In Silico Drug Design for GPCRs: Big Data for Small Molecule Discovery
Share This Topical Collection
Editor
 Dr. Vsevolod Katritch
Dr. Vsevolod Katritch
Dr. Vsevolod Katritch
grade
E-Mail
Website
Collection Editor
The Bridge Institute, University of Southern California, Los Angeles, CA 90032, USA
Interests: structure–function of GPCRs; integrative modeling; rational ligand design; virtual screening; machine learning; allosteric, bitopic, and photoswitchable ligands; chemical probes; drug discovery
Topical Collection Information
Dear Colleagues,
The current drug discovery process increasingly relies on data-driven computational approaches. Among many aspects of computational revolution, one can name the following: of high-resolution structural information and molecular modeling capabilities for the target receptors, (2) improvement of virtual screening and rational design algorithms, (3) rapid growth of virtual combinatorial libraries, supported by fast parallel synthesis of compounds, (4) application of machine learning approaches to the analysis of chemogenomics information and predictions of multitarget ligand activity profiles, and (5) advanced computational approaches for predicting affinities and potencies of derivatives. The exponentially growing data sources and improved tools, supported by access to highly parallel GPU and cloud computing, are poised to revolutionize the field in both academia and industry, making data-driven computational analysis and design the backbone of drug discovery.
This Topic Collection of Biomolecules will showcase recent examples of computationally intensive data-driven approaches that contribute to the different aspects of small molecule drug discovery for GPCR targets. The main goal is to compile articles that describe new or improved algorithms with practical applications for hit discovery and optimization.
Dr. Vsevolod Katritch
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Biomolecules is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- virtual ligand screening
- computer-assisted drug discovery
- structure-based design
- chemogenomics
- machine learning
Published Papers (9 papers)
Open AccessEditor’s ChoiceArticle
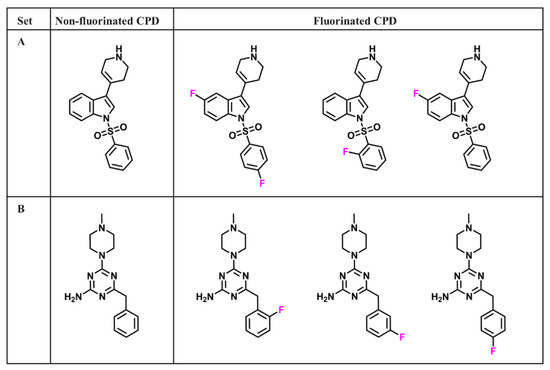
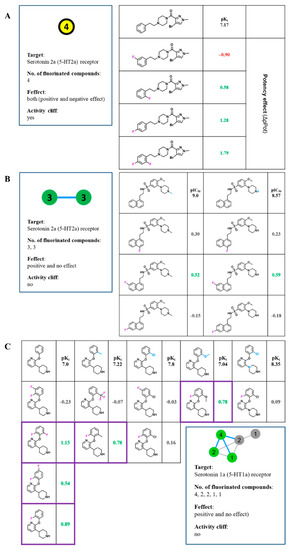
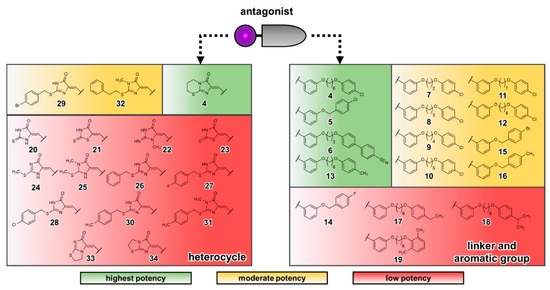
Data-Driven Analysis of Fluorination of Ligands of Aminergic G Protein Coupled Receptors
by
Wojciech Pietruś, Rafał Kurczab, Dagmar Stumpfe, Andrzej J. Bojarski and Jürgen Bajorath
Cited by 2 | Viewed by 1672
Abstract
Currently, G protein-coupled receptors are the targets with the highest number of drugs in many therapeutic areas. Fluorination has become a common strategy in designing highly active biological compounds, as evidenced by the steadily increasing number of newly approved fluorine-containing drugs. Herein, we
[...] Read more.
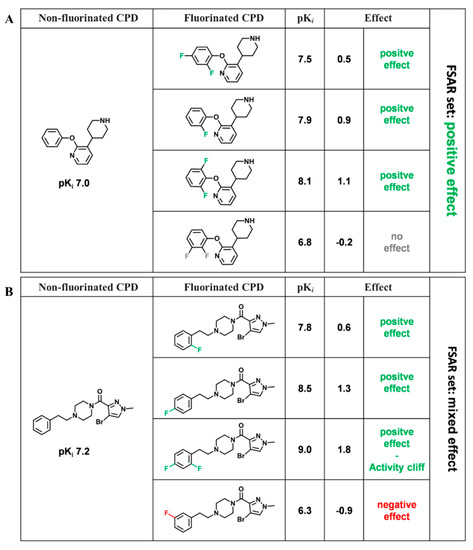
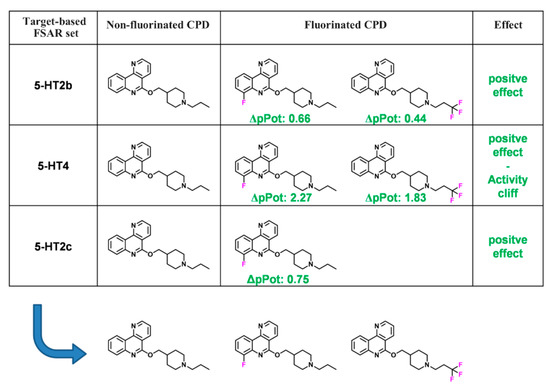
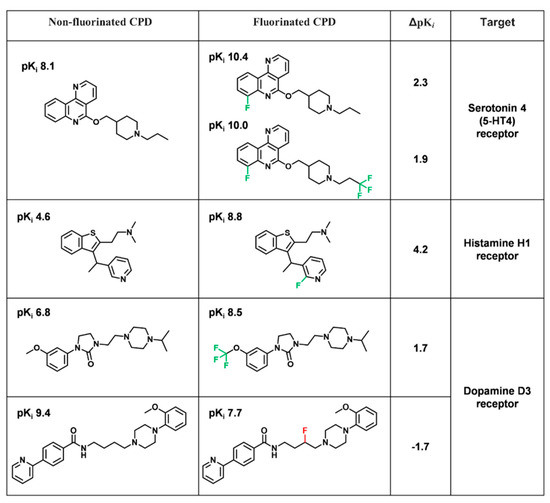
Currently, G protein-coupled receptors are the targets with the highest number of drugs in many therapeutic areas. Fluorination has become a common strategy in designing highly active biological compounds, as evidenced by the steadily increasing number of newly approved fluorine-containing drugs. Herein, we identified in the ChEMBL database and analysed 1554 target-based FSAR sets (non-fluorinated compounds and their fluorinated analogues) comprising 966 unique non-fluorinated and 2457 unique fluorinated compounds active against 33 different aminergic GPCRs. Although a relatively small number of activity cliffs (defined as a pair of structurally similar compounds showing significant differences of activity −ΔpPot > 1.7) was found in FSAR sets, it is clear that appropriately introduced fluorine can increase ligand potency more than 50-fold. The analysis of matched molecular pairs (MMPs) networks indicated that the fluorination of the aromatic ring showed no clear trend towards a positive or negative effect on affinity; however, a favourable site for a positive potency effect of fluorination was the ortho position. Fluorination of aliphatic fragments more often led to a decrease in biological activity. The results may constitute the rules of thumb for fluorination of aminergic receptor ligands and provide insights into the role of fluorine substitutions in medicinal chemistry.
