Lipase-Catalyzed Kinetic Resolution of Aryltrimethylsilyl Chiral Alcohols

Abstract
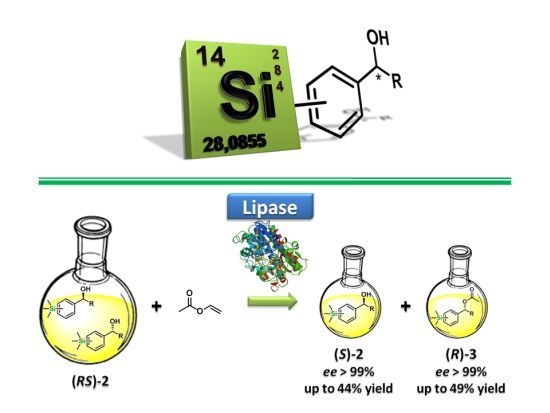
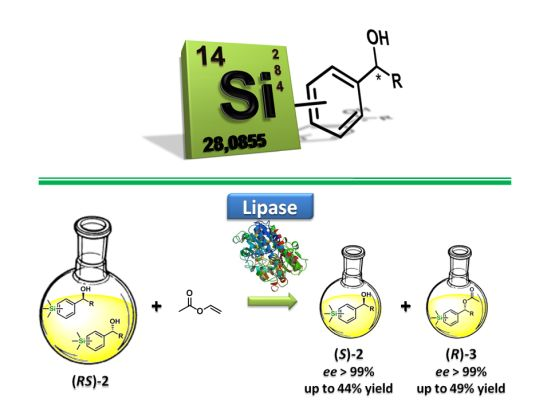
:
1. Introduction
2. Results and Discussion
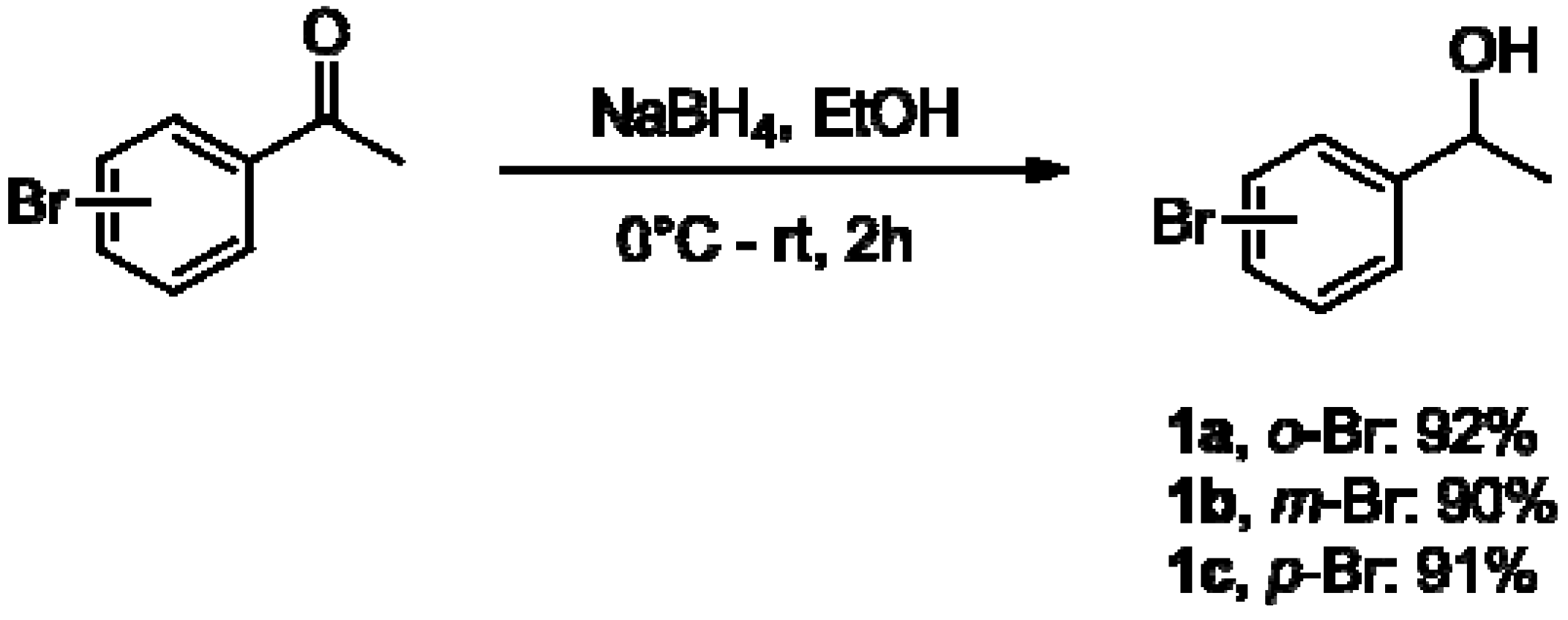
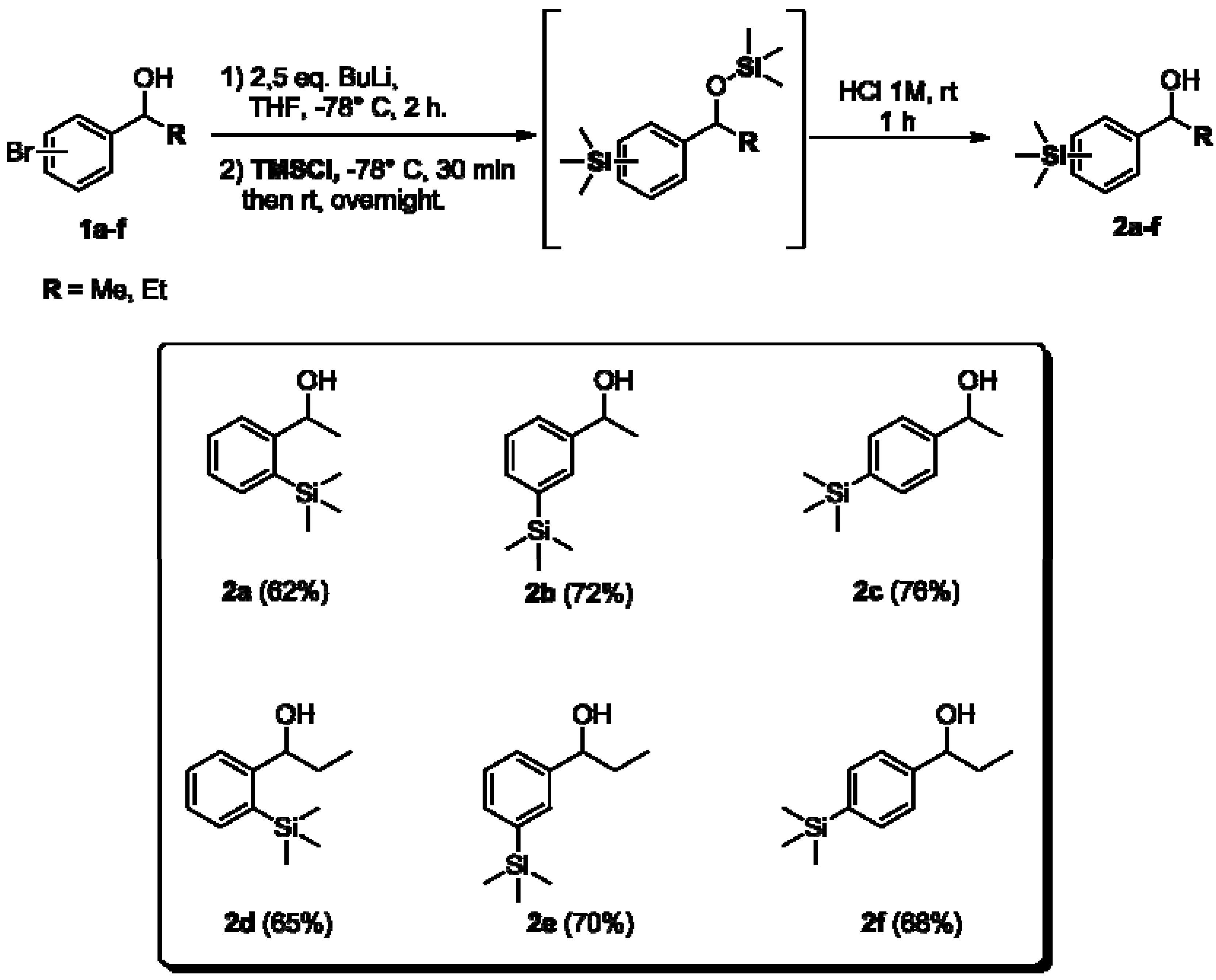
2.1. Synthesis of Aryltrimethylsilyl Chiral Alcohols 2a–f



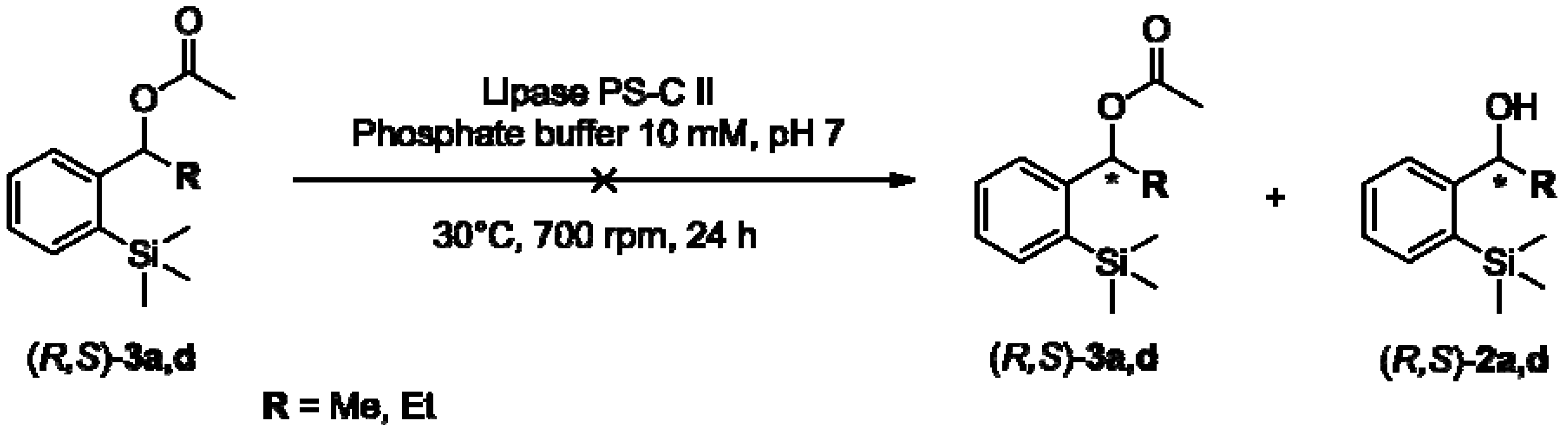
2.2. Enzymatic Kinetic Resolution of the Aryltrimethylsilyl Chiral Alcohols 2a–f

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Lipase (source) | ee (%) b | c (%) c | E d | |
| (S)-2c | (R)-3c | ||||
| 1 | Mucor javanicus (Amano M) | – | – | – | – |
| 2 | Candida cylindracea (Fluka) | – | – | – | – |
| 3 | Candida antarctica (CAL-B, Novozym 435) | 74 | >99 | 43 | >200 |
| 4 | Pseudomonas cepacia (Amano PS) | 48 | >99 | 33 | >200 |
| 5 | Aspergillus niger (Amano A) | – | – | – | – |
| 6 | Pseudomonas cepacia (Amano PS-D I) | 98 | >99 | 49 | >200 |
