
The Contribution of Endothelial-Mesenchymal Transition to Atherosclerosis

Abstract
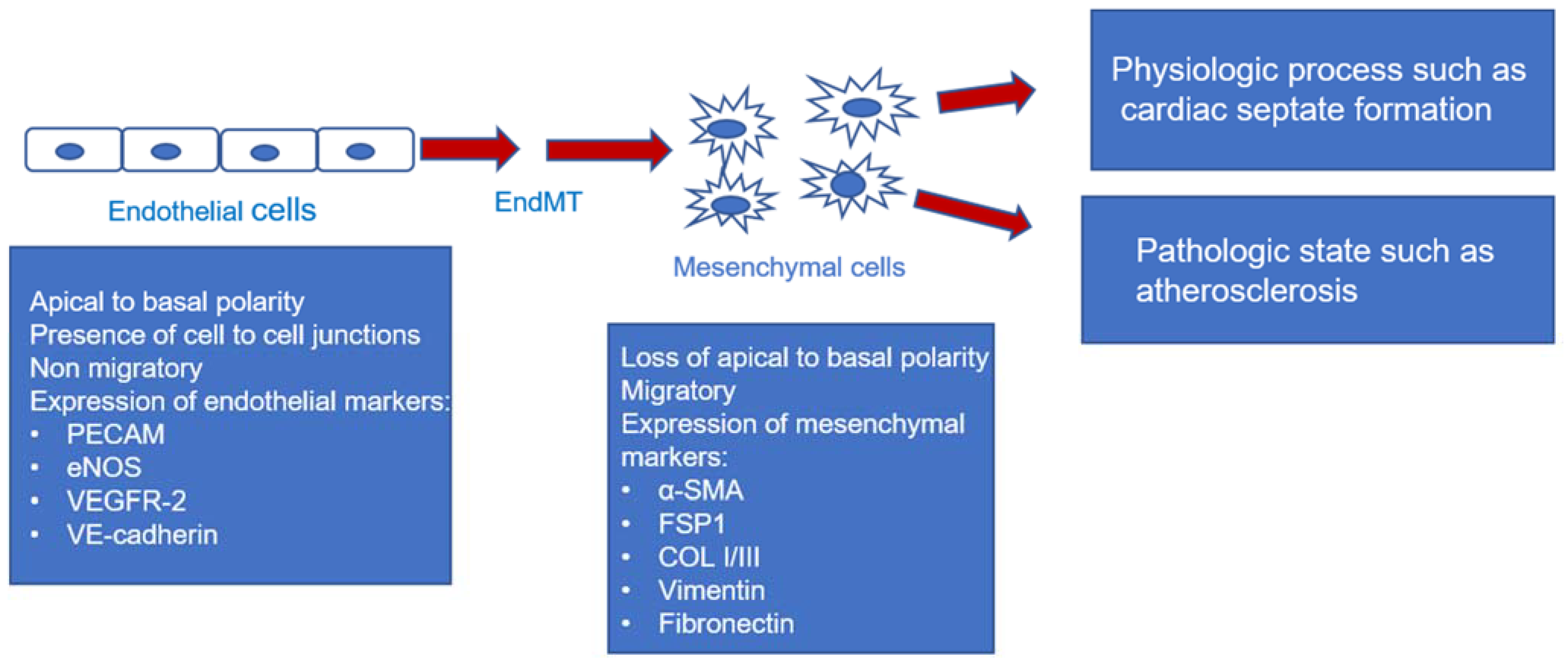
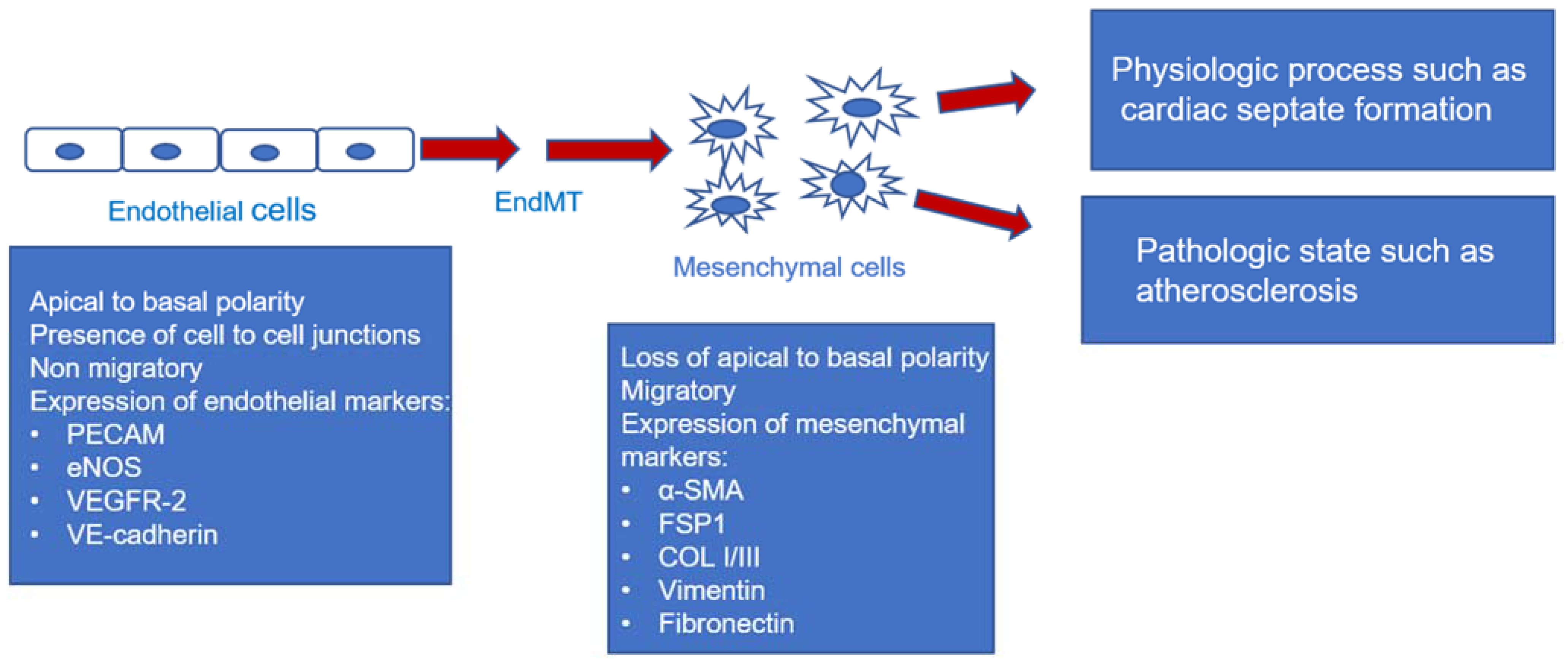
:1. Introduction
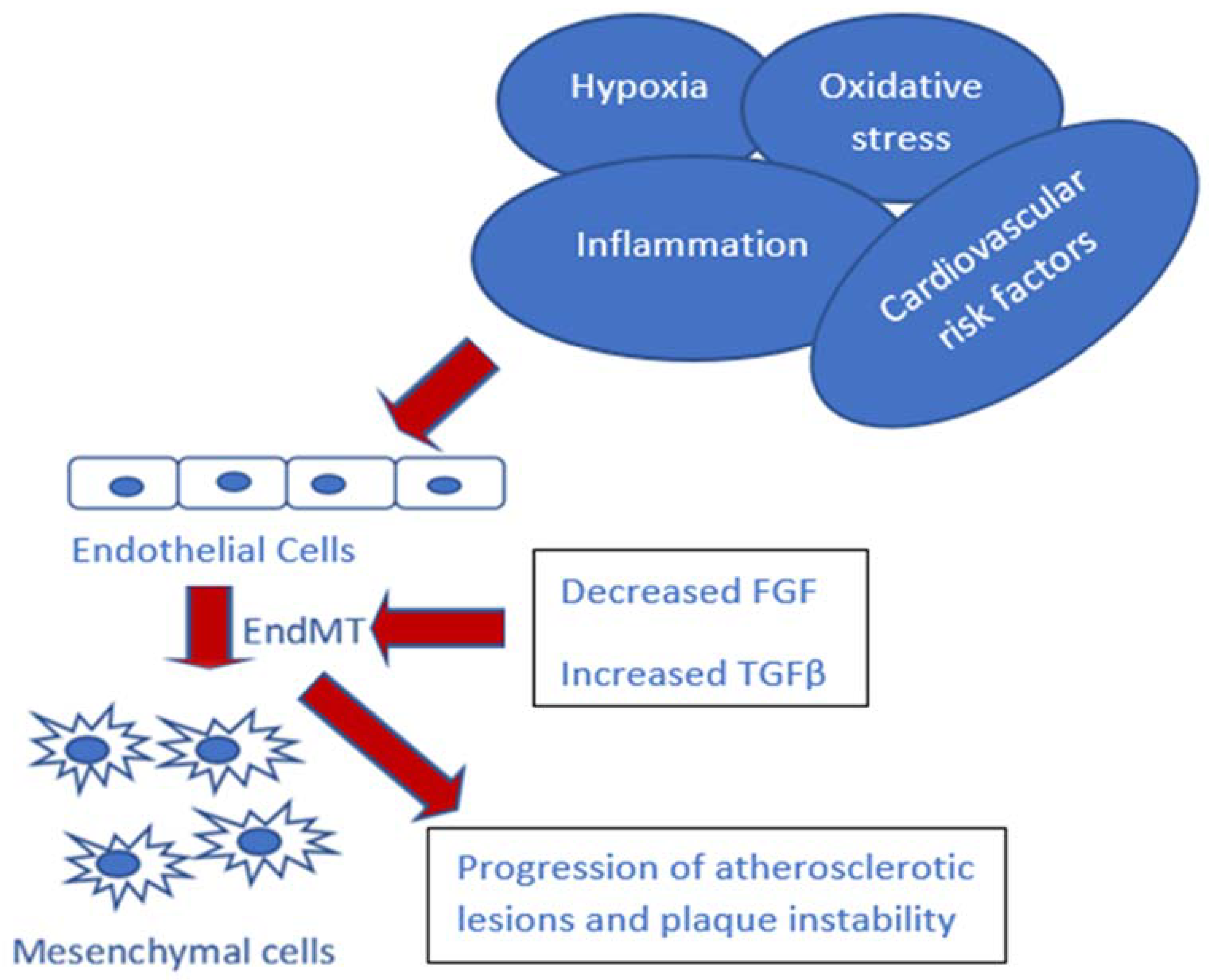
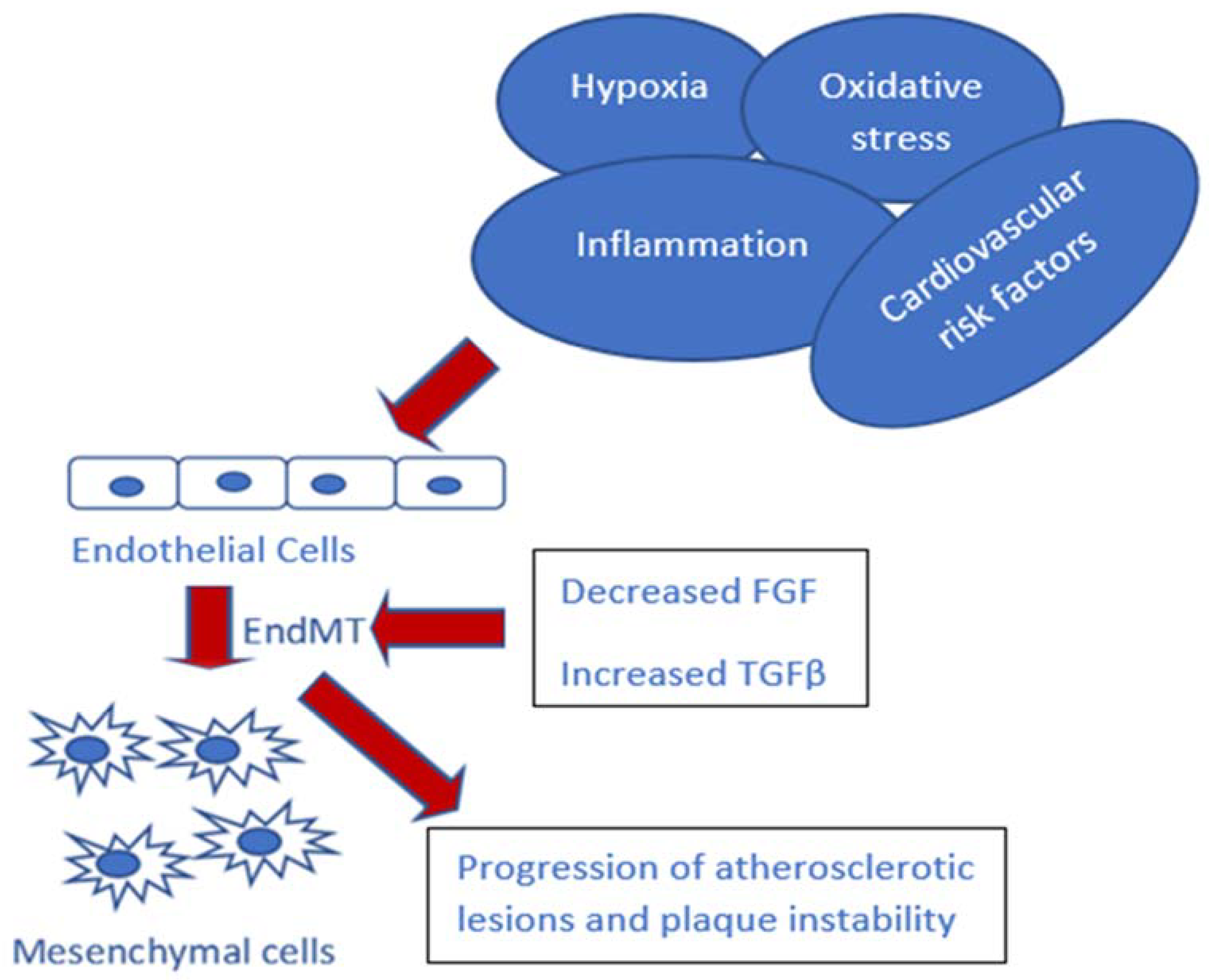
2. Hypoxia and EndMT in Atherosclerosis
3. TGF-β Signaling and EndMT in Atherosclerosis
4. Inflammation and EndMT in Atherosclerosis
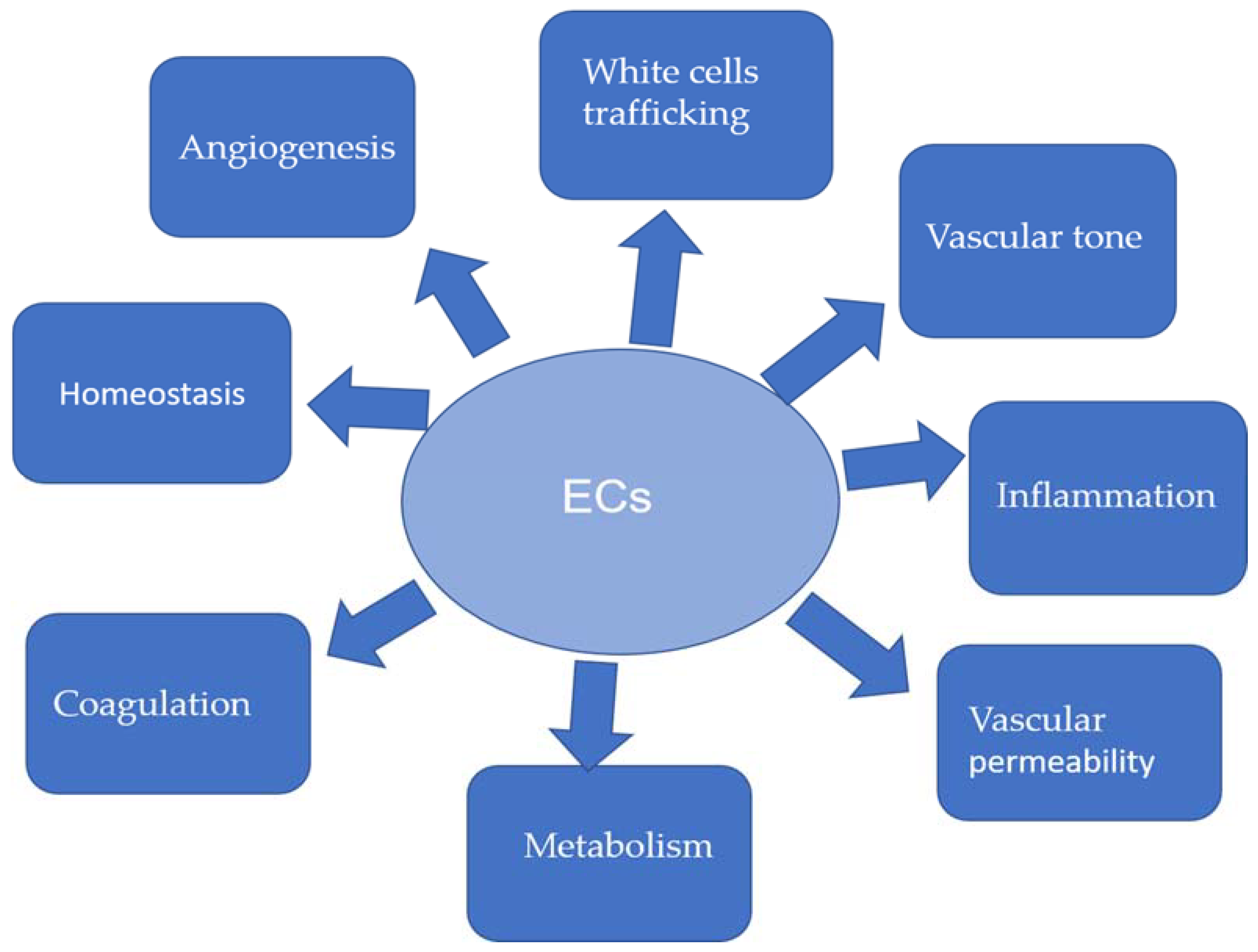
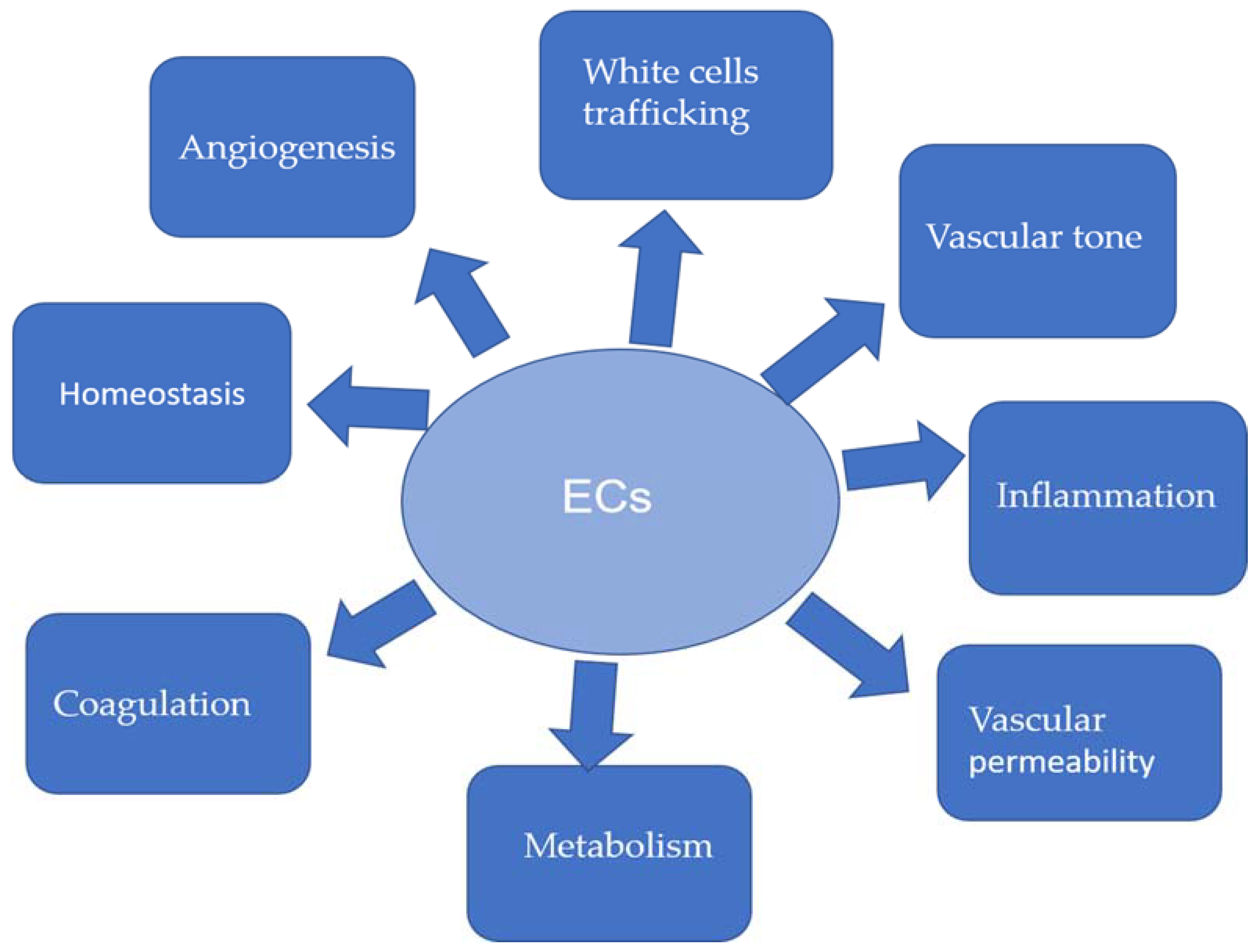
5. Endothelium Dysfunction and EndMT in Atherosclerosis
6. Environment Factors in Atherosclerosis
7. Mouse Models for Atherosclerosis
8. Conclusions and Further Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV-8 | adeno-associated-virus-8 |
| ACh | acetylcholine |
| AMPK | adenosine monophosphate-activated protein kinase |
| ApoE | apolipoprotein E |
| α-SMA | alpha-smooth muscle actin |
| ND | normal diet |
| COL1A1 | collagen type I alpha 1 chain |
| CD31 | cluster of differentiation 31 |
| CETP | cholesteryl ester transfer protein |
| CRP | C-reactive protein |
| DCs | dendritic cells |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EndMT | endothelial to mesenchymal transition |
| EPC | endothelial progenitor cell |
| eNOS | endothelial nitric oxide synthase |
| ERK | extracellular-signal-regulated kinases |
| FGF | fibroblast growth factor |
| FGFR | fibroblast growth factor receptor |
| FN | fibronectin |
| FSP1 | fibroblast-specific protein 1 |
| HCAECs | human coronary artery endothelial cells |
| HCMECs | human cutaneous microvascular endothelial cells |
| HDL | high-density lipoprotein |
| HIFs | hypoxia-inducible factors |
| HUVECs | human umbilical vein endothelial cells |
| ICAM | intercellular adhesion molecule |
| IGF | insulin-like growth factor |
| IL | interleukin |
| LDL | low-density lipoprotein |
| LDLr | low-density lipoprotein receptor |
| MAPK | mitogen-activated protein kinase |
| MMP | matrix metalloproteinase |
| Ntn1 | netrin 1 |
| NLRP3 | nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 |
| PAH | pulmonary arterial hypertension |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| PDCD4 | programmed cell death 4 |
| PECAM | platelet-endothelial cell adhesion molecule |
| PDGF | platelet-derived growth factor |
| PDR | proliferative diabetic retinopathy |
| PGC1α | proliferator-activated receptor gamma coactivator 1-alpha TGF-β transforming growth factor-β |
| PPAR | peroxisome proliferator-activated receptor |
| ROS | reactive oxygen species |
| siRNA | small interfering RNA |
| SLUG | snail family zinc finger 2 |
| SMC | Smooth muscle cell |
| TGFβR1 | transforming growth factor-β receptor 1 |
| TNF-α | tumor necrosis factor-α |
| TWIST | bHLH transcription factor 1 |
| UNC5B | Unc-5 netrin receptor B |
| VCAM | vascular cell adhesion molecule |
| VE-Cadherin | vascular endothelial-cadherin |
| VEGF | vascular endothelial growth factor |
| VEGFR-2 | vascular endothelial growth factor 2 receptor |
| VSMC | vascular smooth muscle cell |
| WD | Western diet |
References
