



Genes 2021, 12(6), 831; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060831 - 28 May 2021
Cited by 9 | Viewed by 4030
Abstract
►
Show Figures
Heritable Connective Tissue Disorders (HCTD) show an overlap in the physical features that can evolve in childhood. It is unclear to what extent children with HCTD experience burden of disease. This study aims to quantify fatigue, pain, disability and general health with standardized
[...] Read more.
Heritable Connective Tissue Disorders (HCTD) show an overlap in the physical features that can evolve in childhood. It is unclear to what extent children with HCTD experience burden of disease. This study aims to quantify fatigue, pain, disability and general health with standardized validated questionnaires. Methods. This observational, multicenter study included 107 children, aged 4–18 years, with Marfan syndrome (MFS), 58%; Loeys-Dietz syndrome (LDS), 7%; Ehlers-Danlos syndromes (EDS), 8%; and hypermobile Ehlers-Danlos syndrome (hEDS), 27%. The assessments included PROMIS Fatigue Parent–Proxy and Pediatric self-report, pain and general health Visual-Analogue-Scales (VAS) and a Childhood Health Assessment Questionnaire (CHAQ). Results. Compared to normative data, the total HCTD-group showed significantly higher parent-rated fatigue T-scores (M = 53 (SD = 12), p = 0.004, d = 0.3), pain VAS scores (M = 2.8 (SD = 3.1), p < 0.001, d = 1.27), general health VAS scores (M = 2.5 (SD = 1.8), p < 0.001, d = 2.04) and CHAQ disability index scores (M = 0.9 (SD = 0.7), p < 0.001, d = 1.23). HCTD-subgroups showed similar results. The most adverse sequels were reported in children with hEDS, whereas the least were reported in those with MFS. Disability showed significant relationships with fatigue (p < 0.001, rs = 0.68), pain (p < 0.001, rs = 0.64) and general health (p < 0.001, rs = 0.59). Conclusions. Compared to normative data, children and adolescents with HCTD reported increased fatigue, pain, disability and decreased general health, with most differences translating into very large-sized effects. This new knowledge calls for systematic monitoring with standardized validated questionnaires, physical assessments and tailored interventions in clinical care.
Full article
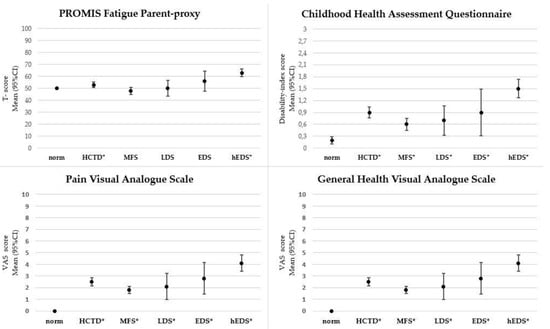
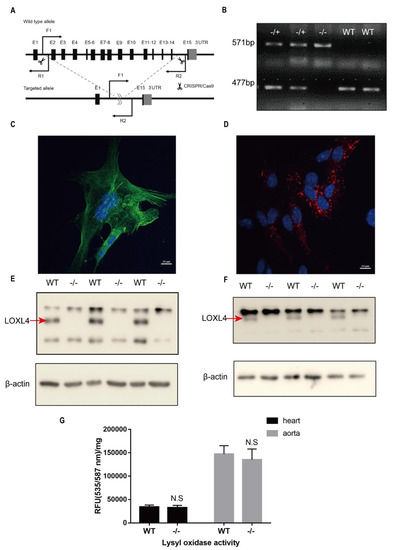
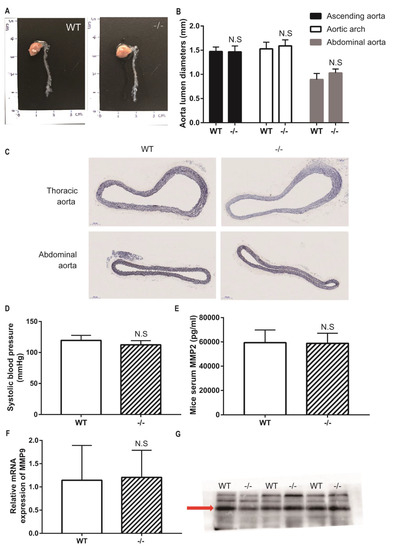
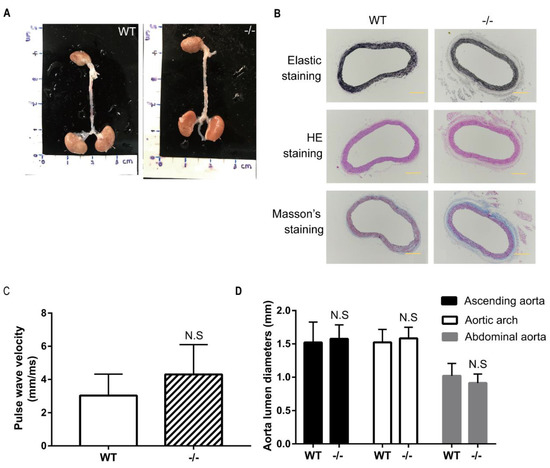
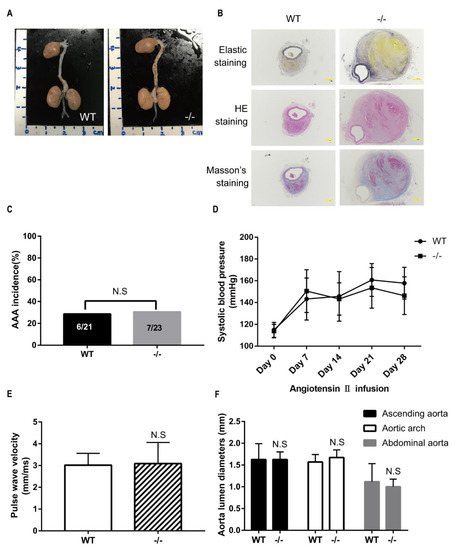
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}