| 7 | Candida rugosa (Amano, type VII) | – | – | – | – |
| 8 | Pseudomonas cepacia (Amano PS-C II) | >99 | >99 | 50 | >200 |
| 9 | Mucor meihei (Sigma) | – | – | – | – |
| 10 | Pseudomonas fluorescens (Amano AK) | 58 | >99 | 37 | >200 |
| 11 | Porcine pancreas (Sigma, type II) | – | – | – | – |
| Entry | Substrate (mmol) | Amano PS-C II (mg) | Time (h) | ee (%) b | c (%) c | E d | |
|---|---|---|---|---|---|---|---|
| (S)-2c | (R)-3c | ||||||
| 1 | 0.05 | 2 | 6 | >99 | >99 | 50 | >200 |
| 2 | 0.05 | 2 | 3 | >99 | >99 | 50 | >200 |
| 3 | 0.05 | 1 | 6 | >99 | >99 | 50 | >200 |
| 4 | 0.05 | 1 | 3 | >99 | >99 | 50 | >200 |
| 5 | 0.05 | 1 | 1.5 | 86 | >99 | 46 | >200 |
| 6 | 0.1 | 1 | 6 | >99 | >99 | 50 | >200 |
| 7 | 0.1 | 1 | 3 | 83 | >99 | 45 | >200 |
| 8 | 0.1 | 1 | 1.5 | 48 | >99 | 33 | >200 |
| Entry | Substrate | Time (h) | ee (%) b | c (%) c | E d | |
|---|---|---|---|---|---|---|
| (S)-2 | (R)-3 | |||||
| 1 | (RS)-2a | 3 | – | – | – | – |