- Agnelli, G.; Belch, J.J.F.; Baumgartner, I.; Giovas, P.; Hoffmann, U. Morbidity and mortality associated with atherosclerotic peripheral artery disease: A systematic review. Atherosclerosis 2020, 293, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Obara, H.; Matsubara, K.; Kitagawa, Y. Acute Limb Ischemia. Ann. Vasc. Dis. 2018, 11, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Whelton, S.P.; Deal, J.A.; Zikusoka, M.; Jacobson, L.P.; Sarkar, S.; Palella, F.J., Jr.; Kingsley, L.; Budoff, M.; Witt, M.D.; Brown, T.T.; et al. Associations between lipids and subclinical coronary atherosclerosis. AIDS 2019, 33, 1053–1061. [Google Scholar] [CrossRef]
- Viola, J.; Soehnlein, O. Atherosclerosis—A matter of unresolved inflammation. Semin. Immunol. 2015, 27, 184–193. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Verma, I.; Syngle, A.; Krishan, P. Predictors of endothelial dysfunction and atherosclerosis in rheumatoid arthritis in Indian population. Indian Heart J. 2017, 69, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardio Vasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Martin, L.V.; Jurand, A. The absence of teratogenic effects of some analgesics used in anaesthesia. Additional evidence from a mouse model. Anaesthesia 1992, 47, 473–476. [Google Scholar] [CrossRef]
- Stary, H.C. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am. J. Clin. Nutr. 2000, 72, 1297S–1306S. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Insull, W., Jr. The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Wolf, M.P.; Hunziker, P. Atherosclerosis: Insights into vascular pathobiology and outlook to novel treatments. J. Cardiovasc. Transl. Res. 2020, 13, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmke, A.; Casper, J.; Nordlohne, J.; David, S.; Haller, H.; Zeisberg, E.M.; Vietinghoff, S.v. Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. FASEB J. 2019, 33, 2278–2289. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhao, D.; Yuan, P.; Li, J.; Yun, Y.; Cui, Y.; Zhang, T.; Ma, J.; Sun, L.; Ma, H.; et al. Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease. Acta Cardiol. Sin. 2020, 36, 183–194. [Google Scholar] [CrossRef]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardio Vasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Clere, N.; Renault, S.; Corre, I. Endothelial-to-Mesenchymal Transition in Cancer. Front. Cell Dev. Biol. 2020, 8, 747. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, X.; Hu, R.; Dai, A. TGF-beta1-induced SMAD2/3/4 activation promotes RELM-beta transcription to modulate the endothelium-mesenchymal transition in human endothelial cells. Int. J. Biochem. Cell Biol. 2018, 105, 52–60. [Google Scholar] [CrossRef]
- Qin, W.; Zhang, L.; Li, Z.; Xiao, D.; Zhang, Y.; Zhang, H.; Mokembo, J.N.; Monayo, S.M.; Jha, N.K.; Kopylov, P.; et al. Endothelial to mesenchymal transition contributes to nicotine-induced atherosclerosis. Theranostics 2020, 10, 5276–5289. [Google Scholar] [CrossRef] [PubMed]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Corrigendum: Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2017, 8, 14710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef]
- Jeong, D.; Lee, M.A.; Li, Y.; Yang, D.K.; Kho, C.; Oh, J.G.; Hong, G.; Lee, A.; Song, M.H.; LaRocca, T.J.; et al. Matricellular Protein CCN5 Reverses Established Cardiac Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 1556–1568. [Google Scholar] [CrossRef]
