Newborn Screening in Japan
Printed Edition Available!
A printed edition of this Special Issue is available
here.
Share This Topical Collection
Editors
 Prof. Dr. Toshihiro Tajima
Prof. Dr. Toshihiro Tajima
Prof. Dr. Toshihiro Tajima
E-Mail
Collection Editor
Department of Pediatrics, Jichi Children's Medical Center Tochigi, Shimotsuke, Japan
Interests: conegnital adrenal hyperplasia; congenital hypothyroidism
 Prof. Dr. Seiji Yamaguchi
Prof. Dr. Seiji Yamaguchi
Prof. Dr. Seiji Yamaguchi
E-Mail
Website
Collection Editor
Department of Pediatrics, Shimane University School of Medicine, 89-1 En-ya-cho, Izumo Shimane 693-8501 Japan
Interests: screening for organic acidemias and fatty acid oxidation defects; expanded newborn screening; new treatment option of bezafibrate for fatty acid; molecular study on fatty acid oxidation defects oxidation defects
Topical Collection Information
Dear Colleagues,
The Japanese Society for Neonatal Screening has made the International Journal of Neonatal Screening its official journal. We would like to thank everyone who made this possible.
We take this opportunity to announce the "Topical Collection" with the theme of newborn screening in Japan. This collection will include reviews on the current status and progress of newborn screening in Japan, as well as the latest original research on newborn screening. We would like this Topical Collection to stimulate intellectual curiosity in our readers. In the future, the Japanese Society for Neonatal Screening will disseminate information through the International Journal of Neonatal Screening and promote wide-ranging communication with everyone involved in newborn screening around the world.
Prof. Dr. Toshihiro Tajima
Prof. Dr. Seiji Yamaguchi
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. International Journal of Neonatal Screening is an international peer-reviewed open access quarterly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 1600 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Published Papers (18 papers)
Open AccessArticle
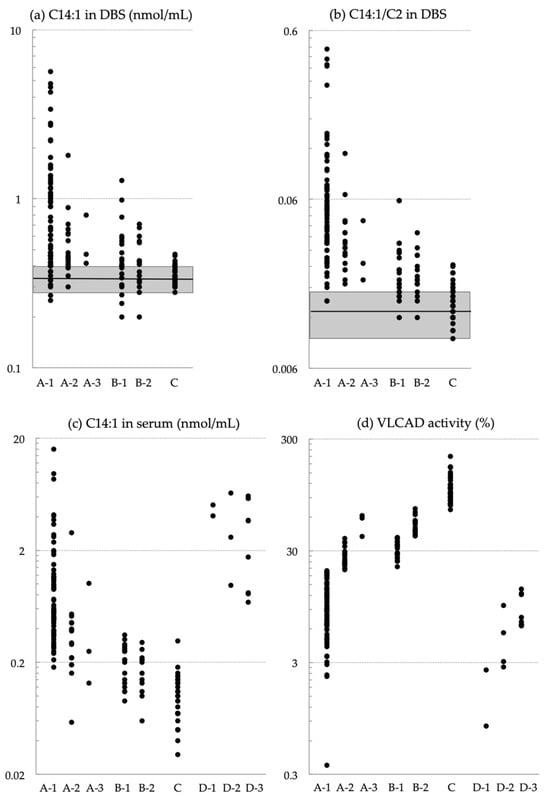
Using the C14:1/Medium-Chain Acylcarnitine Ratio Instead of C14:1 to Reduce False-Positive Results for Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency in Newborn Screening in Japan
by
Go Tajima, Junko Aisaki, Keiichi Hara, Miyuki Tsumura, Reiko Kagawa, Fumiaki Sakura, Hideo Sasai, Miori Yuasa, Yosuke Shigematsu and Satoshi Okada
Viewed by 885
Abstract
Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency is a long-chain fatty acid oxidation disorder that manifests as either a severe phenotype associated with cardiomyopathy, a hypoglycemic phenotype, or a myopathic phenotype. As the hypoglycemic phenotype can cause sudden infant death, VLCAD deficiency is included in
[...] Read more.
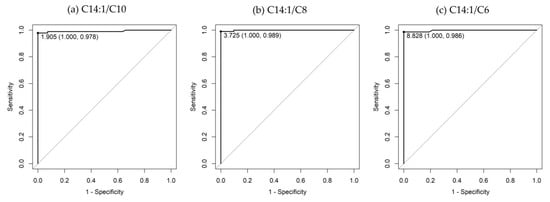
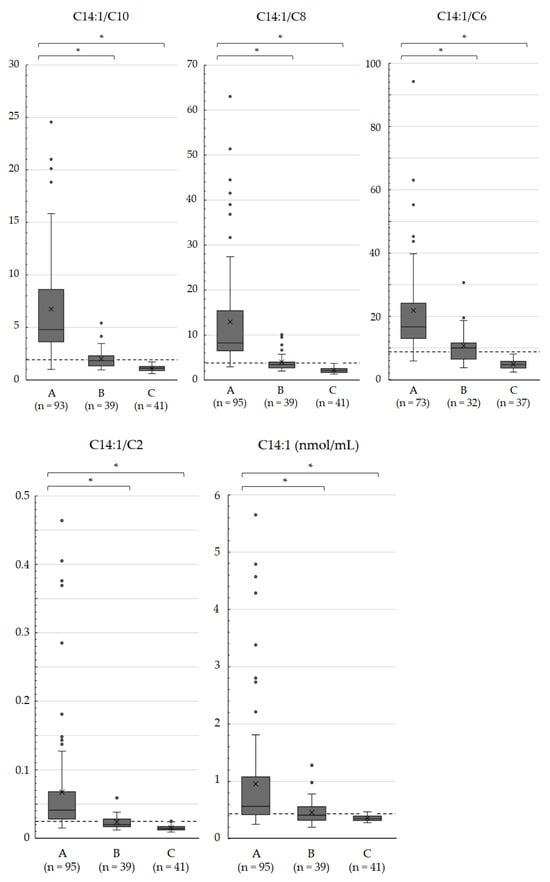
Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency is a long-chain fatty acid oxidation disorder that manifests as either a severe phenotype associated with cardiomyopathy, a hypoglycemic phenotype, or a myopathic phenotype. As the hypoglycemic phenotype can cause sudden infant death, VLCAD deficiency is included in newborn screening (NBS) panels in many countries. The tetradecenoylcarnitine (C14:1) level in dried blood specimens is commonly used as a primary marker for VLCAD deficiency in NBS panels. Its ratio to acetylcarnitine (C2) and various other acylcarnitines is used as secondary markers. In Japan, tandem mass spectrometry-based NBS, initially launched as a pilot study in 1997, was introduced to the nationwide NBS program in 2013. In the present study, we evaluated levels of acylcarnitine with various chain lengths (C18 to C2), free carnitine, and their ratios in 175 infants who tested positive for VLCAD deficiency with C14:1 and C14:1/C2 ratios. Our analyses indicated that the ratios of C14:1 to medium-chain acylcarnitines (C10, C8, and C6) were the most effective markers in reducing false-positive rates. Their use with appropriate cutoffs is expected to improve NBS performance for VLCAD deficiency.