| 2 | (RS)-2a | 16 | – | – | – | – |
| 3 | (RS)-2b | 3 | 87 | >99 | 47 | >200 |
| 4 | (RS)-2b | 16 | >99 | >99 | 50 | >200 |
| 5 | (RS)-2d | 3 | – | – | – | – |
| 6 | (RS)-2d | 16 | – | – | – | – |
| 7 | (RS)-2e | 3 | 54 | >99 | 35 | >200 |
| 8 | (RS)-2ee | 24 | >99 | >99 | 50 | >200 |
| 9 | (RS)-2f | 3 | 96 | >99 | 49 | >200 |
| 10 | (RS)-2f | 16 | >99 | >99 | 50 | >200 |


| Entry | Substrate | Isolated yield % (ee %) b | |
|---|---|---|---|
| 1 | (RS)-2b |  |  |
| 2 | (RS)-2cc |  |  |
| 3 | (RS)-2ed |  |  |
| 4 | (RS)-2f |  |  |
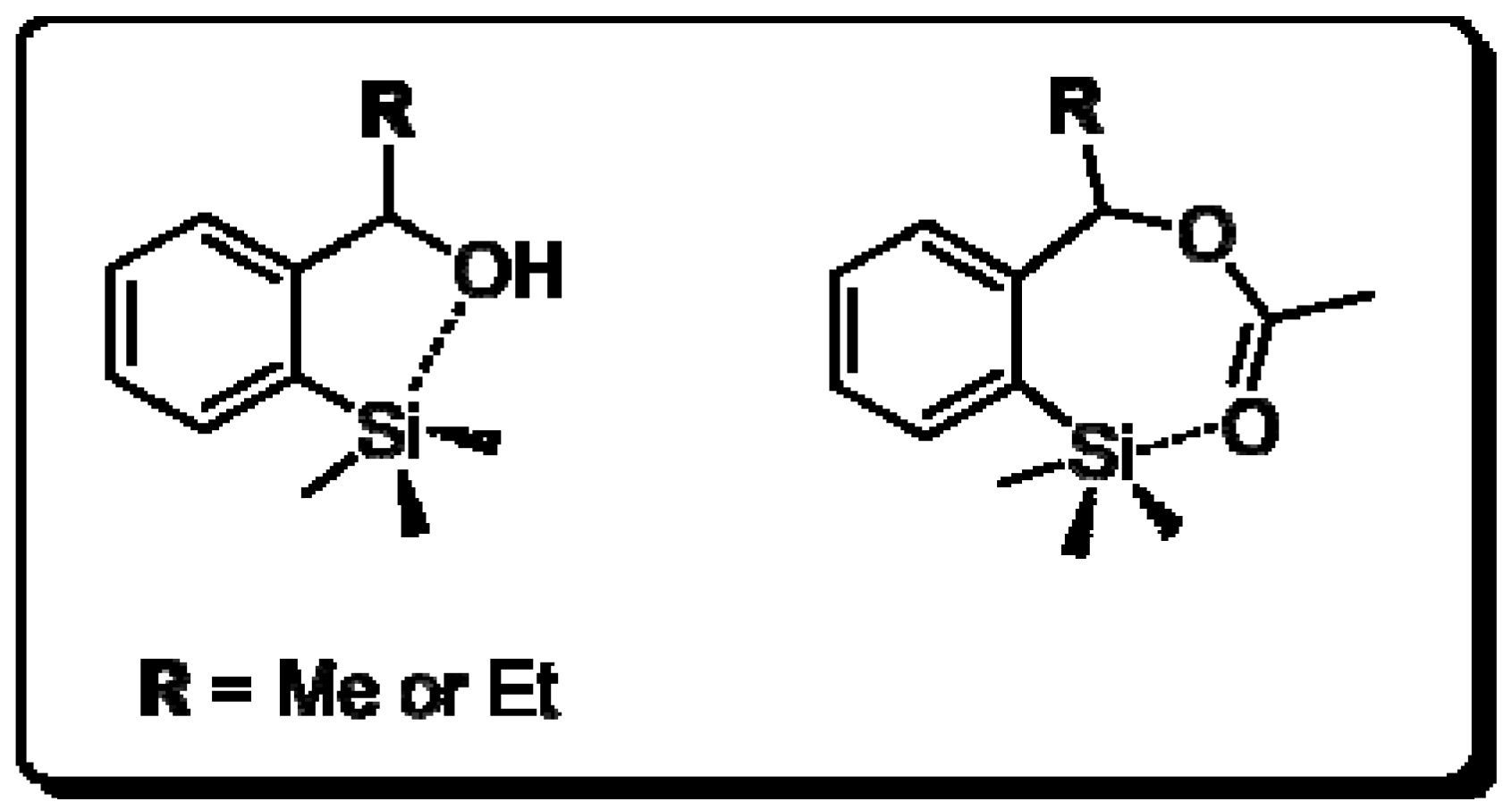
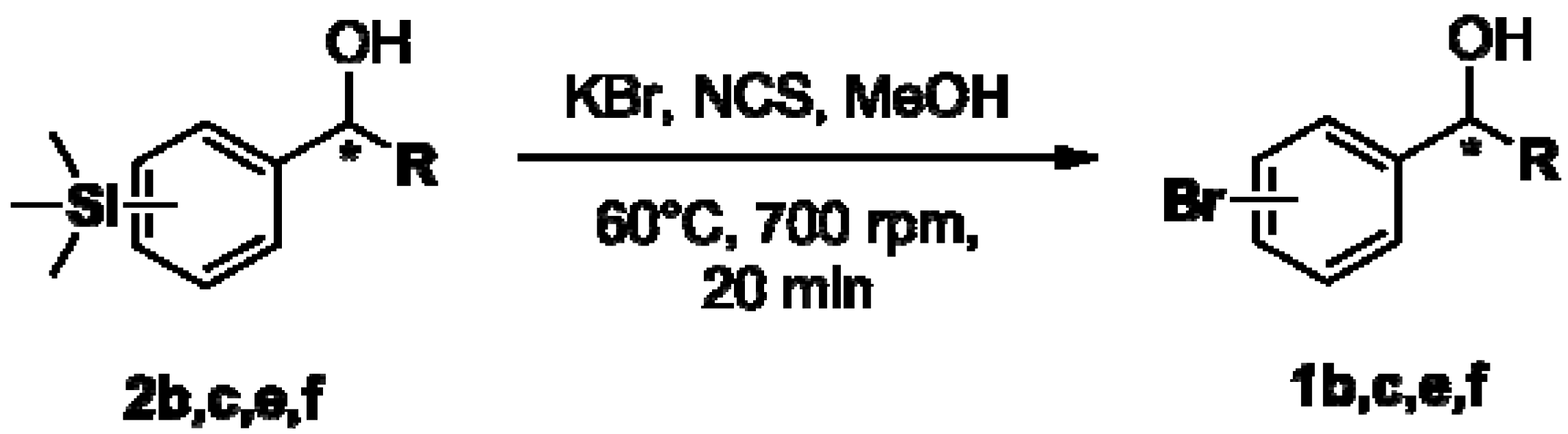
2.3. Determination of Absolute Configuration of Enantioenriched Aryltrimethylsilyl Chiral Alcohols (2b, 2c, 2e and 2f)

3. Experimental
3.1. General Methods
3.2. Synthetic Procedures
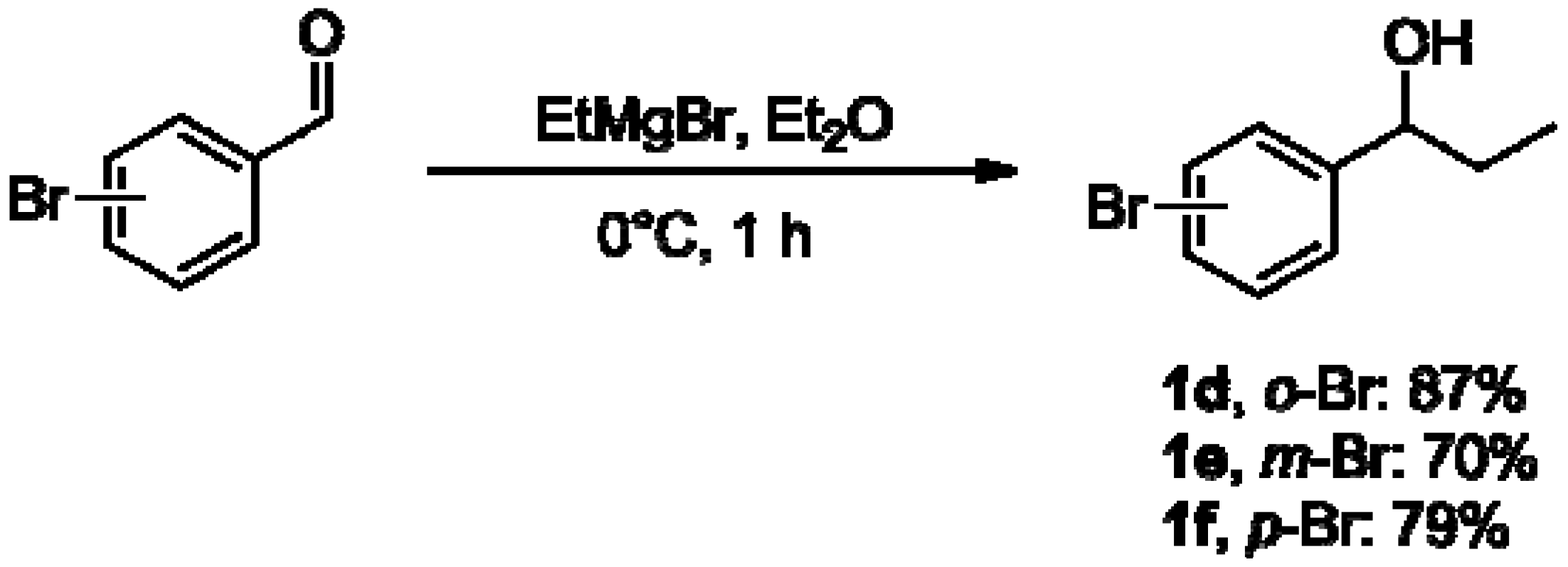
3.2.1. General Procedure for the Preparation of 1-(Bromophenyl)ethanols 1a–c [35,47]