- Moonen, J.R.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; van Kooten, T.G.; van Luyn, M.J.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardio Vasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Chen, J.; Sun, L.; Xu, Y. RhoJ promotes hypoxia induced endothelial-to-mesenchymal transition by activating WDR5 expression. J. Cell Biochem. 2018, 119, 3384–3393. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Hulshoff, M.S.; Wilhelmi, T.; Zeisberg, M.; Zeisberg, E.M. Hypoxia-induced endothelial-mesenchymal transition is associated with RASAL1 promoter hypermethylation in human coronary endothelial cells. FEBS Lett. 2016, 590, 1222–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.B.; Fisher, E.A. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ. Res. 2011, 109, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, X.; Zhu, Y.; Chen, Q.; Li, B.; Zhang, F. Protective effects of acetylcholine on hypoxia-induced endothelial-to-mesenchymal transition in human cardiac microvascular endothelial cells. Mol. Cell Biochem. 2020, 473, 101–110. [Google Scholar] [CrossRef]
- Ramkhelawon, B.; Yang, Y.; van Gils, J.M.; Hewing, B.; Rayner, K.J.; Parathath, S.; Guo, L.; Oldebeken, S.; Feig, J.L.; Fisher, E.A.; et al. Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: Mechanism for macrophage retention and survival. Arterioscler Thromb. Vasc. Biol. 2013, 33, 1180–1188. [Google Scholar] [CrossRef] [Green Version]
- van Gils, J.M.; Derby, M.C.; Fernandes, L.R.; Ramkhelawon, B.; Ray, T.D.; Rayner, K.J.; Parathath, S.; Distel, E.; Feig, J.L.; Alvarez-Leite, J.I.; et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat. Immunol. 2012, 13, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-Inducible Factor-1alpha Expression in Macrophages Promotes Development of Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 2016, 36, 1782–1790. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail Is a Direct Target of Hypoxia-inducible Factor 1alpha (HIF1alpha) in Hypoxia-induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Niu, W.; Dong, H.Y.; Liu, M.L.; Luo, Y.; Li, Z.C. Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.Y.; Simons, M. When endothelial cells go rogue. EMBO Mol. Med. 2016, 8, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Toma, I.; McCaffrey, T.A. Transforming growth factor-beta and atherosclerosis: Interwoven atherogenic and atheroprotective aspects. Cell Tissue Res. 2012, 347, 155–175. [Google Scholar] [CrossRef] [Green Version]
- Pardali, E.; Ten Dijke, P. TGFbeta signaling and cardiovascular diseases. Int. J. Biol. Sci. 2012, 8, 195–213. [Google Scholar] [CrossRef]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, M.; Sakkers, T.R.; de Jager, S.C.A.; Pasterkamp, G.; Goumans, M.J. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vascul. Pharmacol. 2018, 106, 1–8. [Google Scholar] [CrossRef]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef]