Full article
►▼
Show Figures
Open AccessArticle
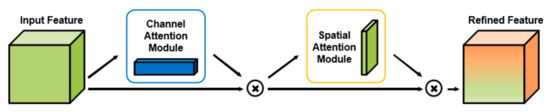
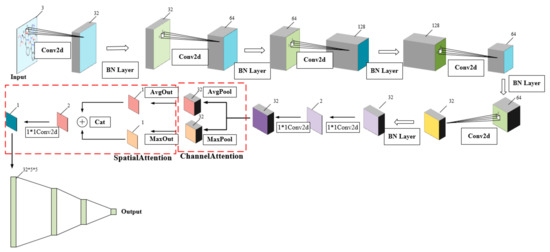
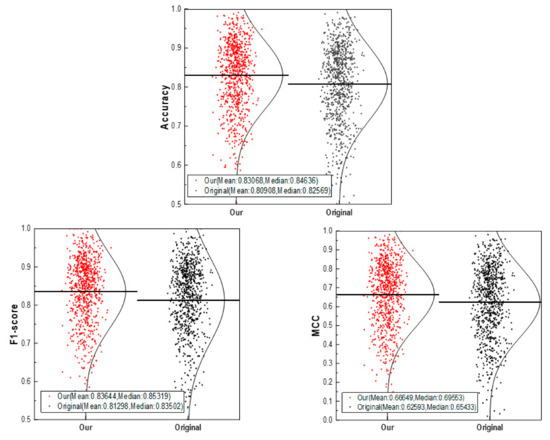
CSConv2d: A 2-D Structural Convolution Neural Network with a Channel and Spatial Attention Mechanism for Protein-Ligand Binding Affinity Prediction
by
Xun Wang, Dayan Liu, Jinfu Zhu, Alfonso Rodriguez-Paton and Tao Song
Cited by 13 | Viewed by 2721
Abstract
The binding affinity of small molecules to receptor proteins is essential to drug discovery and drug repositioning. Chemical methods are often time-consuming and costly, and models for calculating the binding affinity are imperative. In this study, we propose a novel deep learning method,
[...] Read more.
The binding affinity of small molecules to receptor proteins is essential to drug discovery and drug repositioning. Chemical methods are often time-consuming and costly, and models for calculating the binding affinity are imperative. In this study, we propose a novel deep learning method, namely CSConv2d, for protein-ligand interactions’ prediction. The proposed method is improved by a DEEPScreen model using 2-D structural representations of compounds as input. Furthermore, a channel and spatial attention mechanism (CS) is added in feature abstractions. Data experiments conducted on ChEMBLv23 datasets show that CSConv2d performs better than the original DEEPScreen model in predicting protein-ligand binding affinity, as well as some state-of-the-art DTIs (drug-target interactions) prediction methods including DeepConv-DTI, CPI-Prediction, CPI-Prediction+CS, DeepGS and DeepGS+CS. In practice, the docking results of protein (PDB ID: 5ceo) and ligand (Chemical ID: 50D) and a series of kinase inhibitors are operated to verify the robustness.
Full article
►▼
Show Figures
Open AccessArticle
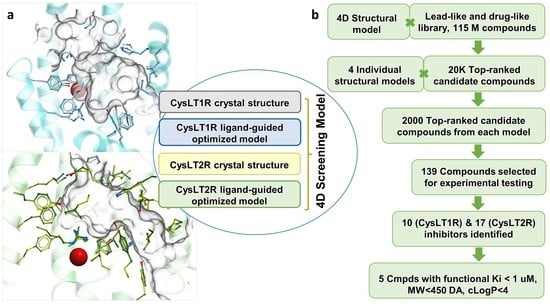
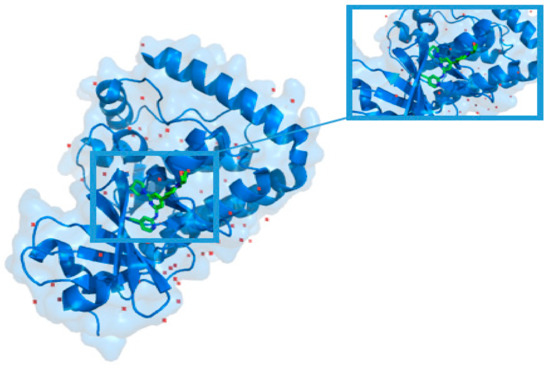
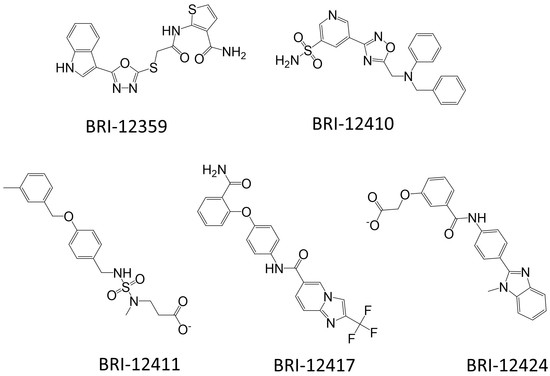
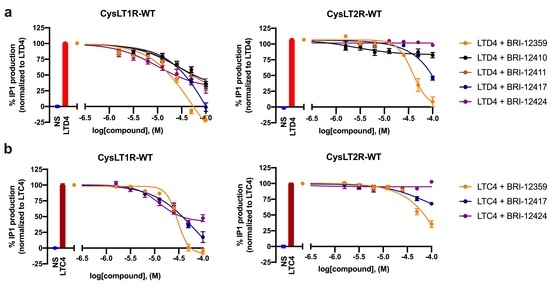
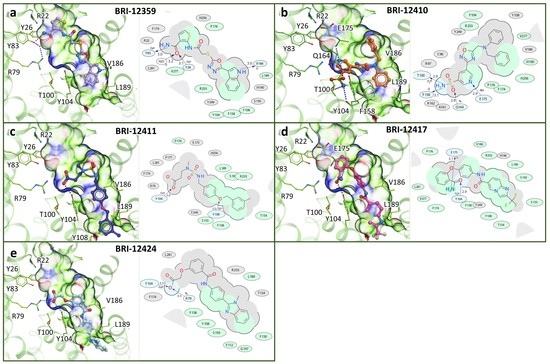
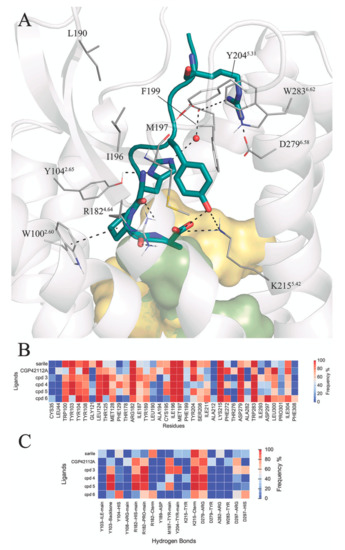
Structure-Based Virtual Screening of Ultra-Large Library Yields Potent Antagonists for a Lipid GPCR
by
Arman A. Sadybekov, Rebecca L. Brouillette, Egor Marin, Anastasiia V. Sadybekov, Aleksandra Luginina, Anastasiia Gusach, Alexey Mishin, Élie Besserer-Offroy, Jean-Michel Longpré, Valentin Borshchevskiy, Vadim Cherezov, Philippe Sarret and Vsevolod Katritch