3.2.2. General Procedure for the Preparation of 1-(Bromophenyl)propanols 1d–f [49]
3.2.3. General Procedure for the Preparation of Aryltrimethylsilyl Chiral Alcohols 2a–f [11,50]
3.2.4. General Procedure for the Preparation of Aryltrimethylsilyl Chiral Acetates 3a–f [51]
3.3. Determination of the Enantiomeric Excess (ee)
- (2a): tR = 19.8 min for (S) and tR = 20.9 min for (R);
- (2b): tR = 19.6 min for (S) and tR = 20.1 min for (R);
- (2c): tR = 26.5 min for (S) and tR = 25.6 min for (R);
- (2d): tR = 21.6 min for (S) and tR = 22.6 min for (R);
- (2e): tR = 25.0 min for (S) and tR = 24.4 min for (R);
- (2f): tR = 31.2 min for (S) and tR = 30.3 min for (R);
- (3a): tR = 16.0 min for (S) and tR = 16.2 min for (R);
- (3b): tR = 16.2 min for (S) and tR = 16.8 min for (R);
- (3c): tR = 23.7 min for (S) and tR = 25.9 min for (R);
- (3d): tR = 63.4 min for (S) and tR = 63.6 min for (R) (oven: 50 °C to 150 °C, 1 °C/min);
- (3e): tR = 22.0 min for (S) and tR = 20.4 min for (R);