- Luxan, G.; D’Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- Bosada, F.M.; Devasthali, V.; Jones, K.A.; Stankunas, K. Wnt/beta-catenin signaling enables developmental transitions during valvulogenesis. Development 2016, 143, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, C.M.; Cheek, J.; Chakraborty, S.; Yutzey, K.E. Wnt signaling in heart valve development and osteogenic gene induction. Dev. Biol. 2010, 338, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebner, S.; Cattelino, A.; Gallini, R.; Rudini, N.; Iurlaro, M.; Piccolo, S.; Dejana, E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004, 166, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.T.; Ferreira Melo, F.U.; Malta, T.M.; Rodrigues, E.S.; Placa, J.R.; Silva, W.A., Jr.; Panepucci, R.A.; Covas, D.T.; de Oliveira Rodrigues, C.; Kashima, S. Endothelial cells from different anatomical origin have distinct responses during SNAIL/TGF-beta2-mediated endothelial-mesenchymal transition. Am. J. Transl. Res. 2018, 10, 4065–4081. [Google Scholar]
- Lin, Q.Q.; Zhao, J.; Zheng, C.G.; Chun, J. Roles of notch signaling pathway and endothelial-mesenchymal transition in vascular endothelial dysfunction and atherosclerosis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6485–6491. [Google Scholar] [CrossRef]
- Yang, X.; Liaw, L.; Prudovsky, I.; Brooks, P.C.; Vary, C.; Oxburgh, L.; Friesel, R. Fibroblast growth factor signaling in the vasculature. Curr. Atheroscler Rep. 2015, 17, 509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Shi, S.; Srivastava, S.P.; Kitada, M.; Nagai, T.; Nitta, K.; Kohno, M.; Kanasaki, K.; Koya, D. FGFR1 is critical for the anti-endothelial mesenchymal transition effect of N-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway. Cell Death Dis. 2017, 8, e2965. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Tellides, G.; Simons, M. Fibroblast growth factor receptor 1 is a key inhibitor of TGFbeta signaling in the endothelium. Sci. Signal. 2014, 7, ra90. [Google Scholar] [CrossRef]
- Bolt, H.M.; Filser, J.G.; Stormer, F. Inhalation pharmacokinetics based on gas uptake studies. V. Comparative pharmacokinetics of ethylene and 1,3-butadiene in rats. Arch. Toxicol. 1984, 55, 213–218. [Google Scholar] [CrossRef]
- Qi, M.; Xin, S. FGF signaling contributes to atherosclerosis by enhancing the inflammatory response in vascular smooth muscle cells. Mol. Med. Rep. 2019, 20, 162–170. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Tellides, G.; Simons, M. Smooth muscle FGF/TGFbeta cross talk regulates atherosclerosis progression. EMBO Mol. Med. 2016, 8, 712–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, T.; Kanellakis, P.; Pomilio, G.; Jennings, G.; Bobik, A.; Agrotis, A. Inhibition of fibroblast growth factor receptor signaling attenuates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb. Vasc. Biol. 2006, 26, 1845–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Dudley, A.C. Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 2017, 241, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Sterpetti, A.V. Inflammatory Cytokines and Atherosclerotic Plaque Progression. Therapeutic Implications. Curr. Atheroscler Rep. 2020, 22, 75. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Kopp, J.B. TGF-beta and fibrosis. Microbes Infect. 1999, 1, 1349–1365. [Google Scholar] [CrossRef]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, F.; Wang, Y.; Gistera, A.; Roy, J.; Paulsson-Berne, G.; Hedin, U.; Lerman, A.; Hansson, G.K.; Herrmann, J.; et al. Inflammasome-Driven Interleukin-1alpha and Interleukin-1beta Production in Atherosclerotic Plaques Relates to Hyperlipidemia and Plaque Complexity. JACC Basic Transl. Sci. 2019, 4, 304–317. [Google Scholar] [CrossRef]
