulation of vascular tone by endothelial cells. The EDHF pathway (represented in orange) promotes an increase in intracellular Ca2+, activating K+ channels, and increasing K+ efflux. In SMC, it activates K+ channels causing a hyperpolarization of SMC, leading to the closure of voltage-sensitive Ca2+ channels (kCa), and a relaxation of SMC. In ECs, AA is converted to PGG2, which is converted back to PGH2. PGH2 will form PGI2. PGI2 is released and activates G-protein coupled IP receptors, increasing cAMP and activating PKA (red). Subsequently, this decreases Ca2+ concentration, decreases Ca2+-Cam binding, and leads to SMC relaxation (black). PGI2 can also bind to the IP receptor present on the endothelial cell, leading to an activation of eNOS, increases cGMP, and activates PKG, causing vasorelaxation. In ECs, L-arginine (L-arg) is converted to NO and L-citrulline via eNOS (green mechanism). After diffusing into the membrane, it activates guanylyl cyclase (GC), which in turn activates PKG and increases cGMP. This increase leads to a decrease in Ca2+, a decrease in Ca2+-CaM binding and decreases the phosphorylation of myosin light chains (MLCK) (common black mechanism). When bradykinin binds to the B2 receptor of the endothelial cell, it activates a Gq protein, which activates PLC and releases IP3, increasing Ca2+ production and leading to activation of eNOS, producing NO (blue pathway). If it binds to the B2 receptor of SMC, it will activate the Gq protein, activating PLC, releasing IP3 and DAG. This release will activate PKC which increases Ca2+ concentration and causes vasocontraction (common black pathway). Thus, similarly to PGI2, the TXA2 formation pathway is similar in ECs. After PGH2 is formed, it is converted into TXA2 (pink pathway). In SMC, TXA2 binds to the TP receptor, activating a Gq protein activating PLC, releasing IP3 and DAG. This release will activate PKC which increases Ca2+ concentration and causes vasocontraction (common black pathway). In ECs, Ang I is converted to Ang II, by ACE. Ang II will bind to the AT1 receptor (gray pathway) present in SMC, activating a Gq protein activating PLC, releasing IP3 and DAG. This release will activate PKC which increases Ca2+ concentration and causes vasocontraction (common black pathway). Ang II can also bind to the AT2 receptor present in ECs, thus activating the B2 receptor, and