- (3f): tR = 26.3 min for (S) and tR = 27.1 min for (R);
3.4. Enzymatic Procedures
3.4.1. General Procedure for Small Scale Enzymatic Reactions
3.4.2. General Procedure for Preparative-Scale Enzymatic Reactions
- (S)-2b: isolated yield = 44%; enantiomeric excess > 99%; [α]D20 = -37.0 (c = 1.0; CHCl3).
- (R)-3b: isolated yield = 49%; enantiomeric excess > 99%; [α]D20 = +70.8 (c = 1.0; CHCl3).
- (S)-2c: isolated yield = 31%; enantiomeric excess > 99%; [α]D20 = -31.2 (c = 1.0; CHCl3).
- (R)-3c: isolated yield = 46%; enantiomeric excess > 99%; [α]D20 = +93.9 (c = 1.0; CHCl3).
- (S)-2e: isolated yield = 38%; enantiomeric excess > 99%; [α]D20 = -32.9 (c = 1.0; CHCl3).
- (R)-3e: isolated yield = 49%; enantiomeric excess > 99%; [α]D20 = +76.8 (c = 1.0; CHCl3).
- (S)-2f: isolated yield = 39%; enantiomeric excess > 99%; [α]D20 = -34.1 (c = 1.0; CHCl3).
- (R)-3f: isolated yield = 34%; enantiomeric excess > 99%; [α]D20 = +91.3 (c = 1.0; CHCl3).