- Wang, M.; Li, J.; Cai, J.; Cheng, L.; Wang, X.; Xu, P.; Li, G.; Liang, X. Overexpression of MicroRNA-16 Alleviates Atherosclerosis by Inhibition of Inflammatory Pathways. Biomed. Res. Int. 2020, 2020, 8504238. [Google Scholar] [CrossRef]
- Chen, P.Y.; Schwartz, M.A.; Simons, M. Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front. Cardio Vasc. Med. 2020, 7, 53. [Google Scholar] [CrossRef]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, Y.; Kondo, K.; Momiyama, Y. The Protective Role of Sestrin2 in Atherosclerotic and Cardiac Diseases. Int. J. Mol. Sci. 2021, 22, 1200. [Google Scholar] [CrossRef]
- Lee, E.S.; Boldo, L.S.; Fernandez, B.O.; Feelisch, M.; Harmsen, M.C. Suppression of TAK1 pathway by shear stress counteracts the inflammatory endothelial cell phenotype induced by oxidative stress and TGF-beta1. Sci. Rep. 2017, 7, 42487. [Google Scholar] [CrossRef]
- Mahmoud, M.M.; Kim, H.R.; Xing, R.; Hsiao, S.; Mammoto, A.; Chen, J.; Serbanovic-Canic, J.; Feng, S.; Bowden, N.P.; Maguire, R.; et al. TWIST1 Integrates Endothelial Responses to Flow in Vascular Dysfunction and Atherosclerosis. Circ. Res. 2016, 119, 450–462. [Google Scholar] [CrossRef]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Inflammation in Atherosclerosis-No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef]
- Ma, K.L.; Liu, J.; Ni, J.; Zhang, Y.; Lv, L.L.; Tang, R.N.; Ni, H.F.; Ruan, X.Z.; Liu, B.C. Inflammatory stress exacerbates the progression of cardiac fibrosis in high-fat-fed apolipoprotein E knockout mice via endothelial-mesenchymal transition. Int. J. Med. Sci. 2013, 10, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Du, X.; Yang, Y.; Liu, X.; Liu, X.; Zhang, N.; Li, Y.; Jiang, X.; Jiang, Y.; Yang, Z. Inhibition of miR-122 reduced atherosclerotic lesion formation by regulating NPAS3-mediated endothelial to mesenchymal transition. Life Sci. 2021, 265, 118816. [Google Scholar] [CrossRef]
- Pathak, A.; Singh, S.K.; Thewke, D.P.; Agrawal, A. Conformationally Altered C-Reactive Protein Capable of Binding to Atherogenic Lipoproteins Reduces Atherosclerosis. Front. Immunol. 2020, 11, 1780. [Google Scholar] [CrossRef]
- Sena, C.M.; Pereira, A.M.; Seica, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Yun, E.; Kook, Y.; Yoo, K.H.; Kim, K.I.; Lee, M.S.; Kim, J.; Lee, A. Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases. Biomedicines 2020, 8, 639. [Google Scholar] [CrossRef]
- Choi, B.J.; Prasad, A.; Gulati, R.; Best, P.J.; Lennon, R.J.; Barsness, G.W.; Lerman, L.O.; Lerman, A. Coronary endothelial dysfunction in patients with early coronary artery disease is associated with the increase in intravascular lipid core plaque. Eur. Heart J. 2013, 34, 2047–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.P.; Fan, Z.X.; Gao, J.; Sun, X.P.; Zhu, G.H.; Zhang, Y.H.; Si, J.; Zuo, X.B.; Liu, Z.; Hua, Q.; et al. Influencing factors of vascular endothelial function in patients with non-obstructive coronary atherosclerosis: A 1-year observational study. BMC Cardio Vasc. Disord. 