Cited by 15 | Viewed by 6809
Abstract
Cysteinyl leukotriene G protein-coupled receptors, CysLT1R and CysLT2R, regulate bronchoconstrictive and pro-inflammatory effects and play a key role in allergic disorders, cardiovascular diseases, and cancer. CysLT1R antagonists have been widely used to treat asthma disorders, while CysLT2R is a potential target against uveal
[...] Read more.
Cysteinyl leukotriene G protein-coupled receptors, CysLT1R and CysLT2R, regulate bronchoconstrictive and pro-inflammatory effects and play a key role in allergic disorders, cardiovascular diseases, and cancer. CysLT1R antagonists have been widely used to treat asthma disorders, while CysLT2R is a potential target against uveal melanoma. However, very few selective antagonist chemotypes for CysLT receptors are available, and the design of such ligands has proved to be challenging. To overcome this obstacle, we took advantage of recently solved crystal structures of CysLT receptors and an ultra-large Enamine REAL library, representing a chemical space of 680 M readily available compounds. Virtual ligand screening employed 4D docking models comprising crystal structures of CysLT1R and CysLT2R and their corresponding ligand-optimized models. Functional assessment of the candidate hits yielded discovery of five novel antagonist chemotypes with sub-micromolar potencies and the best Ki = 220 nM at CysLT1R. One of the hits showed inverse agonism at the L129Q constitutively active mutant of CysLT2R, with potential utility against uveal melanoma.
Full article
►▼
Show Figures
Open AccessFeature PaperReview
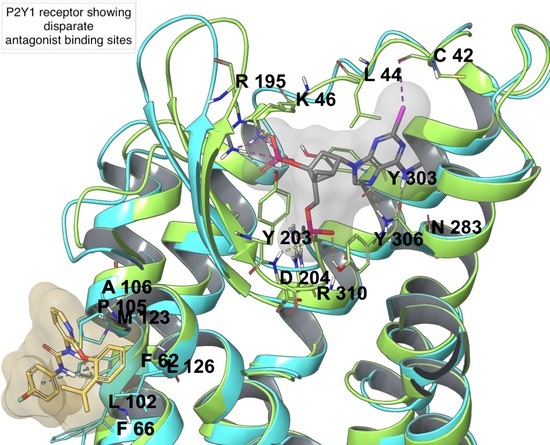
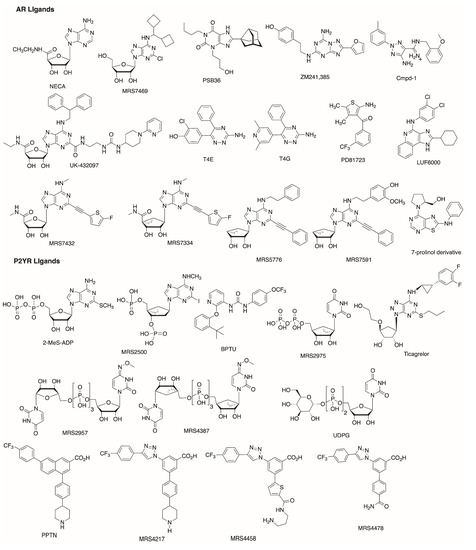
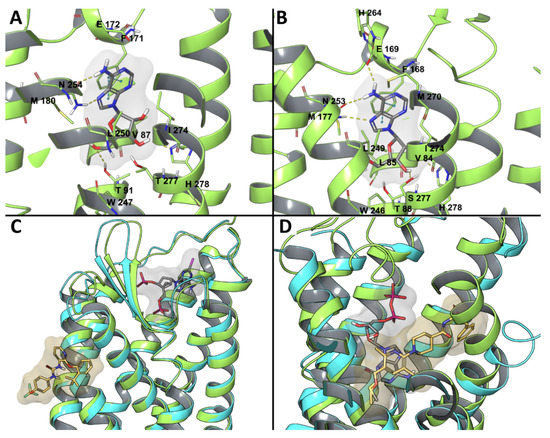
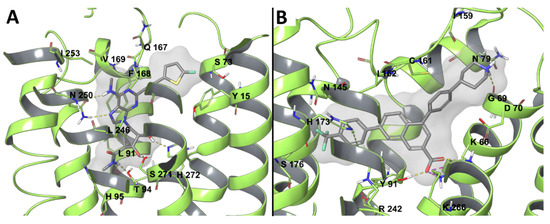
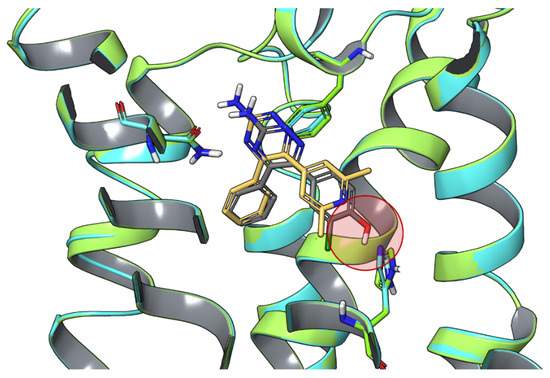
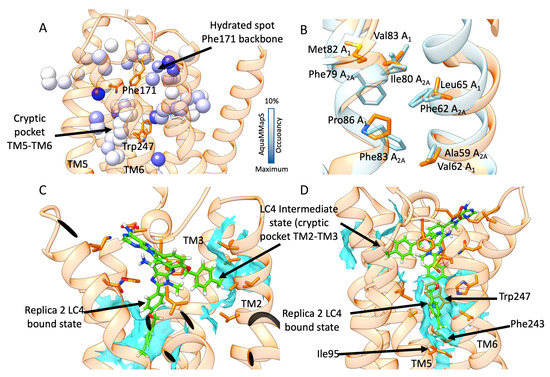
In Silico Drug Design for Purinergic GPCRs: Overview on Molecular Dynamics Applied to Adenosine and P2Y Receptors
by
Veronica Salmaso and Kenneth A. Jacobson
Cited by 16 | Viewed by 4006
Abstract
Molecular modeling has contributed to drug discovery for purinergic GPCRs, including adenosine receptors (ARs) and P2Y receptors (P2YRs). Experimental structures and homology modeling have proven to be useful in understanding and predicting structure activity relationships (SAR) of agonists and antagonists. This review provides
[...] Read more.