3.5. Determination of the Absolute Configuration
3.5.1. General Procedure for Transformation of Enantioenriched Aryltrimethylsilyl Chiral Alcohols 2b, 2c, 2e and 2f to Their Bromide Derivatives [52]
3.5.2. Determination of Absolute Configuration of Enantioenriched Aryltrimethylsilyl Chiral Alcohols 2b, 2c, 2e and 2f
4. Conclusions
Acknowledgments
References and Notes
- Colvin, E.W. Silicon Reagents in Organic Synthesis, 1st ed; Academic Press: San Diego, CA, USA, 1988; pp. 7–137. [Google Scholar]
- Patai, S.; Rappoport, Z. The Chemistry of Organic Silicon Compounds, 1st ed; Wiley: New York, NY, USA, 1998; Volume 2, pp. 1667–1793. [Google Scholar]
- Gilman, H.; Dunn, G.E. Relationships between analgous organic compounds of silicon and carbon. Chem. Rev. 1953, 52, 77–115. [Google Scholar] [CrossRef]
- Burkhard, C.A.; Rochow, E.G.; Booth, H.S.; Hartt, J. The present state of organosilicon chemistry. Chem. Rev. 1947, 41, 97–149. [Google Scholar]
- Yoshida, H.; Sugiura, S.; Kunai, A. Facile synthesis of N-alkyl-N′-arylimidazolium salts via addition of imidazoles to arynes. Org. Lett. 2002, 4, 2767–2769. [Google Scholar] [CrossRef]
- Dockendorff, C.; Sahli, S.; Olsen, M.; Milhau, L.; Lautens, M. Synthesis of dihydronaphthalenes via aryne Diels-Alder reactions: Scope and diastereoselectivity. J. Am. Chem. Soc. 2005, 127, 15028–15029. [Google Scholar] [CrossRef]
- Liu, Z.; Larock, R.C. Highly efficient route to fused polycyclic aromatics via palladium-catalyzed aryne annulation by aryl halides. J. Org. Chem. 2007, 72, 223–232. [Google Scholar] [CrossRef]
- Xie, C.; Liu, L.; Zhang, Y.; Xu, P. Copper-catalyzed Alkyne-aryne and alkyne-alkene-aryne coupling reactions. Org. Lett. 2008, 10, 2393–2396. [Google Scholar] [CrossRef]
- Gebara, K.S.; Casagrande, G.A.; Raminelli, C. An efficient fluoride-mediated O-arylation of sterically hindered halophenols with silylaryl triflates under mild reaction conditions. Tetrahedron Lett. 2011, 52, 2849–2852. [Google Scholar] [CrossRef]
- McAusland, D.; Seo, S.; Pintori, D.G.; Finlayson, J.; Greaney, M.F. The benzyne Fischer-indole reaction. Org. Lett. 2011, 13, 3667–3669. [Google Scholar] [CrossRef]
- Brenzovich, W.E.; Brazeau, J.-F.; Toste, F.D. Gold-catalyzed oxidative coupling reactions with aryltrimethylsilanes. Org. Lett. 2010, 12, 4728–4731. [Google Scholar] [CrossRef]
- Ball, L.T.; Green, M.; Lloyd-Jones, G.C.; Russell, C.A. Arylsilanes: Application to gold-catalyzed oxyarylation of alkenes. Org. Lett. 2010, 12, 4724–4727. [Google Scholar]
- Suzawa, K.; Ueno, M.; Wheatley, A.E.H.; Kondo, Y. Phosphazene base-promoted functionalization of aryltrimethylsilanes. Chem. Commun. 2006, 4850–4852. [Google Scholar]
- Nakao, Y.; Imanaka, H.; Sahoo, A.K.; Yada, A.; Hiyama, T. Alkenyl- and aryl[2-(hydroxymethyl)phenyl]dimethylsilanes: An entry to tetraorganosilicon reagents for the silicon-based cross-coupling reaction. J. Am. Chem. Soc. 2005, 127, 6952–6953. [Google Scholar]
- Nakao, Y.; Imanaka, H.; Chen, J.; Yada, A.; Hiyama, T. Synthesis and cross-coupling reaction of alkenyl[(2-hydroxymethyl)phenyl]dimethylsilanes. J. Organomet. Chem. 2007, 692, 585–603. [Google Scholar] [CrossRef]
- Chen, J.; Tanaka, M.; Sahoo, A.K.; Takeda, M.; Yada, A.; Nakao, Y.; Hiyama, T. Synthesis of biaryls and oligoarenes using aryl[2-(hydroxymethyl)phenyl]dimethylsilanes. Bull. Chem. Soc. Jpn. 2010, 83, 554–569. [Google Scholar] [CrossRef]
- Nakao, Y.; Takeda, M.; Matsumoto, T.; Hiyama, T. Cross-coupling reactions through the intramolecular activation of alkyl(triorgano)silanes. Angew. Chem. Int. Ed. 2010, 49, 4447–4450. [Google Scholar] [CrossRef]
- Denmark, S.E. The interplay of invention, discovery, development, and application in organic synthetic methodology: A case study. J. Org. Chem. 2009, 74, 2915–2927. [Google Scholar] [CrossRef]
- Rauf, W.; Brown, J.M. Catalytic amide-mediated methyl transfer from silanes to alkenes in fujiwara-moritani oxidative coupling. Angew. Chem. Int. Ed. 2008, 47, 4228–4230. [Google Scholar] [CrossRef]
- Tacke, R.; Brakmann, S.; Wuttke, F. Stereoselective microbial reduction of racemic acetyl(t-butyl)methylphenylsilane by Trigonopsis variabilis (DSM 70714) and Corynebacterium dioxydans (ATCC 21766). J. Organomet. Chem. 1991, 403, 29–41. [Google Scholar]