Full article
►▼
Show Figures
Open AccessArticle
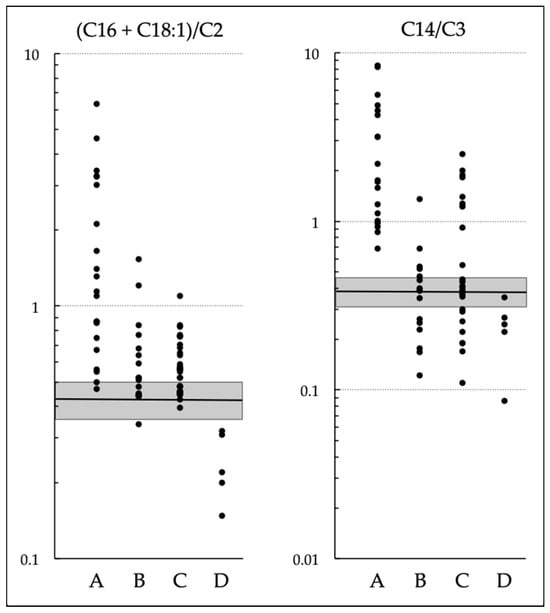
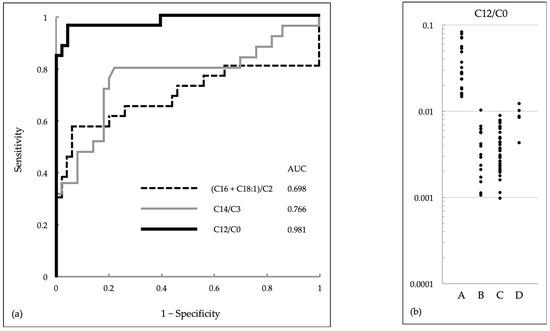
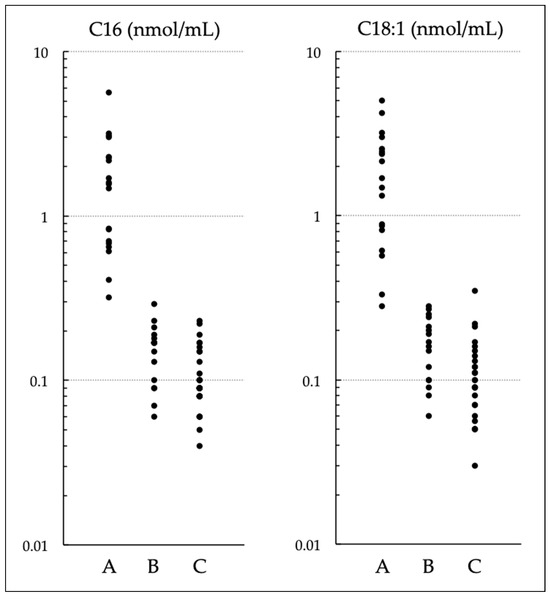
Newborn Screening with (C16 + C18:1)/C2 and C14/C3 for Carnitine Palmitoyltransferase II Deficiency throughout Japan Has Revealed C12/C0 as an Index of Higher Sensitivity and Specificity
by
Go Tajima, Keiichi Hara, Miyuki Tsumura, Reiko Kagawa, Fumiaki Sakura, Hideo Sasai, Miori Yuasa, Yosuke Shigematsu and Satoshi Okada
Viewed by 1422
Abstract
Carnitine palmitoyltransferase (CPT) II deficiency is a long-chain fatty acid oxidation disorder. It manifests as (1) a lethal neonatal form, (2) a hypoglycemic form, or (3) a myopathic form. The second form can cause sudden infant death and is more common among Japanese
[...] Read more.
Carnitine palmitoyltransferase (CPT) II deficiency is a long-chain fatty acid oxidation disorder. It manifests as (1) a lethal neonatal form, (2) a hypoglycemic form, or (3) a myopathic form. The second form can cause sudden infant death and is more common among Japanese people than in other ethnic groups. Our study group had earlier used (C16 + C18:1)/C2 to conduct a pilot newborn screening (NBS) study, and found that the use of C14/C3 for screening yielded lower rates of false positivity; in 2018, as a result, nationwide NBS for CPT II deficiency started. In this study, we evaluated the utility of these ratios in 71 NBS-positive infants and found that the levels of both C14/C3 and (C16 + C18:1)/C2 in patients overlapped greatly with those of infants without the disease. Among the levels of acylcarnitines with various chain lengths (C18 to C2) and levels of free carnitine (C0) as well as their ratios of various patterns, C12/C0 appeared to be a promising index that could reduce false-positive results without missing true-positive cases detected by current indices. Although some cases of the myopathic form may go undetected even with C12/C0, its use will help prevent life-threatening onset of the hypoglycemic form of CPT II deficiency.
Full article
►▼
Show Figures
Open AccessArticle
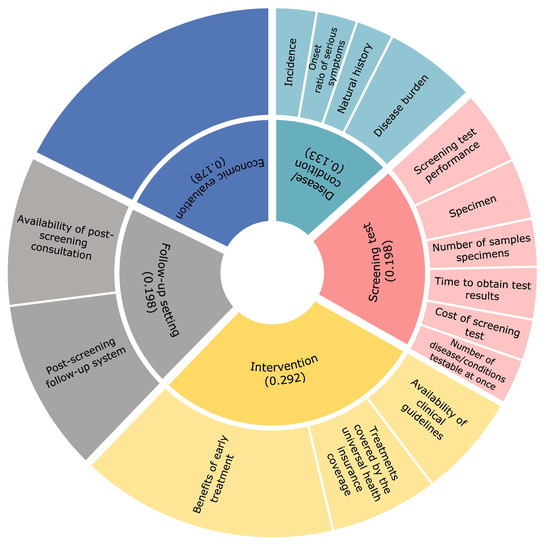
Development of a Model for Quantitative Assessment of Newborn Screening in Japan Using the Analytic Hierarchy Process
by
Keiko Konomura, Eri Hoshino, Kotomi Sakai, Takashi Fukuda and Go Tajima
Viewed by 1270
Abstract
Whether or not conditions should be included in publicly funded newborn screening (NBS) programs should be discussed according to objective and transparent criteria. Certain criteria have been developed for the introduction of NBS programs in the context of individual countries; however, there are
[...] Read more.