Molecular modeling has contributed to drug discovery for purinergic GPCRs, including adenosine receptors (ARs) and P2Y receptors (P2YRs). Experimental structures and homology modeling have proven to be useful in understanding and predicting structure activity relationships (SAR) of agonists and antagonists. This review provides an excursus on molecular dynamics (MD) simulations applied to ARs and P2YRs. The binding modes of newly synthesized A
1AR- and A
3AR-selective nucleoside derivatives, potentially of use against depression and inflammation, respectively, have been predicted to recapitulate their SAR and the species dependence of A
3AR affinity. P2Y
12R and P2Y
1R crystallographic structures, respectively, have provided a detailed understanding of the recognition of anti-inflammatory P2Y
14R antagonists and a large group of allosteric and orthosteric antagonists of P2Y
1R, an antithrombotic and neuroprotective target. MD of A
2AAR (an anticancer and neuroprotective target), A
3AR, and P2Y
1R has identified microswitches that are putatively involved in receptor activation. The approach pathways of different ligands toward A
2AAR and P2Y
1R binding sites have also been explored. A
1AR, A
2AAR, and A
3AR were utilizes to study allosteric phenomena, but locating the binding site of structurally diverse allosteric modulators, such as an A
3AR enhancer LUF6000, is challenging. Ligand residence time, a predictor of in vivo efficacy, and the structural role of water were investigated through A
2AAR MD simulations. Thus, new MD and other modeling algorithms have contributed to purinergic GPCR drug discovery.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
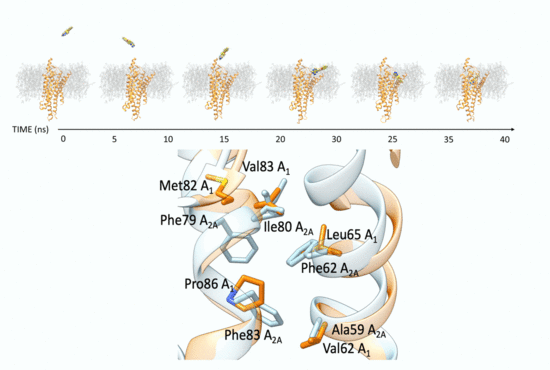
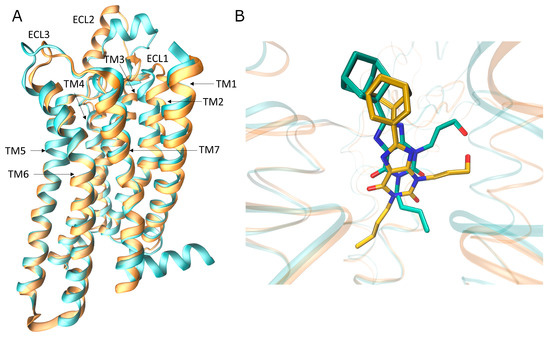
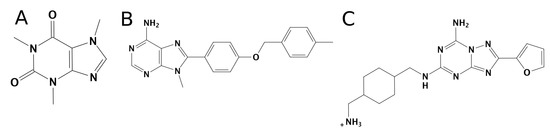
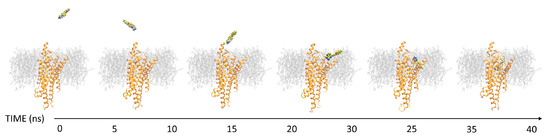
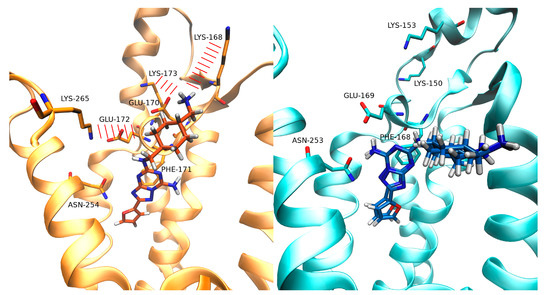
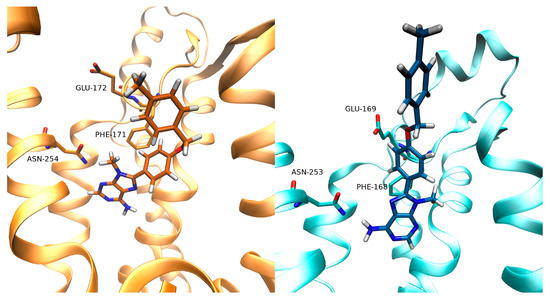
New Insights into Key Determinants for Adenosine 1 Receptor Antagonists Selectivity Using Supervised Molecular Dynamics Simulations
by
Giovanni Bolcato, Maicol Bissaro, Giuseppe Deganutti, Mattia Sturlese and Stefano Moro
Cited by 5 | Viewed by 3499
Abstract
Adenosine receptors (ARs), like many otherGprotein-coupledreceptors (GPCRs), are targets of primary interest indrug design. However, one of the main limits for the development of drugs for this class of GPCRs is the complex selectivity profile usually displayed by ligands. Numerous efforts have been
[...] Read more.