triggering the same mechanism that leads to vasorelaxation (gray pathway). Prepro-ET1 is converted to Big ET1 which, via the ECE, is converted back to ET1 (purple pathway) in the ECs. When ETB receptors are expressed on SMC, an activation of Gq protein and an activation of PKC occur, releasing IP3, and a subsequent release of Ca2+, leading to vasoconstriction (common black pathway). On the other hand, when ET-1, binds to the ETB receptor present on endothelial cells the release of Ca2+ occurs, which will bind to calmodulin to activate calmodulin kinase, responsible for the phosphorylation of eNOS, initiating NO synthesis in the endothelial cell. However, this binding can also trigger prostacyclin production, such as PGI2, which leads to an increase in cAMP and subsequently vasodilation.


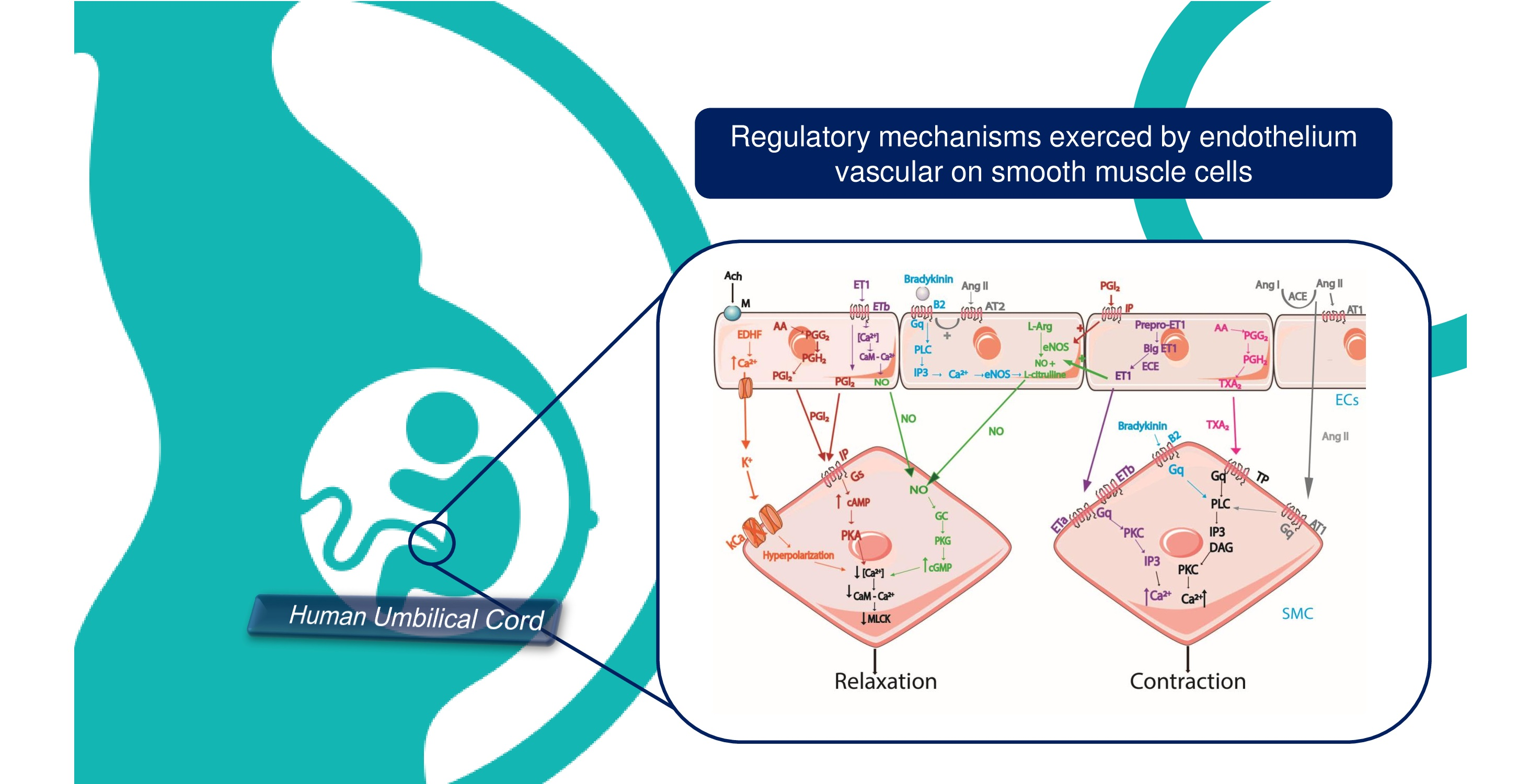{kind=link}
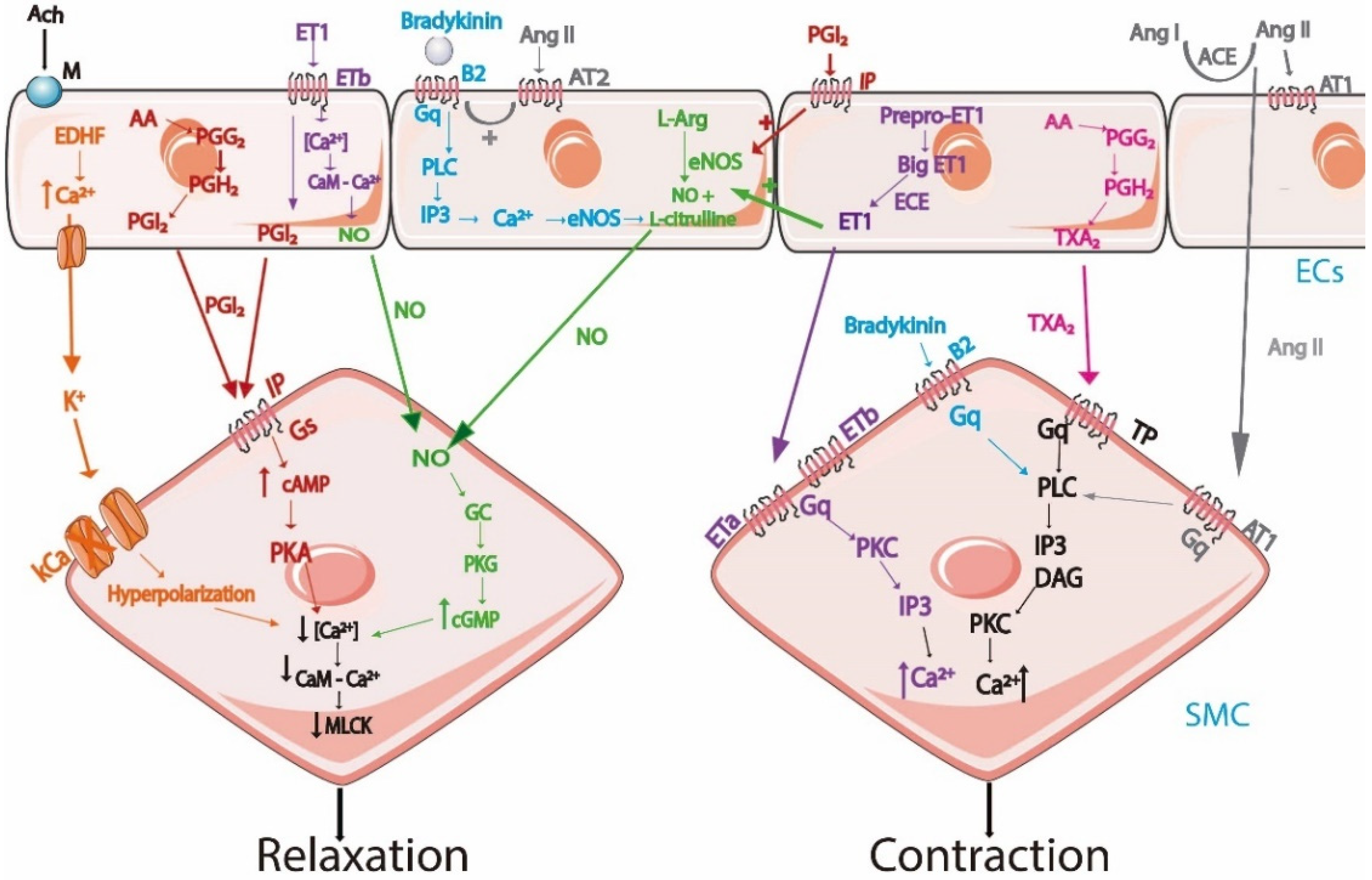{kind=link}
Table 1.
Conditions used for isolation of endothelial cells.
| Studies Performed | Digestive Enzyme | Medium Used | Centrifugation Conditions | Coating | Cells | Protocol Based on | Observations |
|---|---|---|---|---|---|---|---|
| Maruyama et al. (1963) [38] | 0.2% Trypsin | YLH | - | Rat-tail Collagen (coverslip) | HUVECs | - | The cells were cultured in sheets |
| Jaffe et al. (1973) [39] | 0.2% Collagenase | TC 199 | 250 g 10 min | - | HUVECs | Based on Maruyama et al. | ECs differentiation based on morphologic and immunologic criteria |
| Mann et al. (1989) [65] | Collagenase type II | M199 | - | 1% Gelatin | HUVECs | Based on Jaffe et al. | ECs were cultured on microcarrier beads |
| Sobrevia et al. (1995) [66] | Collagenase | M199 | - | - | HUVECs | - | HUC from gestational diabetic pregnancies |
| Marin et al. (2001) [41] | Collagenase A | M199 | 1500 r/min 5 min | 1% Gelatin | HUVECs | - | Three different cords were used to limit the variability of ECs |
| Ulrich-mersenich et al. (2002) [42] | Dispase II and Collagenase tipo IV | M199 | 1500 rpm 15 min | Fibronectin | HUVECS and HUAECS | Based on Ko et al. (1995) | Isolation of ECs and SMC from the same vessel of HUC obtain the best results |
| Larrivee et al. (2005) [48] | Collagenase A | MCDB 131 | 300 g 5 min | 0.2% Gelatin | HUVECs | - | ECs successful cryopreservation |
| Mahabeleshwar et al. (2006) [67] | 0.1% Collagenase | Endothelial cell growth medium | 320 g 10 min | Gelatin | HUVECs | - | Obtention pure cultures of ECs |
