Chemical Methods for Peptide and Protein Production

Abstract
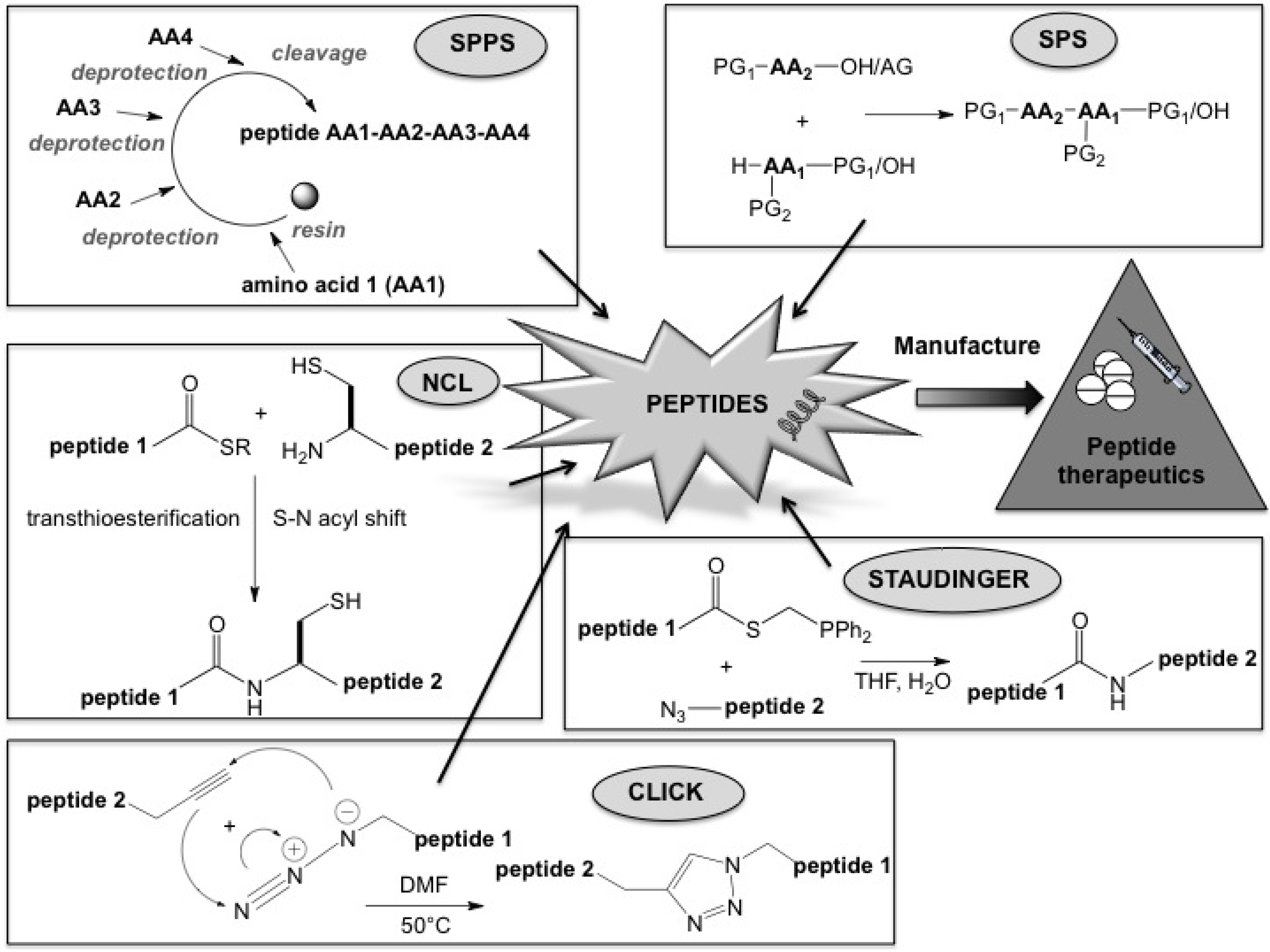
:1. The World of Peptides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Discovery |
|---|---|
| 1901 | First published synthesized dipeptide [2] |
| 1957 | Boc protecting group [12] |
| 1963 | SPPS discovery [3] |
| 1967 | HF cleavage [13] |
| 1968 | First automated solid phase synthesizer |
| 1970 | BHA resin [14]Fmoc protecting group [15] |
| 1973 | Wang resin [16] |
| 1976 | Preparative HPLC to purify peptides synthesized by SPPS |
| 1977 | Orthogonal protection [17] |
| 1987 | Rink resin [18]Sieber resin [19] |
| 1992 | Fast Boc protocol [20] |
| 1994 | NCL for protein and peptide synthesis [21] |
| 1996 | Pseudoprolines [22,23] |

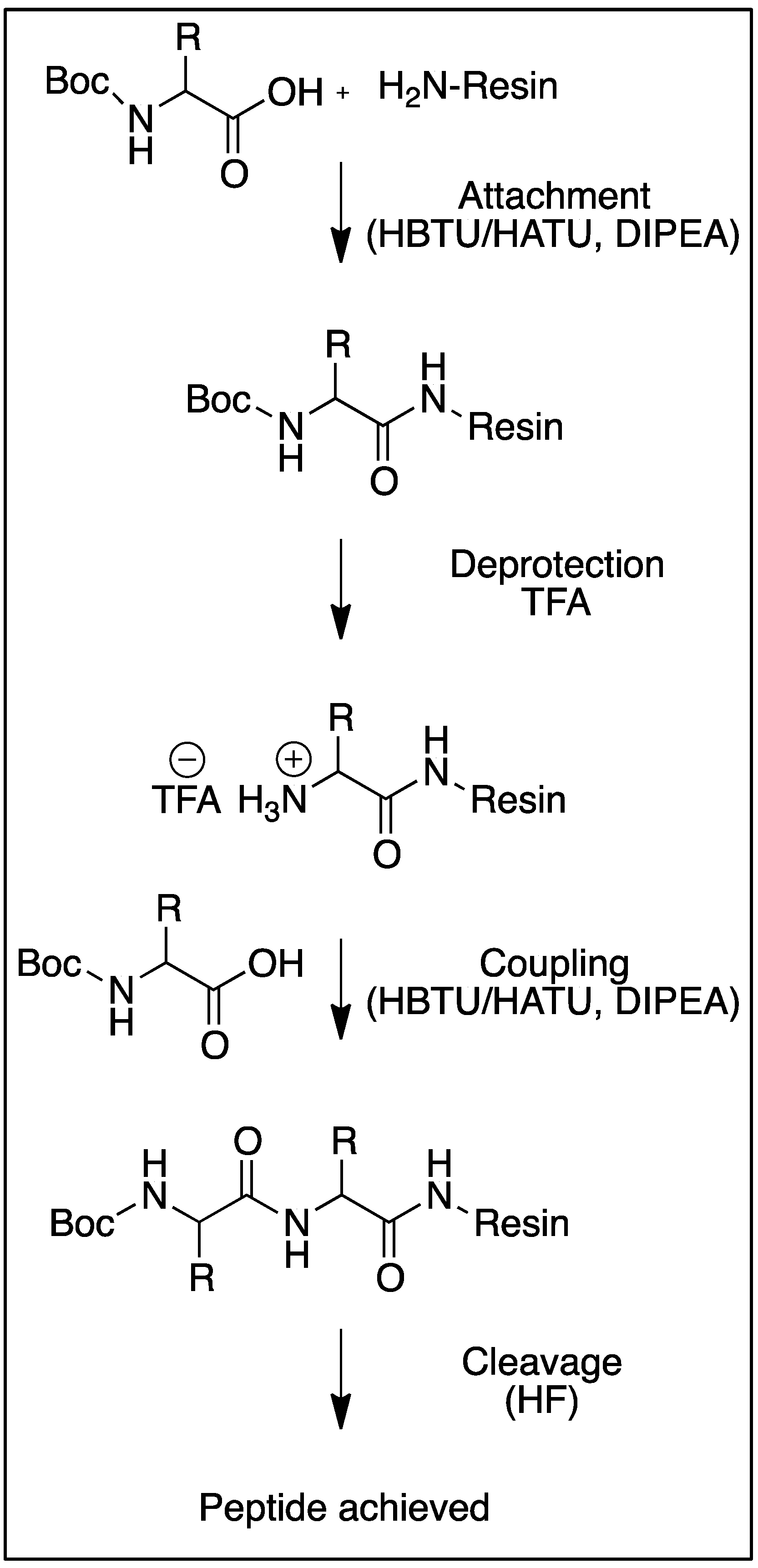
2. Chemical Methods for Peptide Synthesis

| Synthetic Cycle | Reagents | Time & Conditions |
|---|---|---|
| Deprotections | Trifluoroacetic acid (Boc chemistry)/20% piperidine in DMF (Fmoc chemistry) | 1–5 min, 0 Watt, rt (Boc chemistry) or 70 °C (Fmoc chemistry) |
| Couplings | Amino acid, HBTU/HATU/HOBt/HOAt/DIC, DIPEA | 5–15 min, 20 Watt,50–70 °C |