Adenosine receptors (ARs), like many otherGprotein-coupledreceptors (GPCRs), are targets of primary interest indrug design. However, one of the main limits for the development of drugs for this class of GPCRs is the complex selectivity profile usually displayed by ligands. Numerous efforts have been madefor clarifying the selectivity of ARs, leading to the development of many ligand-based models. The structure of the AR subtype A
1 (A
1AR) has been recently solved, providing important structural insights. In the present work, we rationalized the selectivity profile of two selective A
1AR and A
2AAR antagonists, investigating their recognition trajectories obtained by Supervised Molecular Dynamics from an unbound state and monitoring the role of the water molecules in the binding site.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
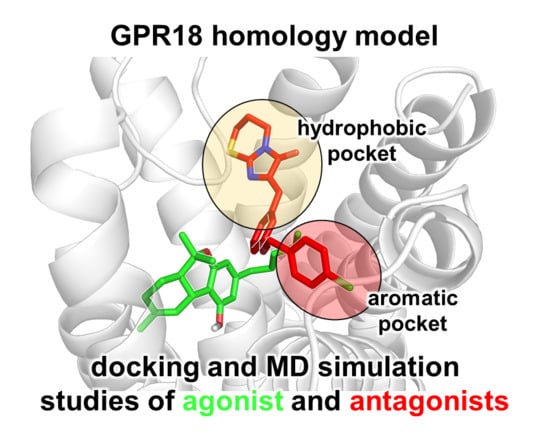
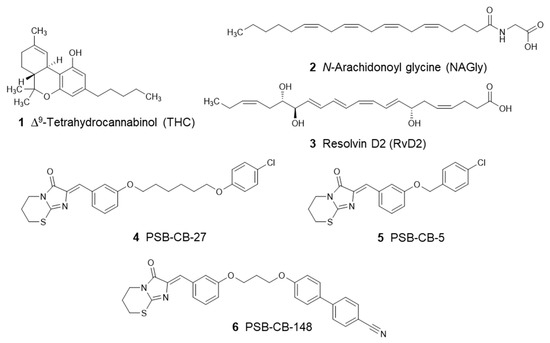
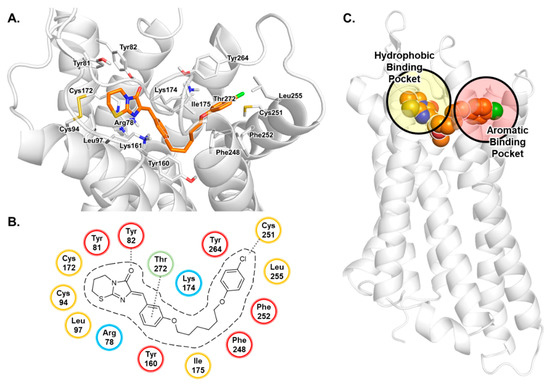
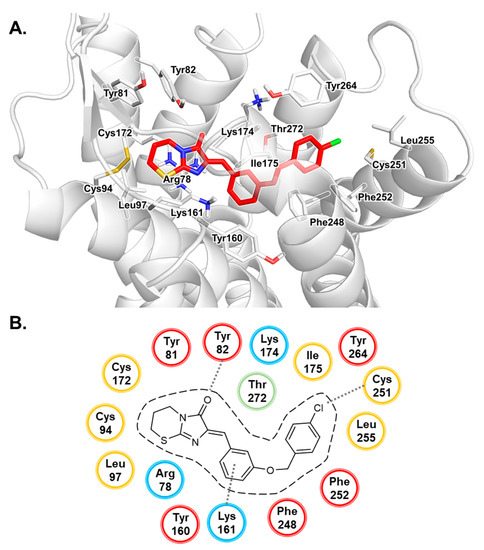
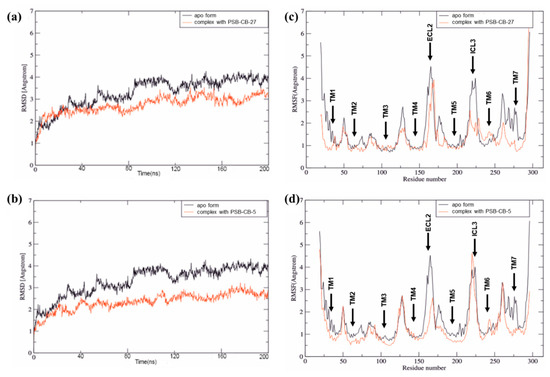
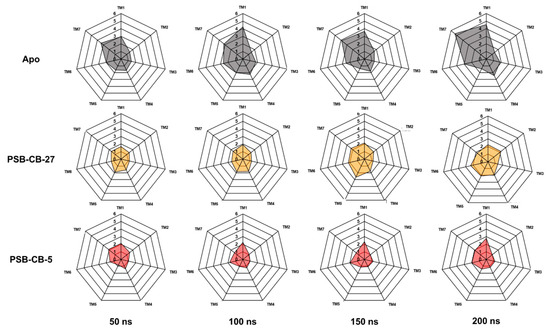
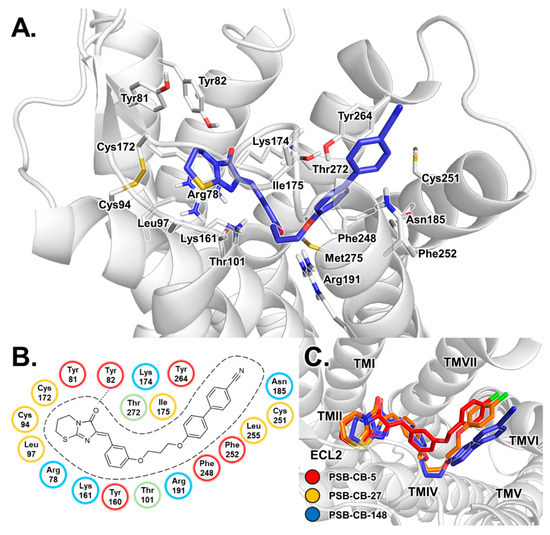
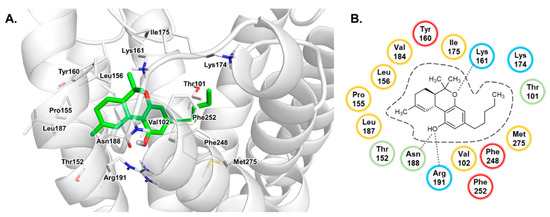
Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18
by
Alexander Neumann, Viktor Engel, Andhika B. Mahardhika, Clara T. Schoeder, Vigneshwaran Namasivayam, Katarzyna Kieć-Kononowicz and Christa E. Müller
Cited by 14 | Viewed by 5444
Abstract
GPR18 is an orphan G protein-coupled receptor (GPCR) expressed in cells of the immune system. It is activated by the cannabinoid receptor (CB) agonist ∆
9-tetrahydrocannabinol (THC). Several further lipids have been proposed to act as GPR18 agonists, but these results still
[...] Read more.