Whether or not conditions should be included in publicly funded newborn screening (NBS) programs should be discussed according to objective and transparent criteria. Certain criteria have been developed for the introduction of NBS programs in the context of individual countries; however, there are no standard selection criteria for NBS programs in Japan. This study aimed to develop a quantitative scoring model to assess newborn screening that incorporates the views of a variety of stakeholders in Japan. The five recommended eligibility criteria for NBS were stratified based on previous studies and expert opinions, using the analytic hierarchy process. We conducted a cross-sectional, web-based questionnaire targeting a wide range of people involved in NBS to investigate pairwise comparisons of the evaluation items between February and April of 2022. There were 143 respondents. Most of our respondents (44.1%) were physicians. Fifty-eight respondents (40.6%) had been engaged in NBS-related research or work for more than 10 years. The distribution of allocation points was the highest for ‘intervention’, ‘screening test’, ‘follow-up setting’, ’economic evaluation’, and ’disease/condition’, in that order. The algorithm in this study will guide decision makers in collecting and evaluating objective data, thus enabling transparent discussions to occur.
Full article
►▼
Show Figures
Open AccessArticle
Changes in the Incidence of Infantile Spinal Muscular Atrophy in Shikoku, Japan between 2011 and 2020
by
Kentaro Okamoto, Hisahide Nishio, Takahiro Motoki, Toshihiro Jogamoto, Kaori Aibara, Yoichi Kondo, Kentaro Kawamura, Yukihiko Konishi, Chiho Tokorodani, Ritsuo Nishiuchi and Mariko Eguchi
Cited by 1 | Viewed by 2697
Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder. Al-though there was no cure for SMA, newly developed therapeutic drugs (nusinersen, onasemnogene abeparvovec, and risdiplam) have been proven effective for the improvement of motor function and prevention of respiratory insufficiency of infants
[...] Read more.
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder. Al-though there was no cure for SMA, newly developed therapeutic drugs (nusinersen, onasemnogene abeparvovec, and risdiplam) have been proven effective for the improvement of motor function and prevention of respiratory insufficiency of infants with SMA. Nusinersen was introduced in Japan in 2017 and onasemnogene abeparvovec in 2020. We hypothesized that the introduction of these drugs might influence the incidence of SMA (more precisely, increase the diagnosis rate of SMA) in Japan. To test this hypothesis, we conducted a second epidemiological study of infantile SMA using questionnaires in Shikoku, Japan between October 2021 and February 2022. The incidence of infantile SMA during the period 2016–2020 was 7.08 (95% confidence interval [CI] 2.45–11.71) per 100,000 live births. According to our previous epidemiological study, the incidence of infantile SMA during 2011–2015 was 2.70 (95% CI 0.05–5.35) per 100,000 live births. The increased incidence of infantile SMA suggests that the widespread news in Japan regarding the introduction of therapeutic agents, nusinersen and onasemnogene abeparvovec, raised clinicians’ awareness about SMA, leading to increased and earlier diagnosis of SMA in Shikoku.
Full article
Open AccessEditorial
Newborn Screening in Japan—2021
by
Toshihiro Tajima
Cited by 4 | Viewed by 2879
Abstract
Japan’s Newborn Mass Screening (NBS) was started in 1977 for amino acid metabolism disorders (phenylketonuria (PKU), homocystinuria, maple syrup urine, histidineemia (discontinued in 1993)) and galactosemia at the national level as a national project [...]
Full article
Open AccessArticle
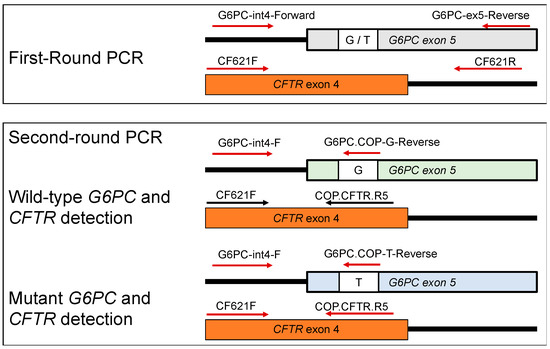
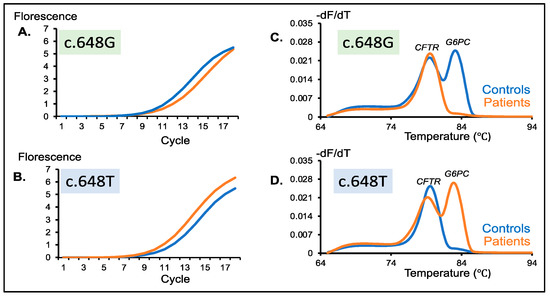
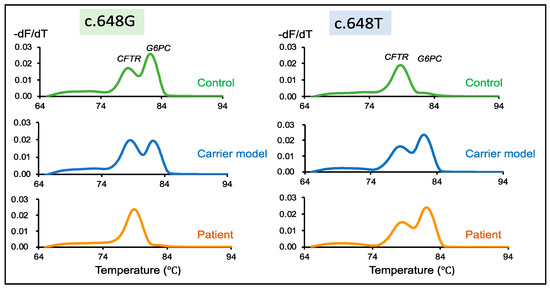
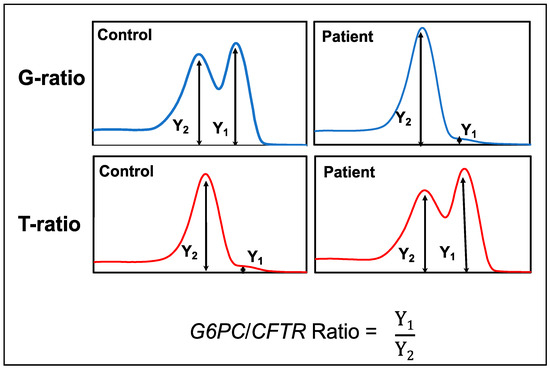
DBS Screening for Glycogen Storage Disease Type 1a: Detection of c.648G>T Mutation in G6PC by Combination of Modified Competitive Oligonucleotide Priming-PCR and Melting Curve Analysis
by
Emma Tabe Eko Niba, Yogik Onky Silvana Wijaya, Hiroyuki Awano, Naoko Taniguchi, Yasuhiro Takeshima, Hisahide Nishio and Masakazu Shinohara
Cited by 2 | Viewed by 2841
Abstract
Glycogen storage disease type Ia (GSDIa) is an autosomal recessive disorder caused by glucose-6-phosphatase (G6PC) deficiency. GSDIa causes not only life-threatening hypoglycemia in infancy, but also hepatocellular adenoma as a long-term complication. Hepatocellular adenoma may undergo malignant transformation to hepatocellular carcinoma. New treatment
[...] Read more.