- Yamanaka, H.; Kawamoto, T.; Tanaka, A. Efficient preparation of optically active p-trimethylsilylphenylalanine by using cell-free extract of Blastobacter sp. A17p-4. J. Ferment. Bioeng. 1997, 84, 181–184. [Google Scholar]
- Zani, P. Biotransformations of organosilicon compounds: enantioselective reduction of acyl silanes by means of baker’s yeast. J. Mol. Catal. B: Enzym. 2001, 11, 279–285. [Google Scholar] [CrossRef]
- Li, N.; Zong, M.-H.; Peng, H.-S.; Wu, H.-C.; Liu, C. (R)-Oxynitrilase-catalyzed synthesis of (R)-2-trimethylsilyl-2-hydroxyl-ethylcyanide. J. Mol. Catal. B: Enzym. 2003, 22, 7–12. [Google Scholar] [CrossRef]
- Maraite, A.; Ansorge-Schumacher, M. B.; Ganchegui, B.; Leitner, W.; Grogan, G. On the biocatalytic cleavage of silicon-oxygen bonds: A substrate structural approach to investigating the cleavage of protecting group silyl ethers by serine-triad hydrolases. J. Mol. Catal. B: Enzym. 2009, 56, 24–28. [Google Scholar] [CrossRef]
- Frampton, M.B.; Simionescu, R.; Dudding, T.; Zelisko, P.M. The enzymatic cleavage of Si–O bonds: A kinetic analysis of the biocatalyzed hydrolysis of phenyltrimethoxysilane. J. Mol. Catal. B: Enzym. 2010, 66, 105–112. [Google Scholar] [CrossRef]
- Bassindale, A.R.; Brandstadt, K.F.; Lane, T.H.; Taylor, P.G. Enzyme-catalysed siloxane bond formation. J. Inorg. Biochem. 2003, 96, 401–406. [Google Scholar] [CrossRef]
- Abbate, V.; Bassindale, A.R.; Brandstadt, K.F.; Lawson, R.; Taylor, P.G. Enzyme mediated silicon–oxygen bond formation; the use of Rhizopus oryzae lipase, lysozyme and phytase under mild conditions. Dalton Trans. 2010, 39, 9361–9368. [Google Scholar] [CrossRef]
- Abbate, V.; Bassindale, A.R.; Brandstadt, K.F.; Taylor, P.G. A large scale enzyme screen in the search for new methods of silicon-oxygen bond formation. J. Inog. Biochem. 2011, 105, 268–275. [Google Scholar] [CrossRef]
- Djerourou, A.-H.; Blanco, L. Synthesis of optically active 2-sila-1,3-propanediols derivatives by enzymatic transesterification. Tetrahedron Lett. 1991, 32, 6325–6326. [Google Scholar] [CrossRef]
- Fukui, T.; Kawamoto, T.; Tanaka, A. Enzymatic preparation of optically active silylmethanol derivatives having a stereogenic silicon atom by hydrolase-catalyzed enantioselective esterification. Tetrahedron: Asymmetry 1994, 5, 73–82. [Google Scholar] [CrossRef]
- Burgess, K.; Jennings, L.D. Enantioselective esterifications of unsaturated alcohols mediated by a lipase prepared from Pseudomonas sp. J. Am. Chem. Soc. 1991, 113, 6129–6139. [Google Scholar] [CrossRef]
- Allevi, P.; Ciuffreda, P.; Anastasia, M. Lipase catalysed resolution of (R)- and (S)-l-trimethylsilyl-l-alkyn-3-ols—Useful intermediates for the synthesis of optically active γ-lactones. Tetrahedron: Asymmetry 1997, 8, 93–99. [Google Scholar] [CrossRef]
- Ratnayake, A.S.; Hemscheidt, T. Olefin cross-metathesis as a tool in natural product degradation. The stereochemistry of (+)-Falcarindiol. Org. Lett. 2002, 4, 4667–4669. [Google Scholar] [CrossRef]
- Brawn, R.A.; Panek, J.S. Preparation and use of enantioenriched allenylsilanes for the stereoselective synthesis of homopropargylic ethers. Org. Lett. 2007, 9, 2689–2692. [Google Scholar] [CrossRef]
- Andrade, L.H.; Omori, A.T.; Porto, A.L.M.; Comasseto, J.V. Asymmetric synthesis of arylselenoalcohols by means of the reduction of organoseleno acetophenones by whole fungal cells. J. Mol. Catal. B: Enzym. 2004, 29, 47–54. [Google Scholar] [CrossRef]
- Bianchi, D.; Battistel, E.; Bosetti, A.; Cesti, P.; Fekete, Z. Effects of chemical modification on stereoselectivity of Pseudomonas cepacia lipase. Tetrahedron: Asymmetry 1993, 4, 777–782. [Google Scholar] [CrossRef]
- Ignatyev, I.S. Intramolecular coordination of silicon in silyl formates: Spectroscopic evidence confirmed by ab initio calculations. J. Mol. Struct. 1991, 245, 139–145. [Google Scholar] [CrossRef]
- Tamao, K.; Asahara, M.; Sun, G.-R.; Kawachi, A. Synthesis, structure, and reactivity of 1,ω-bis(pseudo-pentacoordinated) 1,ω-difluoro-oligosilanes bearing 8-(dimethylamino)-1-naphthyl groups. J. Organomet. Chem. 1999, 574, 193–205. [Google Scholar]