GPR18 is an orphan G protein-coupled receptor (GPCR) expressed in cells of the immune system. It is activated by the cannabinoid receptor (CB) agonist ∆
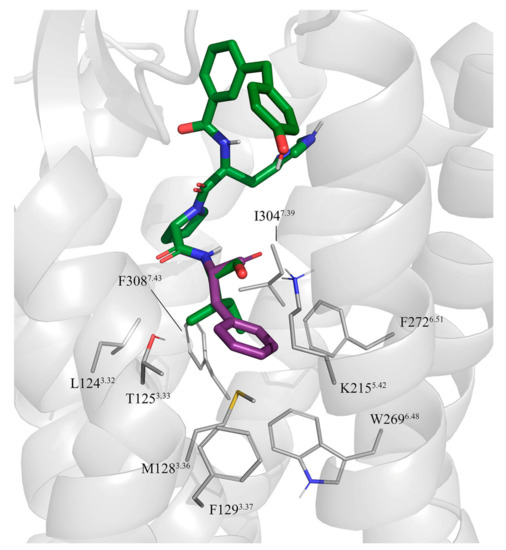
9-tetrahydrocannabinol (THC). Several further lipids have been proposed to act as GPR18 agonists, but these results still require unambiguous confirmation. In the present study, we constructed a homology model of the human GPR18 based on an ensemble of three GPCR crystal structures to investigate the binding modes of the agonist THC and the recently reported antagonists which feature an imidazothiazinone core to which a (substituted) phenyl ring is connected via a lipophilic linker. Docking and molecular dynamics simulation studies were performed. As a result, a hydrophobic binding pocket is predicted to accommodate the imidazothiazinone core, while the terminal phenyl ring projects towards an aromatic pocket. Hydrophobic interaction of Cys251 with substituents on the phenyl ring could explain the high potency of the most potent derivatives. Molecular dynamics simulation studies suggest that the binding of imidazothiazinone antagonists stabilizes transmembrane regions TM1, TM6 and TM7 of the receptor through a salt bridge between Asp118 and Lys133. The agonist THC is presumed to bind differently to GPR18 than to the distantly related CB receptors. This study provides insights into the binding mode of GPR18 agonists and antagonists which will facilitate future drug design for this promising potential drug target.
Full article
►▼
Show Figures
Open AccessArticle
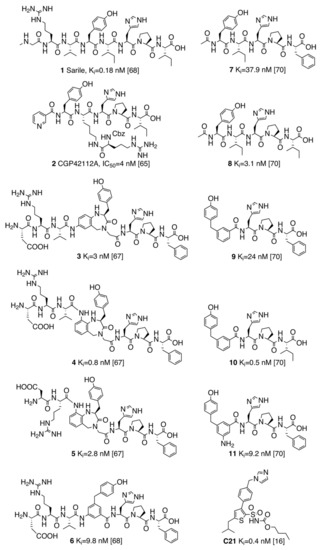
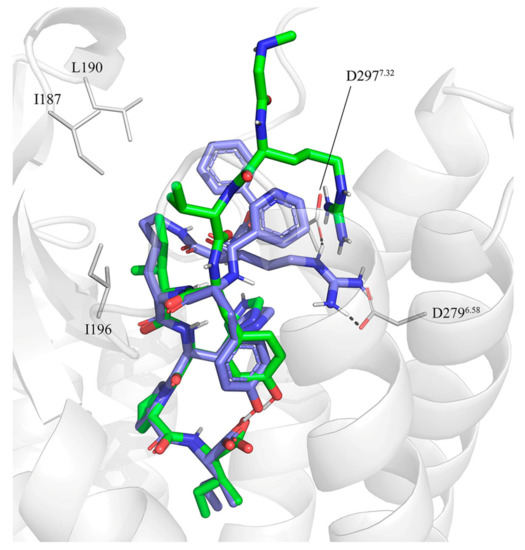
Evolution of Angiotensin Peptides and Peptidomimetics as Angiotensin II Receptor Type 2 (AT2) Receptor Agonists
by
Silvana Vasile, Anders Hallberg, Jessica Sallander, Mathias Hallberg, Johan Åqvist and Hugo Gutiérrez-de-Terán
Cited by 13 | Viewed by 4572
Abstract
Angiotensin II receptor type 1 and 2 (AT1R and AT2R) are two G-protein coupled receptors that mediate most biological functions of the octapeptide Angiotensin II (Ang II). AT2R is upregulated upon tissue damage and its activation by selective AT2R agonists has become a
[...] Read more.
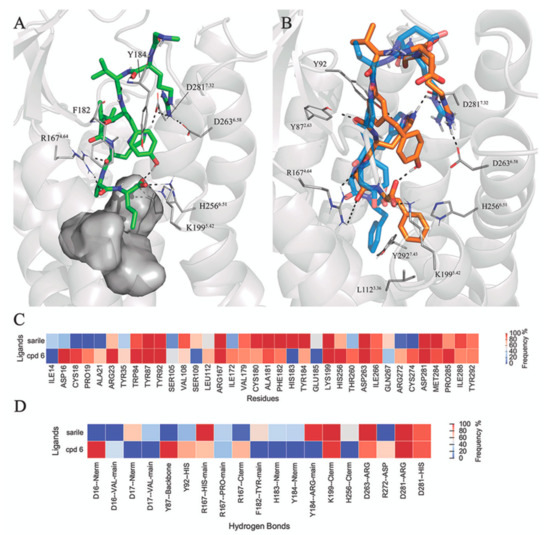
Angiotensin II receptor type 1 and 2 (AT1R and AT2R) are two G-protein coupled receptors that mediate most biological functions of the octapeptide Angiotensin II (Ang II). AT2R is upregulated upon tissue damage and its activation by selective AT2R agonists has become a promising approach in the search for new classes of pharmaceutical agents. We herein analyzed the chemical evolution of AT2R agonists starting from octapeptides, through shorter peptides and peptidomimetics to the first drug-like AT2R-selective agonist, C21, which is in Phase II clinical trials and aimed for idiopathic pulmonary fibrosis. Based on the recent crystal structures of AT1R and AT2R in complex with sarile, we identified a common binding model for a series of 11 selected AT2R agonists, consisting of peptides and peptidomimetics of different length, affinity towards AT2R and selectivity versus AT1R. Subsequent molecular dynamics simulations and free energy perturbation (FEP) calculations of binding affinities allowed the identification of the bioactive conformation and common pharmacophoric points, responsible for the key interactions with the receptor, which are maintained by the drug-like agonists. The results of this study should be helpful and facilitate the search for improved and even more potent AT2R-selective drug-like agonists.