Glycogen storage disease type Ia (GSDIa) is an autosomal recessive disorder caused by glucose-6-phosphatase (G6PC) deficiency. GSDIa causes not only life-threatening hypoglycemia in infancy, but also hepatocellular adenoma as a long-term complication. Hepatocellular adenoma may undergo malignant transformation to hepatocellular carcinoma. New treatment approaches are keenly anticipated for the prevention of hepatic tumors. Gene replacement therapy (GRT) is a promising approach, although early treatment in infancy is essential for its safety and efficiency. Thus, GRT requires screening systems for early disease detection. In this study, we developed a screening system for GSDIa using dried blood spots (DBS) on filter paper, which can detect the most common causative mutation in the East-Asian population, c.648G>T in the
G6PC gene. Our system consisted of nested PCR analysis with modified competitive oligonucleotide priming (mCOP)-PCR in the second round and melting curve analysis of the amplified products. Here, we tested 54 DBS samples from 50 c.648G (wild type) controls and four c.648T (mutant) patients. This system, using DBS samples, specifically amplified and clearly detected wild-type and mutant alleles from controls and patients, respectively. In conclusion, our system will be applicable to newborn screening for GSDIa in the real world.
Full article
►▼
Show Figures
Open AccessReview
The Discovery of GALM Deficiency (Type IV Galactosemia) and Newborn Screening System for Galactosemia in Japan
by
Atsuo Kikuchi, Yoichi Wada, Toshihiro Ohura and Shigeo Kure
Cited by 6 | Viewed by 3820
Abstract
The Leloir pathway, which consists of highly conserved enzymes, metabolizes galactose. Deficits in three enzymes in this pathway, namely galactose-1-phosphate uridylyltransferase (GALT), galactokinase (GALK1), and UDP-galactose-4′-epimerase (GALE), are associated with genetic galactosemia. We recently identified patients with galactosemia and biallelic variants in
GALM
[...] Read more.
The Leloir pathway, which consists of highly conserved enzymes, metabolizes galactose. Deficits in three enzymes in this pathway, namely galactose-1-phosphate uridylyltransferase (GALT), galactokinase (GALK1), and UDP-galactose-4′-epimerase (GALE), are associated with genetic galactosemia. We recently identified patients with galactosemia and biallelic variants in
GALM, encoding galactose epimerase (GALM), an enzyme that is directly upstream of GALK1. GALM deficiency was subsequently designated as type IV galactosemia. Currently, all the published patients with biallelic
GALM variants were found through newborn screening in Japan. Here, we review GALM deficiency and describe how we discovered this relatively mild but not rare disease through the newborn screening system in Japan.
Full article
►▼
Show Figures
Open AccessReview
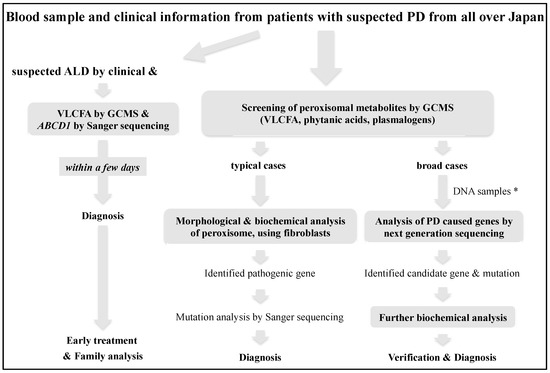
Advanced Diagnostic System and Introduction of Newborn Screening of Adrenoleukodystrophy and Peroxisomal Disorders in Japan
by
Nobuyuki Shimozawa, Shigeo Takashima, Hiroki Kawai, Kazuo Kubota, Hideo Sasai, Kenji Orii, Megumi Ogawa and Hidenori Ohnishi
Cited by 11 | Viewed by 2891
Abstract
We established a diagnostic system for adrenoleukodystrophy (ALD) and peroxisomal disorders (PD) over 35 years ago in Japan, and have diagnosed 237 families with ALD and more than 100 cases of PD other than ALD using biochemical and molecular analyses. In particular, since
[...] Read more.
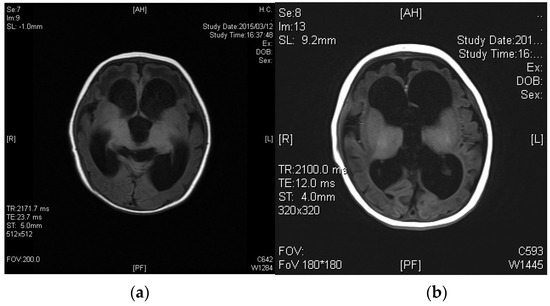
We established a diagnostic system for adrenoleukodystrophy (ALD) and peroxisomal disorders (PD) over 35 years ago in Japan, and have diagnosed 237 families with ALD and more than 100 cases of PD other than ALD using biochemical and molecular analyses. In particular, since the only treatment for the cerebral form of ALD is hematopoietic stem cell transplantation at an early stage of onset, we have developed a protocol for the rapid diagnosis of ALD that can provide the measurements of the levels of very-long-chain fatty acids in the serum and genetic analysis within a few days. In addition, to improve the prognosis of patients with ALD, we are working on the detection of pre-symptomatic patients by familial analysis from the proband, and the introduction of newborn screening. In this review, we introduce the diagnostic and newborn screening approaches for ALD and PD in Japan.
Full article
►▼
Show Figures
Open AccessArticle
Spinal Muscular Atrophy: Diagnosis, Incidence, and Newborn Screening in Japan
by
Tomokazu Kimizu, Shinobu Ida, Kentaro Okamoto, Hiroyuki Awano, Emma Tabe Eko Niba, Yogik Onky Silvana Wijaya, Shin Okazaki, Hideki Shimomura, Tomoko Lee, Koji Tominaga, Shin Nabatame, Toshio Saito, Takashi Hamazaki, Norio Sakai, Kayoko Saito, Haruo Shintaku, Kandai Nozu, Yasuhiro Takeshima, Kazumoto Iijima, Hisahide Nishio and Masakazu Shinoharaadd
Show full author list
remove
Hide full author list
| Viewed by 5063
Abstract
Spinal muscular atrophy (SMA) is a genetic neuromuscular disorder that causes degeneration of anterior horn cells in the human spinal cord and subsequent loss of motor neurons. The severe form of SMA is among the genetic diseases with the highest infant mortality. Although
[...] Read more.
Spinal muscular atrophy (SMA) is a genetic neuromuscular disorder that causes degeneration of anterior horn cells in the human spinal cord and subsequent loss of motor neurons. The severe form of SMA is among the genetic diseases with the highest infant mortality. Although SMA has been considered incurable, newly developed drugs—nusinersen and onasemnogene abeparvovec—improve the life prognoses and motor functions of affected infants. To maximize the efficacy of these drugs, treatments should be started at the pre-symptomatic stage of SMA. Thus, newborn screening for SMA is now strongly recommended. Herein, we provide some data based on our experience of SMA diagnosis by genetic testing in Japan. A total of 515 patients suspected of having SMA or another lower motor neuron disease were tested. Among these patients, 228 were diagnosed as having SMA with survival motor neuron 1 (
SMN1) deletion. We analyzed the distribution of clinical subtypes and ages at genetic testing in the
SMN1-deleted patients, and estimated the SMA incidence based on data from Osaka and Hyogo prefectures, Japan. Our data showed that confirmed diagnosis by genetic testing was notably delayed, and the estimated incidence was 1 in 30,000–40,000 live births, which seemed notably lower than in other countries. These findings suggest that many diagnosis-delayed or undiagnosed cases may be present in Japan. To prevent this, newborn screening programs for SMA (SMA-NBS) need to be implemented in all Japanese prefectures. In this article, we also introduce our pilot study for SMA-NBS in Osaka Prefecture.