- Tamao, K.; Asahara, M.; Saeki, T.; Toshimitsu, A. Enhanced nucleophilicity of ambiphilic silylene and silylenoid bearing 8-(dimethylamino)-1-naphthyl group. J. Organomet. Chem. 2000, 600, 118–123. [Google Scholar]
- Pongor, G.; Kolos, Z.; Szalay, R.; Knausz, D. Pseudo-pentacoordination in silylcarbamates: structure-based prediction of silylating power. J. Mol. Struct. (THEOCHEM) 2005, 714, 87–97. [Google Scholar] [CrossRef]
- Szalay, R.; Pongor, G.; Harmat, V.; Böcskei, Z.; Knausz, D. Surprisingly great difference in reactivity depending upon the ring size: Solvolysis and molecular structure study of some N-trimethylsilylated cyclic ureas. J. Organomet. Chem. 2005, 690, 1498–1506. [Google Scholar] [CrossRef]
- Solís, A.; García, S.; Pérez, H.I.; Manjarrez, N.; Luna, H. Screening of liver acetone powders in the resolution of 1-phenylethanols and 1-phenylpropanols derivatives. Tetrahedron: Asymmetry 2008, 19, 549–553. [Google Scholar] [CrossRef]
- Salvia, N.A.; Chattopadhyay, S. Asymmetric reduction of halo-substituted arylalkanones with Rhizopus arrhizus. Tetrahedron: Asymmetry 2008, 19, 1992–1997. [Google Scholar] [CrossRef]
- Machado, L.L.; Lemos, T.L.G.; de Mattos, M.C.; de Oliveira, M.C.F.; de Gonzalo, G.; Gotor-Fernández, V.; Gotor, V. Immobilized Manihot esculenta preparation as a novel biocatalyst in the enantioselective acetylation of racemic alcohols. Tetrahedron: Asymmetry 2008, 19, 1419–1424. [Google Scholar] [CrossRef]
- Araújo, L.S.; Kagohara, E.; Garcia, T.P.; Pellizari, V.H.; Leandro, H.; Andrade, L.H. Screening of microorganisms producing cold-active oxidoreductases to be applied in enantioselective alcohol oxidation. An antarctic survey. Mar. Drugs 2011, 9, 889–905. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Weissfloch, A.N.E.; Rappaport, A.T.; Cuccia, L.A. A rule to predict which enantiomer of a secondary alcohol reacts faster in reactions catalyzed by cholesterol esterase, lipase from Pseudomonas cepacia, and lipase from Candida rugosa. J. Org. Chem. 1991, 56, 2656–2665. [Google Scholar]
- Andrade, L.H.; Polak, R.; Porto, A.L.M.; Schoenlein-Crusius, I.H.; Comasseto, J.V. Application of bioreduction by microorganisms in the enantioselective synthesis of alpha-substituted-1-phenylethanols. Lett. Org. Chem. 2006, 3, 613–618. [Google Scholar] [CrossRef]
- Nakamura, K.; Matsuda, T. Asymmetric reduction of ketones by the acetone powder of Geotrichum candidum. J. Org. Chem. 1998, 63, 8957–8964. [Google Scholar] [CrossRef]
- Zaidlewicz, M.; Wolan, A. Syntheses with organoboranes. XIII. Synthesis of ω-(4-bromophenyl)alkanoic acids and their borylation. J. Organomet. Chem. 2002, 657, 129–135. [Google Scholar] [CrossRef]
- Achmatowicz, B.; Raubo, P.; Wicha, J. Synthesis of four stereoisomeric tetrose derivatives from propargyl alcohol. One-carbon homologation of vinylsilanes via alpha, beta-epoxy silanes. J. Org. Chem. 1992, 57, 6593–6598. [Google Scholar] [CrossRef]
- Demasi, M.; Felicio, A.L.; Pacheco, A.O.; Leite, H.G.; Lima, C.; Andrade, L.H. Studies on terrein as a new class of proteasome inhibitors. J. Braz. Chem. Soc. 2010, 21, 299–305. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Stone, W.E.; Anderson, K.W. Regiospecific incorporation of bromine and iodine into phenols using (trimethylsilyl)phenol derivatives. J. Org. Chem. 1983, 48, 1542–1544. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Palmeira, D.J.; Abreu, J.C.; Andrade, L.H. Lipase-Catalyzed Kinetic Resolution of Aryltrimethylsilyl Chiral Alcohols. Molecules 2011, 16, 9697-9713. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules16119697
Palmeira DJ, Abreu JC, Andrade LH. Lipase-Catalyzed Kinetic Resolution of Aryltrimethylsilyl Chiral Alcohols. Molecules. 2011; 16(11):9697-9713. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules16119697
Chicago/Turabian StylePalmeira, Dayvson J., Juliana C. Abreu, and Leandro H. Andrade. 2011. "Lipase-Catalyzed Kinetic Resolution of Aryltrimethylsilyl Chiral Alcohols" Molecules 16, no. 11: 9697-9713. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules16119697