3. Chemoselective Ligation Techniques

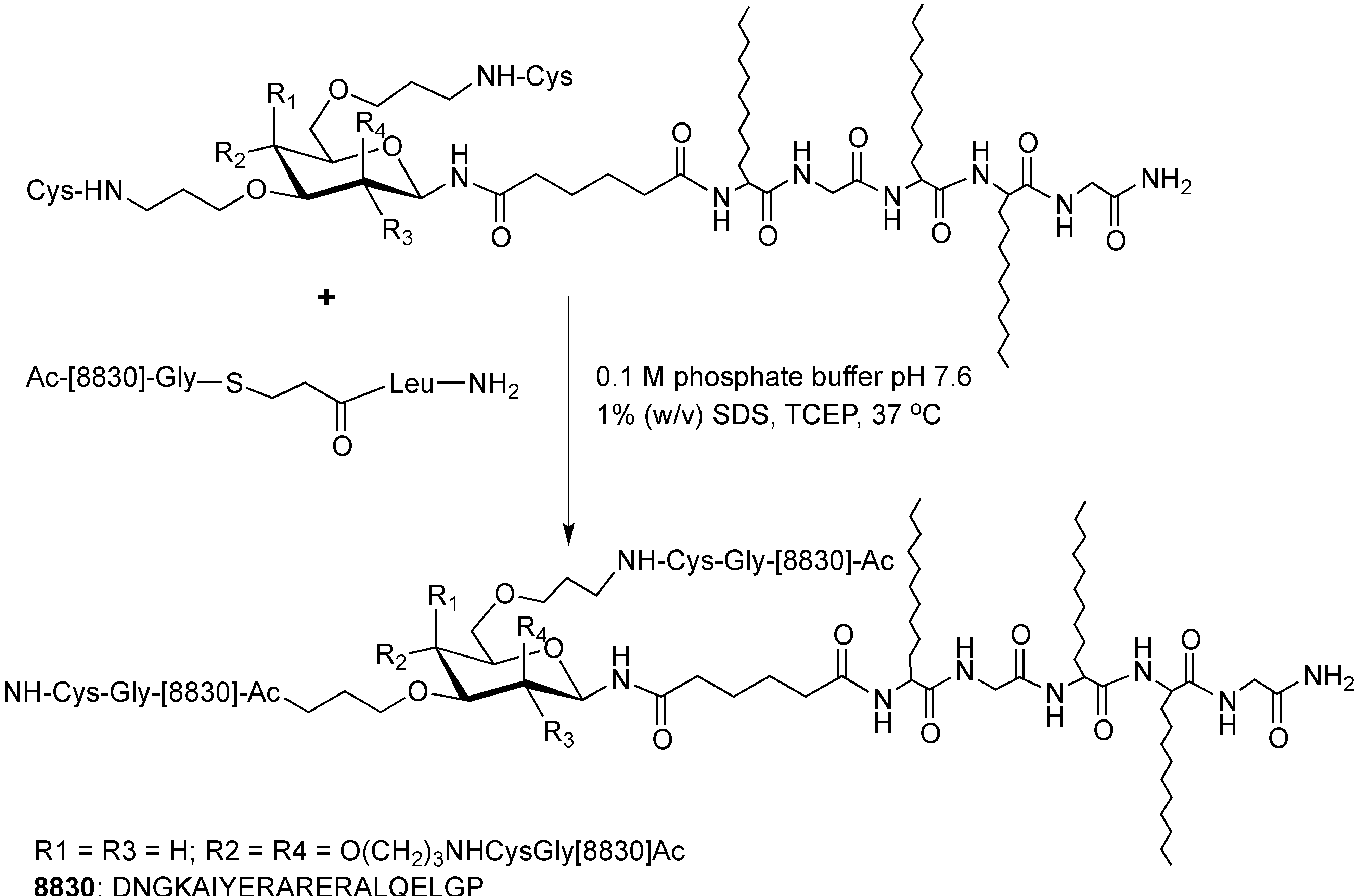
4. Application of Peptides and Proteins in Medicinal Chemistry

5. Peptide Manufacture
| Generic Name | Trade Name | Disease Target | Company |
|---|---|---|---|
| Teriparatide | Forteo | Osteoporosis | Eli Lilly & Co. |
| Exenatide | Byetta | Type 2 diabetes | Amylin/Lilly |
| Enfuvirtide | Fuzeon | HIV | Roche/Trimeris |
| Degarelix | Firmagon | Prostate cancer | Ferring |
| Mifamurtide | Mepact | Bone cancer | Takeda |
| Nesiritide | Natrecor | Heart failure | Johnson & Johnson |
| Goserelin | Zoladex | Breast and Prostate cancer | AstraZeneca |
| Glatiramer | Copaxone | Multiple sclerosis | Teva Pharmaceuticals |
| Octreotide Lanreotide | Sandostatin Somatuline, Angiopeptin | Neuroendocrine tumors | Novartis Pharmaceuticals Ipsen |
| Icatibant | Firazyr | Hereditary angioedema | Jerini |
| Ziconotide | Prialt | Pain | Elan, Azur Pharma |
| Pramlintide | Symlin | Diabetes | Amylin |
| Romiplostim | Nplate | Idiopathic thrombocytopenic purpura | Amgen |
6. Conclusions
Acknowledgments
References