Full article
Open AccessArticle
Development of Second-Tier Liquid Chromatography-Tandem Mass Spectrometry Analysis for Expanded Newborn Screening in Japan
by
Yosuke Shigematsu, Miori Yuasa, Nobuyuki Ishige, Hideki Nakajima and Go Tajima
Cited by 11 | Viewed by 3225
Abstract
To minimize false-positive cases in newborn screening by tandem mass spectrometry in Japan, practical second-tier liquid chromatography-tandem mass spectrometry analyses have been developed using a multimode ODS column with a single set of mobile phases and different gradient elution programs specific to the
[...] Read more.
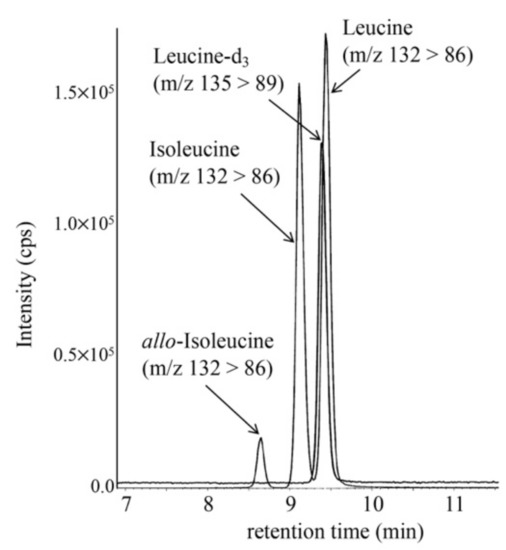
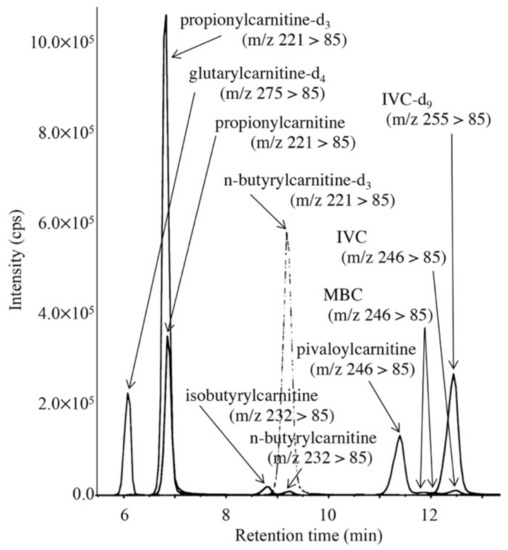
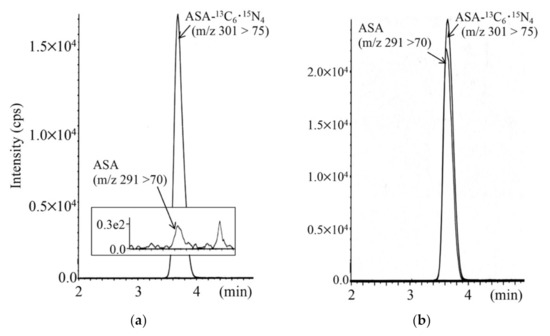
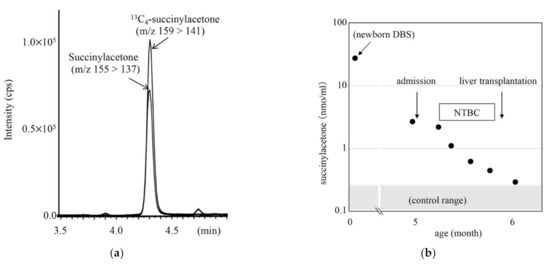
To minimize false-positive cases in newborn screening by tandem mass spectrometry in Japan, practical second-tier liquid chromatography-tandem mass spectrometry analyses have been developed using a multimode ODS column with a single set of mobile phases and different gradient elution programs specific to the analysis of acylcarnitines, acylglycines, amino acids, and organic acids. Most analyses were performed using underivatized samples, except for analysis of methylcitric acid, and careful conditioning of the column was necessary for analyses of organic acids. Our second-tier tests enabled us to measure many metabolites useful for detection of target disorders, including allo-isoleucine, homocysteine, methylmalonic acid, and methylcitric acid. We found that accumulation of 3-hydroxyglutaric acid was specific to glutaric acidemia type I and that the ratio of 3-hydroxyisovaleric acid to 3-hydroxyisovalerylcarnitine was useful to detect newborns of mothers with 3-methylcrotonyl-CoA carboxylase deficiency. Data from the analysis of short-chain acylcarnitine and acylglycine were useful for differential diagnosis in cases positive for C5-OH-acylcarnitine or C5-acylcarnitine.
Full article
►▼
Show Figures
Open AccessArticle
Pilot Study on Neonatal Screening for Methylmalonic Acidemia Caused by Defects in the Adenosylcobalamin Synthesis Pathway and Homocystinuria Caused by Defects in Homocysteine Remethylation
by
Reiko Kagawa, Go Tajima, Takako Maeda, Fumiaki Sakura, Akari Nakamura-Utsunomiya, Keiichi Hara, Yutaka Nishimura, Miori Yuasa, Yosuke Shigematsu, Hiromi Tanaka, Saki Fujihara, Chiyoko Yoshii and Satoshi Okada
Cited by 1 | Viewed by 2490
Abstract
Neonatal screening (NS) for methylmalonic acidemia uses propionylcarnitine (C3) as a primary index, which is insufficiently sensitive at detecting methylmalonic acidemia caused by defects in the adenosylcobalamin synthesis pathway. Moreover, homocystinuria from cystathionine β-synthase deficiency is screened by detecting hypermethioninemia, but methionine levels
[...] Read more.