2020, 20, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, B.; Compagna, R.; Amato, M.; Grande, R.; Butrico, L.; Rossi, A.; Naso, A.; Ruggiero, M.; de Franciscis, S.; Serra, R. Adult vascular wall resident multipotent vascular stem cells, matrix metalloproteinases, and arterial aneurysms. Stem Cells Int. 2015, 2015, 434962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.Y.; Li, C.J.; Hou, M.F.; Chu, P.Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef]
- Varghese, J.F.; Patel, R.; Yadav, U.C.S. Novel Insights in the Metabolic Syndrome-induced Oxidative Stress and Inflammation-mediated Atherosclerosis. Curr. Cardiol. Rev. 2018, 14, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Fukuda, N.; Katakawa, M.; Tsunemi, A.; Tahira, Y.; Matsumoto, T.; Ueno, T.; Soma, M. Effects of an angiotensin II receptor blocker on the impaired function of endothelial progenitor cells in patients with essential hypertension. Am. J. Hypertens 2014, 27, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Perez, L.; Munoz-Durango, N.; Riedel, C.A.; Echeverria, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habas, K.; Shang, L. Alterations in intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in human endothelial cells. Tissue Cell 2018, 54, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, L.; Fedele, G.; Castiglioni, S.; Maier, J.A. Magnesium Deficiency Induces Lipid Accumulation in Vascular Endothelial Cells via Oxidative Stress-The Potential Contribution of EDF-1 and PPARgamma. Int. J. Mol. Sci. 2021, 22, 1050. [Google Scholar] [CrossRef]
- Xu, L.; Wang, S.; Li, B.; Sun, A.; Zou, Y.; Ge, J. A protective role of ciglitazone in ox-LDL-induced rat microvascular endothelial cells via modulating PPARgamma-dependent AMPK/eNOS pathway. J. Cell Mol. Med. 2015, 19, 92–102. [Google Scholar] [CrossRef]
- Yanai, H.; Yoshida, H. Beneficial Effects of Adiponectin on Glucose and Lipid Metabolism and Atherosclerotic Progression: Mechanisms and Perspectives. Int. J. Mol. Sci. 2019, 20, 1190. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.A.; Vestweber, D.; Simons, M. A unifying concept in vascular health and disease. Science 2018, 360, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thome, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faca, V.; Olsen Camara, N.; Kashima, S.; Tadeu Covas, D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-beta2 during Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 458. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, G. Gene/Environment interaction in atherosclerosis: An example of clinical medicine as seen from the evolutionary perspective. Int. J. Hypertens 2010, 2010, 654078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aherrahrou, R.; Guo, L.; Nagraj, V.P.; Aguhob, A.; Hinkle, J.; Chen, L.; Yuhl Soh, J.; Lue, D.; Alencar, G.F.; Boltjes, A.; et al. Genetic Regulation of Atherosclerosis-Relevant Phenotypes in Human Vascular Smooth Muscle Cells. Circ. Res. 2020, 127, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Boua, P.R.; Brandenburg, J.T.; Choudhury, A.; Hazelhurst, S.; Sengupta, D.; Agongo, G.; Nonterah, E.A.; Oduro, A.R.; Tinto, H.; Mathew, C.G.; et al. Novel and Known Gene-Smoking Interactions With cIMT Identified as Potential Drivers for Atherosclerosis Risk in West-African Populations of the AWI-Gen Study. Front. Genet. 2019, 10, 1354. [Google Scholar] [CrossRef] [Green Version]