- Lichtenthaler, F.W. Emil fischer, his personality, his achievements, and his scientific progeny. Eur. J. Org. Chem. 2002, 24, 4095–4122. [Google Scholar] [CrossRef]
- Fischer, E.; Fourneau, E. Ueber einige derivate des glykocolls. Ber. Dtsch. Chem. Ges. 1901, 34, 2868–2877. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Anderson, G.W.; McGregor, A.C. T-butyloxycarbonylamino acids and their use in peptide synthesis. J. Am. Chem. Soc. 1957, 79, 6180–6183. [Google Scholar] [CrossRef]
- Grapsas, I.; Cho, Y.J.; Mobashery, S. N-(tert-butoxycarbonyloxy)-5-norbornene-endo-2, 3-dicarboximide, a reagent for the regioselective introduction of the tert-butoxycarbonyl (boc) protective group at unhindered amines: Application to amino glycoside chemistry. J. Org. Chem. 1994, 59, 1918–1922. [Google Scholar] [CrossRef]
- Pedersen, S.L.; Tofteng, A.P.; Malik, L.; Jensen, K.J. Microwave heating in solid-phase peptide synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. [Google Scholar] [CrossRef]
- Schnölzer, M.; Alewood, P.; Jones, A.; Alewood, D.; Kent, S.B.H. In situ neutralization in boc-chemistry solid phase peptide synthesis. Int. J. Pept. Prot. Res. 2009, 40, 180–193. [Google Scholar] [CrossRef]