Neonatal screening (NS) for methylmalonic acidemia uses propionylcarnitine (C3) as a primary index, which is insufficiently sensitive at detecting methylmalonic acidemia caused by defects in the adenosylcobalamin synthesis pathway. Moreover, homocystinuria from cystathionine β-synthase deficiency is screened by detecting hypermethioninemia, but methionine levels decrease in homocystinuria caused by defects in homocysteine remethylation. To establish NS detection of methylmalonic acidemia and homocystinuria of these subtypes, we evaluated the utility of indices (1) C3 ≥ 3.6 μmol/L and C3/acetylcarnitine (C2) ≥ 0.23, (2) C3/methionine ≥ 0.25, and (3) methionine < 10 μmol/L, by retrospectively applying them to NS data of 59,207 newborns. We found positive results in 116 subjects for index (1), 37 for (2), and 15 for (3). Second-tier tests revealed that for index 1, methylmalonate (MMA) was elevated in two cases, and MMA and total homocysteine (tHcy) were elevated in two cases; for index 2 that MMA was elevated in one case; and for index 3 that tHcy was elevated in one case. Though data were anonymized, two cases identified by index 1 had been diagnosed with maternal vitamin B
12 deficiency during NS. Methylene tetrahydrofolate reductase deficiency was confirmed for the case identified by index 3, which was examined because an elder sibling was affected by the same disease. Based on these data, a prospective NS study is underway.
Full article
►▼
Show Figures
Open AccessReview
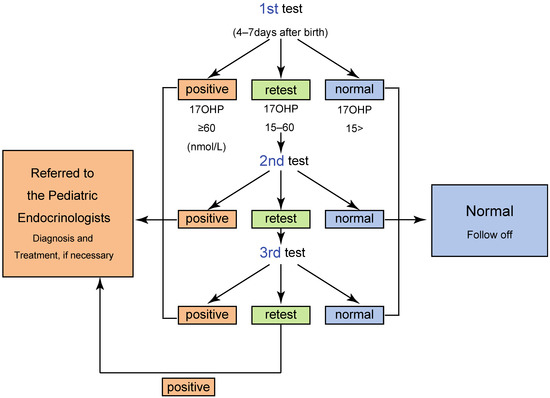
Thirty-Year Lessons from the Newborn Screening for Congenital Adrenal Hyperplasia (CAH) in Japan
by
Atsumi Tsuji-Hosokawa and Kenichi Kashimada
Cited by 11 | Viewed by 3427
Abstract
Congenital adrenal hyperplasia (CAH) is an inherited disorder caused by the absence or severely impaired activity of steroidogenic enzymes involved in cortisol biosynthesis. More than 90% of cases result from 21-hydroxylase deficiency (21OHD). To prevent life-threatening adrenal crisis and to help perform appropriate
[...] Read more.
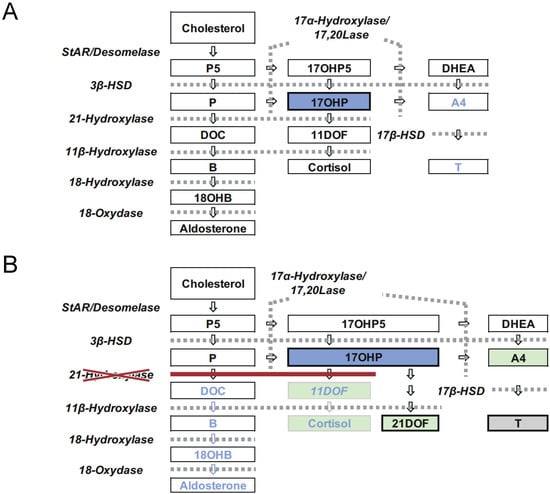
Congenital adrenal hyperplasia (CAH) is an inherited disorder caused by the absence or severely impaired activity of steroidogenic enzymes involved in cortisol biosynthesis. More than 90% of cases result from 21-hydroxylase deficiency (21OHD). To prevent life-threatening adrenal crisis and to help perform appropriate sex assignments for affected female patients, newborn screening (NBS) programs for the classical form of CAH have been introduced in numerous countries. In Japan, the NBS for CAH was introduced in 1989, following the screenings for phenylketonuria and congenital hypothyroidism. In this review, we aim to summarize the experience of the past 30 years of the NBS for CAH in Japan, composed of four parts, 1: screening system in Japan, 2: the clinical outcomes for the patients with CAH, 3: various factors that would impact the NBS system, including timeline, false positive, and LC-MS/MS, 4: Database composition and improvement of the screening program.
Full article
►▼
Show Figures
Open AccessReview
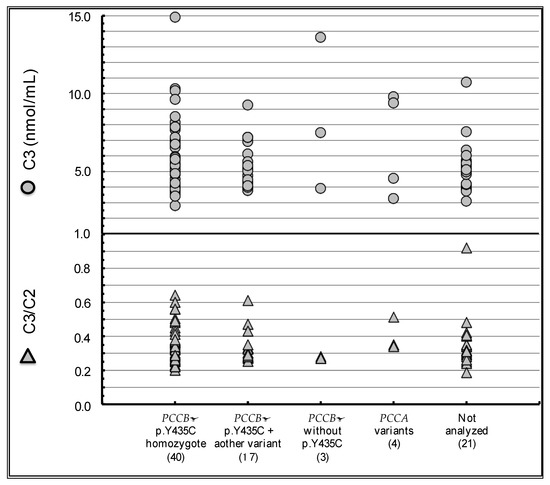
Current Perspectives on Neonatal Screening for Propionic Acidemia in Japan: An Unexpectedly High Incidence of Patients with Mild Disease Caused by a Common PCCB Variant
by
Go Tajima, Reiko Kagawa, Fumiaki Sakura, Akari Nakamura-Utsunomiya, Keiichi Hara, Miori Yuasa, Yuki Hasegawa, Hideo Sasai and Satoshi Okada
Cited by 4 | Viewed by 2681
Abstract
Propionic acidemia (PA) is a disorder of organic acid metabolism which typically presents with acute encephalopathy-like symptoms associated with metabolic acidosis and hyperammonemia during the neonatal period. The estimated incidence of symptomatic PA in Japan is 1/400,000. The introduction of neonatal screening using
[...] Read more.
Propionic acidemia (PA) is a disorder of organic acid metabolism which typically presents with acute encephalopathy-like symptoms associated with metabolic acidosis and hyperammonemia during the neonatal period. The estimated incidence of symptomatic PA in Japan is 1/400,000. The introduction of neonatal screening using tandem mass spectrometry has revealed a far higher disease frequency of approximately 1/45,000 live births due to a prevalent variant of c.1304T>C (p.Y435C) in
PCCB, which codes β-subunit of propionyl-CoA carboxylase. Our questionnaire-based follow-up study reveals that most of these patients remain asymptomatic. However, reports on symptomatic patients exhibiting cardiac complications such as cardiomyopathy and QT prolongation have been increasing. Moreover, there were even cases in which these cardiac complications were the only symptoms related to PA. A currently ongoing study is investigating the risk of cardiac complications in patients with neonatal screening-detected PA caused by this common variant.
Full article
►▼
Show Figures
Open AccessReview
Newborn Screening for Congenital Hypothyroidism in Japan
by
Kanshi Minamitani
Cited by 11 | Viewed by 3116
Abstract
Congenital hypothyroidism (CH) is the most common preventable cause of intellectual impairment or failure to thrive by early identification and treatment. In Japan, newborn screening programs for CH were introduced in 1979, and the clinical guidelines for newborn screening of CH were developed
[...] Read more.