| Baudin et al. (2007) [45] | 0.2% Collagenase | M199 | 750 g 10 min | Fibronectin | HUVECs | Based on Jaffe et al. | HUVECs can keep phenotypic features in free serum medium for up 12 h |
| Crampton et al. (2007) [68] | Collagenase | M199 | 1200 rpm 5 min | Gelatin | HUVECs | - | Used in angiogenesis assay |
| Cheung et al. (2007) [46] | 0.1% Collagenase | M199 | 250 g 10 min Room temperature | Gelatin | HUVECs | - | Confluent T25 flask was obtained in 4–5 days. Cells beyond the 6th passage should be discarded. |
| Martin de llano et al. (2007) [11] | Collagenase type I and Dispase | M200 or M231 | 10 g 10 min | Fibronectin | HUVECs and HUAECs | - | Birth weight does not have influence on the time request to obtain grown cultures |
| Casanello et al. (2009) [69] | Collagenase | M199 | - | - | HUVECs | Based on Sobrevia et al. | ECs maintain the protein expression until 5th passage |
| Kadam et al. (2009) [57] | Dispase II and Collagenase type IV | M199 | 1500 rpm 15 min | Fibronectin | HUVECS and hUCMSCs | Based on Ulrich-mersenich et al. | Two-steps protocol for the simultaneous isolation of ECs and hUCMSCs |
| Krause et al. (2012) [70] | Collagenase | M199 | - | - | HUVECs and HUAECs | Based on Casanello et al. | ECs are responsive to hypoxia |
| Pang et al. (2012) [71] | Collagenase | M199 | 1000 g 5 min | - | HUVECs | Based on Jaffe et al. | ECs are responsive to cytokine stimulation |
| Siow et al. (2012) [36] | Collagenase | M199 | 1000 rpm 5 min | Gelatin | HUVECs | Based on Jaffe et al. | Confluent culture of ECs after 2–3 days will start to detach and decrease viability |
| Lattuada et al. (2013) [72] | 0.1% Collagenase A | M199 | 463 g 15 min | - | HUVECs | Based on Jaffe et al. | Gravitational field-flow fractionation is the new method to easy isolate ECs from HUC |
| Lei et al. (2016) [73] | 0.2% Collagenasee | M199 | 800 g 5 min | Gelatin | HUVECs | - | Used a new instrument for insertion of HUC |
| Krause et al. (2016) [74] | Collagenase | M199 | - | - | HUAECs | Based on Krause et al. (2012) | ECs pure culture can be used for protein expression |
| Amrithraj et al. (2017) [51] | Collagenase | EGM-2 | 1200 rpm 5 min | Gelatin | HUVECs | Based on Crampton et al. | ECs culture from gestational diabetic pregnancies maintain metabolic and molecular imprints of maternal hyperglycemia |