- Zak, I.; Niemiec, P.; Balcerzyk, A.; Krauze, J. Combined “pro-atherosclerotic” variants of the ACE and APOE genes increase the risk of the coronary artery disease associated with the presence of cigarette smoking. Acta Cardiol. 2008, 63, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Lind, P.M.; Lind, L. Are Persistent Organic Pollutants Linked to Lipid Abnormalities, Atherosclerosis and Cardiovascular Disease? A Review. J. Lipid Atheroscler 2020, 9, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Solenkova, N.V.; Newman, J.D.; Berger, J.S.; Thurston, G.; Hochman, J.S.; Lamas, G.A. Metal pollutants and cardiovascular disease: Mechanisms and consequences of exposure. Am. Heart J. 2014, 168, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; O’Hara, L.; Tah, P.; Street, C.; Maras, A.; Ouakil, D.P.; Santosh, P.; Signorini, G.; Singh, S.P.; Tuomainen, H.; et al. A systematic review of the literature on ethical aspects of transitional care between child- and adult-orientated health services. BMC Med. Ethics 2018, 19, 73. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, T.G.S.; Tavares, N.H.; Bastos, M.L.A.; Oliveira, B.B.R.; Araujo, L.F.; Ferreira, M.J.M. Exposure to Chemical and Biological Agents at Work and Cardiovascular Disease in Brazil: A Population-Based Study. J. Occup. Environ. Med. 2021. [Google Scholar] [CrossRef]
- Chowdhury, R.; Ramond, A.; O’Keeffe, L.M.; Shahzad, S.; Kunutsor, S.K.; Muka, T.; Gregson, J.; Willeit, P.; Warnakula, S.; Khan, H.; et al. Environmental toxic metal contaminants and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2018, 362, k3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkhondeh, T.; Afshari, R.; Mehrpour, O.; Samarghandian, S. Mercury and Atherosclerosis: Cell Biology, Pathophysiology, and Epidemiological Studies. Biol. Trace Elem. Res. 2020, 196, 27–36. [Google Scholar] [CrossRef]
- Oppi, S.; Luscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research-Which Is My Line? Front. Cardio Vasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golforoush, P.; Yellon, D.M.; Davidson, S.M. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Res. Cardiol. 2020, 115, 73. [Google Scholar] [CrossRef]
- Jawien, J.; Nastalek, P.; Korbut, R. Mouse models of experimental atherosclerosis. J. Physiol. Pharmacol. 2004, 55, 503–517. [Google Scholar]
- Kumar, S.; Kang, D.W.; Rezvan, A.; Jo, H. Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab. Investig. 2017, 97, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parma, L.; Peters, H.A.B.; Sluiter, T.J.; Simons, K.H.; Lazzari, P.; de Vries, M.R.; Quax, P.H.A. bFGF blockade reduces intraplaque angiogenesis and macrophage infiltration in atherosclerotic vein graft lesions in ApoE3*Leiden mice. Sci. Rep. 2020, 10, 15968. [Google Scholar] [CrossRef] [PubMed]
- In Het Panhuis, W.; Kooijman, S.; Brouwers, B.; Verhoeven, A.; Pronk, A.C.M.; Streefland, T.C.M.; Giera, M.; Schrauwen, P.; Rensen, P.C.N.; Schonke, M. Mild Exercise Does Not Prevent Atherosclerosis in APOE*3-Leiden.CETP Mice or Improve Lipoprotein Profile of Men with Obesity. Obesity 2020, 28 (Suppl. 1), S93–S103. [Google Scholar] [CrossRef]