Congenital hypothyroidism (CH) is the most common preventable cause of intellectual impairment or failure to thrive by early identification and treatment. In Japan, newborn screening programs for CH were introduced in 1979, and the clinical guidelines for newborn screening of CH were developed in 1998, revised in 2014, and are currently undergoing further revision. Newborn screening strategies are designed to detect the elevated levels of thyroid stimulating hormone (TSH) in most areas of Japan, although TSH and free thyroxine (FT4) are often measured simultaneously in some areas. Since 1987, in order not to observe the delayed rise in TSH, additional rescreening of premature neonates and low birth weight infants (<2000 g) at four weeks of life or when their body weight reaches 2500 g has been recommended, despite a normal initial newborn screening. Recently, the actual incidence of CH has doubled to approximately 1:2500 in Japan as in other countries. This increasing incidence is speculated to be mainly due to an increase in the number of mildly affected patients detected by the generalized lowering of TSH screening cutoffs and an increase in the number of preterm or low birth weight neonates at a higher risk of having CH than term infants.
Full article
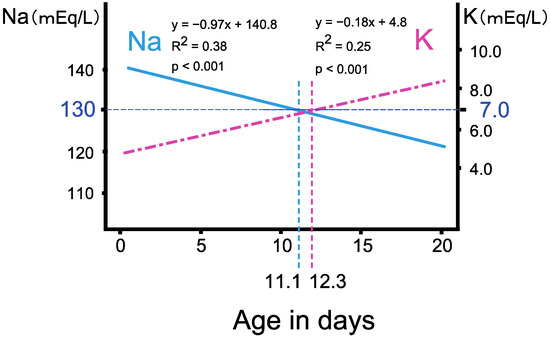
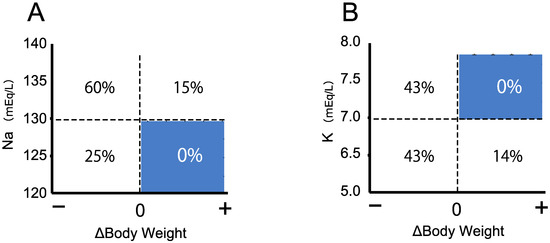
Open AccessArticle
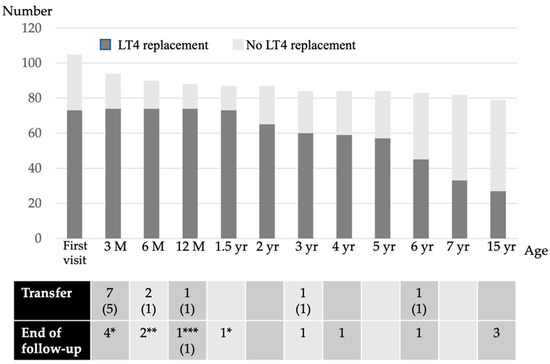
Re-Evaluation of the Prevalence of Permanent Congenital Hypothyroidism in Niigata, Japan: A Retrospective Study
by
Keisuke Nagasaki, Hidetoshi Sato, Sunao Sasaki, Hiromi Nyuzuki, Nao Shibata, Kentaro Sawano, Shota Hiroshima and Tadashi Asami
Cited by 3 | Viewed by 3006
Abstract
Although newborn screening (NBS) for congenital hypothyroidism (CH) in Japan started more than 40 years ago, the prevalence of CH remains unclear. Prevalence estimations among NBS-positive CH individuals include those with transient hypothyroidism and transient hyperthyrotropinemia, and re-evaluation with increasing age is necessary
[...] Read more.
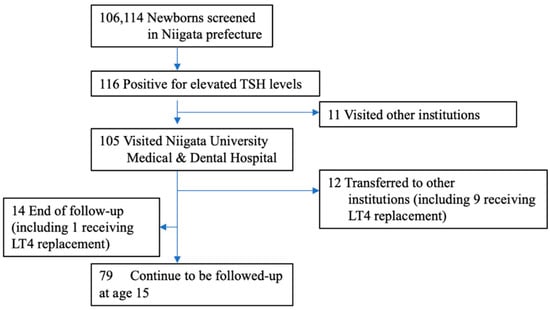
Although newborn screening (NBS) for congenital hypothyroidism (CH) in Japan started more than 40 years ago, the prevalence of CH remains unclear. Prevalence estimations among NBS-positive CH individuals include those with transient hypothyroidism and transient hyperthyrotropinemia, and re-evaluation with increasing age is necessary to clarify the actual incidence. Thus, we re-evaluated the incidence of permanent CH. Of the 106,114 patients who underwent NBS in the Niigata Prefecture, Japan, between April 2002 and March 2006, 116 were examined further due to high thyroid-stimulating hormone levels (>8 mIU/L) and were included in the study. We retrospectively evaluated their levothyroxine sodium (LT4) replacement therapy status from the first visit to 15 years of age. Of the 116 NBS-positive patients, 105 (91%) were initially examined in our department. Of these, 72 (69%) started LT4 replacement therapy on the first visit. Subsequently, 27 patients continued LT4 replacement until 15 years of age after multiple re-evaluations. The prevalence of permanent CH in the Niigata Prefecture during this period was 1 in 2500–3500 children. Ultimately, 62.5% of patients on LT4 replacement discontinued treatment by 15 years of age. This is the first study to clarify the true prevalence of permanent CH in Japan.
Full article
►▼
Show Figures
Open AccessArticle
Long-Term Neurological Outcomes of Adult Patients with Phenylketonuria before and after Newborn Screening in Japan
by
Kenji Yamada, Seiji Yamaguchi, Kazunori Yokoyama, Kikumaro Aoki and Takeshi Taketani
Cited by 4 | Viewed by 2509
Abstract
Japanese newborn screening (NBS) for phenylketonuria (PKU) was initiated in 1977. We surveyed the neurological outcomes of Japanese adult patients with PKU to investigate the long-term effects and of and issues with NBS. Eighty-five patients with PKU aged over 19 years who continued
[...] Read more.