Full article
►▼
Show Figures
Open AccessReview
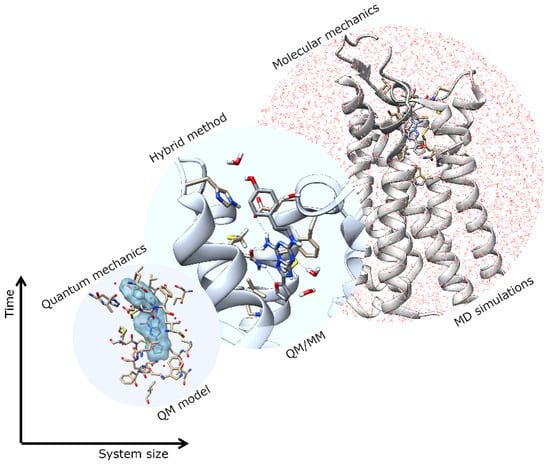
Multiscale Molecular Modeling in G Protein-Coupled Receptor (GPCR)-Ligand Studies
by
Pratanphorn Nakliang, Raudah Lazim, Hyerim Chang and Sun Choi
Cited by 8 | Viewed by 3895
Abstract
G protein-coupled receptors (GPCRs) are major drug targets due to their ability to facilitate signal transduction across cell membranes, a process that is vital for many physiological functions to occur. The development of computational technology provides modern tools that permit accurate studies of
[...] Read more.
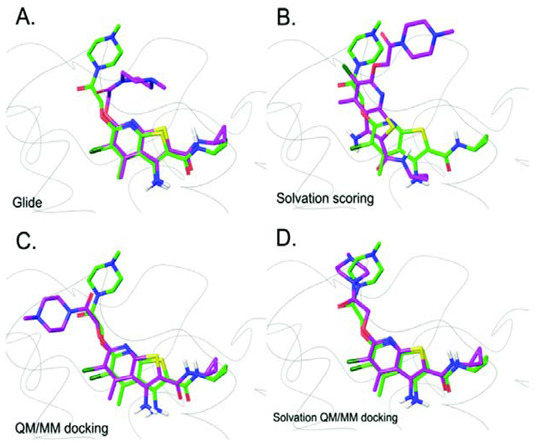
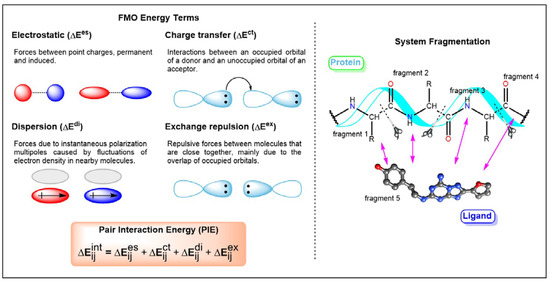
G protein-coupled receptors (GPCRs) are major drug targets due to their ability to facilitate signal transduction across cell membranes, a process that is vital for many physiological functions to occur. The development of computational technology provides modern tools that permit accurate studies of the structures and properties of large chemical systems, such as enzymes and GPCRs, at the molecular level. The advent of multiscale molecular modeling permits the implementation of multiple levels of theories on a system of interest, for instance, assigning chemically relevant regions to high quantum mechanics (QM) level of theory while treating the rest of the system using classical force field (molecular mechanics (MM) potential). Multiscale QM/MM molecular modeling have far-reaching applications in the rational design of GPCR drugs/ligands by affording precise ligand binding configurations through the consideration of conformational plasticity. This enables the identification of key binding site residues that could be targeted to manipulate GPCR function. This review will focus on recent applications of multiscale QM/MM molecular simulations in GPCR studies that could boost the efficiency of future structure-based drug design (SBDD) strategies.
Full article
►▼
Show Figures
Open AccessArticle
Machine Learning to Identify Flexibility Signatures of Class A GPCR Inhibition
by
Joseph Bemister-Buffington, Alex J. Wolf, Sebastian Raschka and Leslie A. Kuhn
Cited by 23 | Viewed by 5249
Abstract
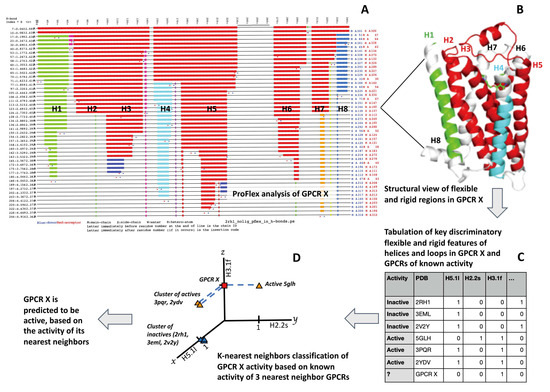
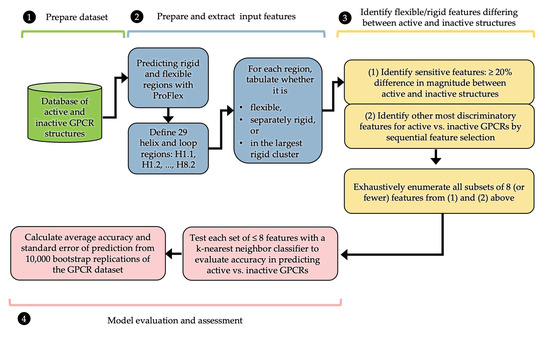
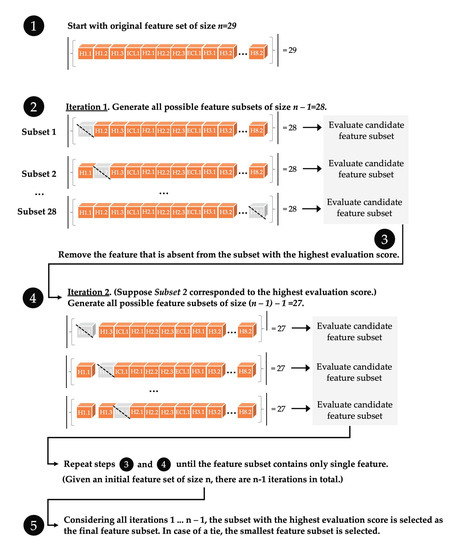
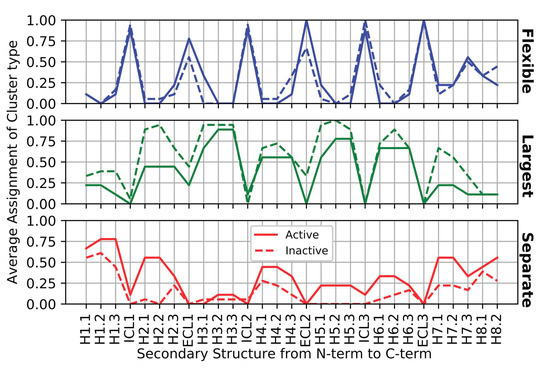
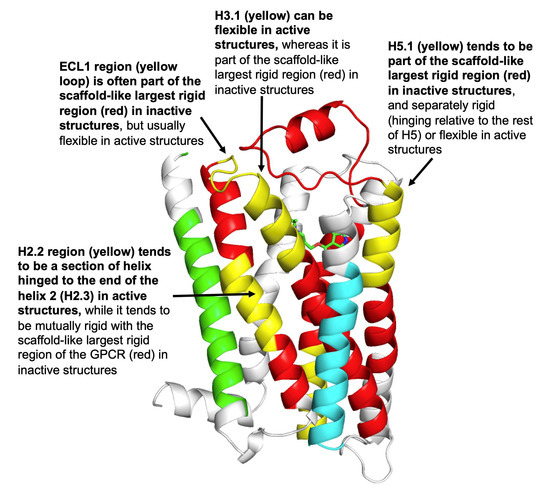
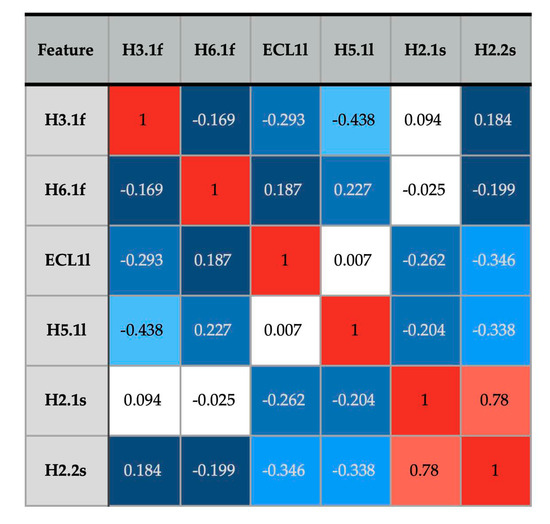
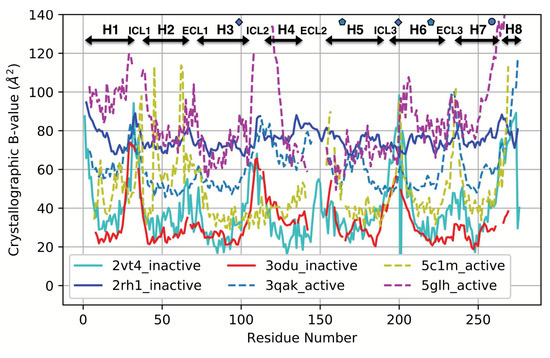
We show that machine learning can pinpoint features distinguishing inactive from active states in proteins, in particular identifying key ligand binding site flexibility transitions in GPCRs that are triggered by biologically active ligands. Our analysis was performed on the helical segments and loops
[...] Read more.
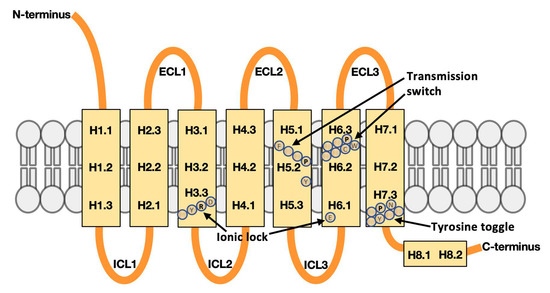