| Brodowaski et al. (2017) [50] | 0.2% Collagenase | EGM | - | - | HUVEC | Based on Jaffe et al. | EC culture from preeclamptic women |
| Di tomo et al. (2017) [55] | Collagenase 1 | M199 | - | 1.5% Gelatin | HUVECs | - | EC culture was obtained with explants |
| Suhaila et al. (2017) [75] | Collagenase type I | M199 | 1500 rpm 5 min | They tested various concentrations of gelatin | HUVECs | - | The 0.2% gelatin obtain the best results |
| Thormodsson et al. (2018) [76] | Collagenase | M199 | 140 g 5 min | - | HUVECS and HUAECs | - | Using NunclonTM Δ T-25 flasks: the cells attach without any problem |
| Yang et al. (2018) [43] | 0.05% Collagenase I | EGM-2 | 250 g 5 min | Fibronectin | HUVECs | Based on Jaffe et al. | The cells were negative for CD34, CD45, and human leukocyte antigen–DR isotype (HLA-DR) |
| Provitera et al. (2019) [54] | 0.1% Collagenase A | M199 | 463 g 15 min | - | HUAECs and HUVECs | Based on Lattuada et al. | Cells were used at P0 |
| Pipino et al. (2020) [49] | Collagenase type1A | DMEM | - | 1.5% Gelatin | HUVECs | Based on Di tomo et al. | Cells between the 3rd and 7th passages were used |
| Psefteli et al. (2021) [52] | 0.2% Collagenase | M199 | - | Gelatin | HUVECs | Based on Jaffe et al. | Cells between P0 and P3 were used for protein expression |
Table 2.
Effects caused by substances released by endothelial cells.
| Vasorelaxation | Vasoconstriction | Both Effects |
|---|---|---|
| NO (guanylyl cyclase) | TXA2 (TP receptor) | ET-1 (ETA and ETB receptor) |
| EDHF (potassium channels) | Bradykinin (B1 and B2 receptor) | |
| PGI2 (IP receptor) | ANGII (AT1 and AT2 receptor) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mangana, C.; Lorigo, M.; Cairrao, E. Implications of Endothelial Cell-Mediated Dysfunctions in Vasomotor Tone Regulation. Biologics 2021, 1, 231-251. https://0-doi-org.brum.beds.ac.uk/10.3390/biologics1020015
AMA Style
Mangana C, Lorigo M, Cairrao E. Implications of Endothelial Cell-Mediated Dysfunctions in Vasomotor Tone Regulation. Biologics. 2021; 1(2):231-251. https://0-doi-org.brum.beds.ac.uk/10.3390/biologics1020015
Chicago/Turabian StyleMangana, Carolina, Margarida Lorigo, and Elisa Cairrao. 2021. "Implications of Endothelial Cell-Mediated Dysfunctions in Vasomotor Tone Regulation" Biologics 1, no. 2: 231-251. https://0-doi-org.brum.beds.ac.uk/10.3390/biologics1020015