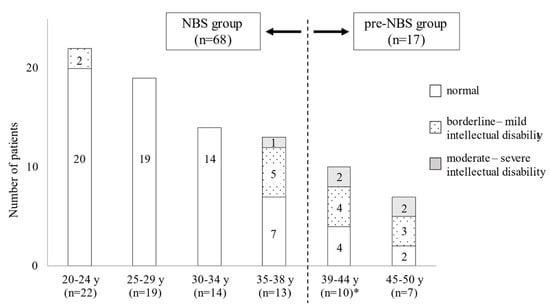
Japanese newborn screening (NBS) for phenylketonuria (PKU) was initiated in 1977. We surveyed the neurological outcomes of Japanese adult patients with PKU to investigate the long-term effects and of and issues with NBS. Eighty-five patients with PKU aged over 19 years who continued to be treated with a phenylalanine-free amino acid formula were investigated by administering questionnaires regarding clinical characteristics, such as mental ability, education status, and therapeutic condition. Of the 85 subjects, 68 patients were detected by NBS (NBS group), while the other 17 were clinically diagnosed before the initiation of NBS (pre-NBS group). Further, 10 of the 68 NBS patients presented intellectual and/or psychiatric disabilities, 5 of whom had a history of treatment discontinuation; in contrast, 12 of the 17 pre-NBS patients presented with neuropsychiatric symptoms. Regarding social outcomes, almost all patients in the NBS group could live an independent life, while over half of the patients in the pre-NBS group were not employed or lived in nursing-care facilities. Neurological outcomes are obviously improved by NBS in Japan. However, some patients, even those detected by NBS, developed neuropsychiatric symptoms due to treatment disruption. Lifelong and strict management is essential to maintain good neurological and social prognoses for patients with PKU.
Full article
►▼
Show Figures
Open AccessArticle
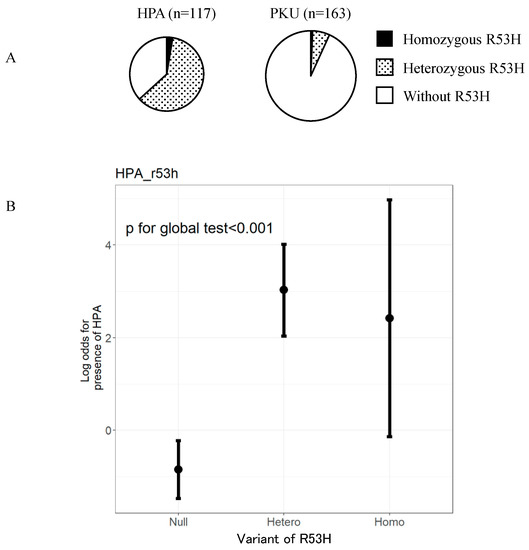
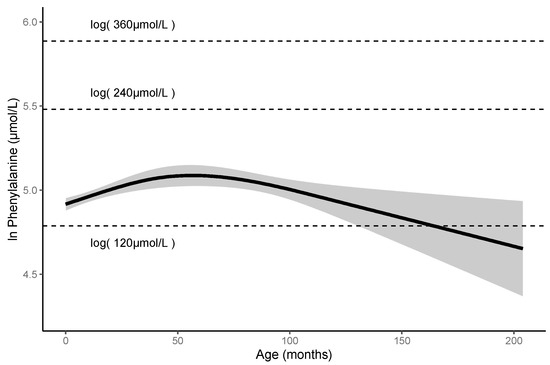
Clinical and Genetic Characteristics of Patients with Mild Hyperphenylalaninemia Identified by Newborn Screening Program in Japan
by
Shino Odagiri, Daijiro Kabata, Shogo Tomita, Satoshi Kudo, Tomoko Sakaguchi, Noriko Nakano, Kouji Yamamoto, Haruo Shintaku and Takashi Hamazaki
Cited by 5 | Viewed by 2836
Abstract
Phenylketonuria (PKU) and hyperphenylalaninemia (HPA), both identified in newborn screening, are attributable to variants in
PAH. Reportedly, the p.R53H(c.158G>A) variant is common in patients with HPA in East Asia. Here, we aimed to define the association between p.R53H and HPA phenotype, and
[...] Read more.
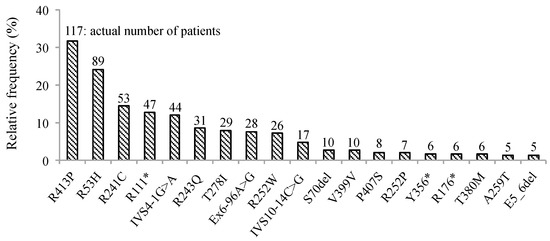
Phenylketonuria (PKU) and hyperphenylalaninemia (HPA), both identified in newborn screening, are attributable to variants in
PAH. Reportedly, the p.R53H(c.158G>A) variant is common in patients with HPA in East Asia. Here, we aimed to define the association between p.R53H and HPA phenotype, and study the long-term outcome of patients with HPA carrying p.R53H. We retrospectively reviewed the genotype in 370 patients detected by newborn screening, and identified the phenotype in 280 (117, HPA; 163, PKU). p.R413P(c.1238G>C) was the most frequently found (
n = 117, 31.6%) variant, followed by
p.R53H (
n = 89, 24.1%). The odds ratio for heterozygous p.R53H to cause HPA was 48.3 (95% CI 19.410–120.004). Furthermore, we assessed the non-linear association between the phenylalanine (Phe) value and elapsed time using the follow-up data of the blood Phe levels of 73 patients with HPA carrying p.R53H. The predicted levels peaked at 161.9 μmol (95% CI 152.088–172.343) at 50–60 months of age and did not exceed 360 μmol/L during the 210-month long observation period. The findings suggest that patients with HPA, carrying p.R53H, do not need frequent Phe monitoring as against those with PKU. Our study provides convincing evidence to determine clinical management of patients detected through newborn screening in Japan.
Full article
►▼
Show Figures
Open AccessArticle
Long-Term Outcomes of Adult Patients with Homocystinuria before and after Newborn Screening
by
Kenji Yamada, Kazunori Yokoyama, Kikumaro Aoki, Takeshi Taketani and Seiji Yamaguchi
Cited by 10 | Viewed by 2815
Abstract
Background: Homocystinuria (HCU) is a rare inherited metabolic disease. In Japan, newborn screening (NBS) for HCU (cystathionine β-synthase deficiency) was initiated in 1977. We compared the outcomes between patients detected by NBS (NBS group) and clinically detected patients (non-NBS group). Methods: We administered
[...] Read more.
Background: Homocystinuria (HCU) is a rare inherited metabolic disease. In Japan, newborn screening (NBS) for HCU (cystathionine β-synthase deficiency) was initiated in 1977. We compared the outcomes between patients detected by NBS (NBS group) and clinically detected patients (non-NBS group). Methods: We administered questionnaires about clinical symptoms and social conditions to 16 attending physicians of 19 adult HCU patients treated with methionine-free formula. Results: Eighteen patients (nine patients each in the NBS and non-NBS groups) participated. The frequency of patients with ocular, vascular, central nervous system, and skeletal symptoms in the NBS group was lower than that in the non-NBS group. Intellectual disability was observed in one and eight patients in the NBS and non-NBS groups, respectively. Concerning their social conditions, all patients in the NBS group were employed or still attending school, while only two patients in the non-NBS group were employed. Three of the four patients who discontinued treatment presented some symptoms, even in the NBS group. Conclusion: The social and intellectual outcomes of adult Japanese patients with HCU detected by NBS were favorable. However, even in the patients in the NBS group, some symptoms might not be preventable without continuous treatment.
Full article







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}