- Perrotta, P.; Van der Veken, B.; Van Der Veken, P.; Pintelon, I.; Roosens, L.; Adriaenssens, E.; Timmerman, V.; Guns, P.J.; De Meyer, G.R.Y.; Martinet, W. Partial Inhibition of Glycolysis Reduces Atherogenesis Independent of Intraplaque Neovascularization in Mice. Arterioscler Thromb. Vasc. Biol. 2020, 40, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Yang, J.; Cheng, J.; Sui, W.; Cheng, C.; Li, H.; Zhang, M.; Zhang, J.; Xu, X.; Ma, J.; et al. Angiotensin-(1-7) mitigated angiotensin II-induced abdominal aortic aneurysms in apolipoprotein E-knockout mice. Br. J. Pharmacol. 2020, 177, 1719–1734. [Google Scholar] [CrossRef]
- Libby, P.; Bornfeldt, K.E.; Tall, A.R. Atherosclerosis: Successes, Surprises, and Future Challenges. Circ. Res. 2016, 118, 531–534. [Google Scholar] [CrossRef] [Green Version]
- McPherson, R.; Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. Circ. Res. 2016, 118, 564–578. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Model | Characterization | Lipid Change | Plaque Development | Advantages & Disadvantages |
|---|---|---|---|---|
| ApoE−/− | Knockout of ApoE gene | LDL↑, HDL↓ | Aortic plaques | Form atherosclerosis with ND |
| LDLr−/− | Knockout of LDLr gene | LDL↑ | Fibrous plaques | Close to human lipid profile; Complex formation needs WD; No automatic ruptured plaques |
| ApoE−/− LDLr−/− | Knockout of both ApoE and LDLr genes | LDL↑ | Fibrosis plaque rupture | Form atherosclerosis with ND; plaque rupture and/or thrombosis are not observed |
| ApoE*3-Leiden.CETP | ApoE*3-Leiden mutation plus the expression of human CETP gene | HDL↑ | Extensive atherosclerosis | A good model for age-related study; Lesion formation needs WD |
| PCSK9-AAV | Adeno-associated virus injection to control gain-of-function PCSK9 mutant | LDL↑ | Quick development of atherosclerosis plaques | Fast formation of the atherosclerotic lesion with WD |
| ApoE−/− with angiotensin II infusion | Infusion of angiotensin II into ApoE−/− mice | LDL↑ | Atherosclerosis plaques rupture | Develops significant atherosclerotic lesions with WD |
| ApoE−/−Fbn1C1039G+/− | ApoE−/− mice with Fbn1 gene mutation (C1039G) | VLDL↑ | Atherosclerosis plaques rupture | Fast development of plaques; Study advanced atherosclerosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Ogbu, S.C.; Musich, P.R.; Thewke, D.P.; Yao, Z.; Jiang, Y. The Contribution of Endothelial-Mesenchymal Transition to Atherosclerosis. Int. J. Transl. Med. 2021, 1, 39-54. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm1010004
Zhang J, Ogbu SC, Musich PR, Thewke DP, Yao Z, Jiang Y. The Contribution of Endothelial-Mesenchymal Transition to Atherosclerosis. International Journal of Translational Medicine. 2021; 1(1):39-54. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm1010004
Chicago/Turabian StyleZhang, Jinyu, Stella C. Ogbu, Phillip R. Musich, Douglas P. Thewke, Zhiqiang Yao, and Yong Jiang. 2021. "The Contribution of Endothelial-Mesenchymal Transition to Atherosclerosis" International Journal of Translational Medicine 1, no. 1: 39-54. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm1010004