We show that machine learning can pinpoint features distinguishing inactive from active states in proteins, in particular identifying key ligand binding site flexibility transitions in GPCRs that are triggered by biologically active ligands. Our analysis was performed on the helical segments and loops in 18 inactive and 9 active class A G protein-coupled receptors (GPCRs). These three-dimensional (3D) structures were determined in complex with ligands. However, considering the flexible versus rigid state identified by graph-theoretic ProFlex rigidity analysis for each helix and loop segment with the ligand removed, followed by feature selection and k-nearest neighbor classification, was sufficient to identify four segments surrounding the ligand binding site whose flexibility/rigidity accurately predicts whether a GPCR is in an active or inactive state. GPCRs bound to inhibitors were similar in their pattern of flexible versus rigid regions, whereas agonist-bound GPCRs were more flexible and diverse. This new ligand-proximal flexibility signature of GPCR activity was identified without knowledge of the ligand binding mode or previously defined switch regions, while being adjacent to the known transmission switch. Following this proof of concept, the ProFlex flexibility analysis coupled with pattern recognition and activity classification may be useful for predicting whether newly designed ligands behave as activators or inhibitors in protein families in general, based on the pattern of flexibility they induce in the protein.
Full article
►▼
Show Figures
Planned Papers
The below list represents only planned manuscripts. Some of these
manuscripts have not been received by the Editorial Office yet. Papers
submitted to MDPI journals are subject to peer-review.
1.
Type: Article
Tentative Title: Multiple component reaction based dynamic libraries: an application on G Protein-Coupled Receptors
Author: Hugo Gutierrez de Teran et al.
2.
Type: Article
Tentative Title: Molecular dynamics simulation of ligand recognition at purinergic G protein-coupled receptors
Author: Kenneth Jacobson et al.
3.
Type: Review
Tentative Title: Computational methods to predict allosteric binding sites in GPCRs
Authors: Almudena Pino-Angeles, Irina G. Tikhonova
4.
Type: Article
Authors: Stefano Moro, et al.
Affiliation: Molecular Modeling Section, Department of Pharmaceutical and Pharmacological Sciences, University of Padova, 35131 Padova, Italy
5.
Type: Article
Authors: Irina Kufareva, et al.
Affiliation: University of California, San Diego, Skaggs School of Pharmacy and Pharmaceutical Sciences, La Jolla, CA 92093, USA
6.
Type: Article
Authors: Masha Niv, et al.
Affiliation: The Institute of Biochemistry, Food Science and Nutrition, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot 76100, Israel
7.
Type: Review
Authors: Nagarajan Vaidehi, et al.
Affiliation: Department of Computational & Quantitative Medicine, Beckman Research Institute of the City of Hope, Duarte, CA, USA
8.
Type: Review
Authors: Andrea Townsend-Nicholson, Alexander Heifetz et al.
Affiliation: Institute of Structural and Molecular Biology, University College London, UK
Title: Exploring efficacy at dopamine D2 receptor by molecular dynamics simulations
Authors: Jesús Giraldo; et al.
Affiliation: Laboratory of Molecular Neuropharmacology and Bioinformatics, Unitat de Bioestadística, Institut de Neurociències, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
Title: Multiscale modeling in exploring GPCRs drug design
Authors: Sun Choi; Pratanphorn Nakliang; Donghyuk Suh; Hyerim Chang
Affiliation: College of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, Korea



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}