- King, D.S.; Fields, C.G.; Fields, G.B. A cleavage method which minimizes side reactions following fmoc solid phase peptide synthesis. Int. J. Pept. Prot. Res. 2009, 36, 255–266. [Google Scholar] [CrossRef]
- Berman, A.L.; Kolker, E.; Trifonov, E.N. Underlying order in protein sequence organization. P. Natl. A. Sci. USA 1994, 91, 4044–4047. [Google Scholar] [CrossRef]
- Kemp, D.; Leung, S.L.; Kerkman, D.J. Models that demonstrate peptide bond formation by prior thiol capture i. Capture by disulfide formation. Tetrahedron Lett. 1981, 22, 181–184. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Kiso, Y. Application of the on intramolecular acyl migration reaction in medicinal chemistry. Curr. Med. Chem. 2007, 14, 2813–2823. [Google Scholar] [CrossRef]
- Carpino, L.A. Oxidative reactions of hydrazines. Iv. Elimination of nitrogen from 1, 1-disubstituted-2-arenesulfonhydrazides1–4. J. Am. Chem. Soc. 1957, 79, 4427–4431. [Google Scholar] [CrossRef]
- Sakakibara, S.; Kishida, Y.; Nishizawa, R.; Shimonishi, Y. Use of anhydrous hydrogen fluoride in peptide synthesis. Procedures for the syntheses of simple peptides. Bull. Chem. Soc. Jpn. 1968, 41, 438. [Google Scholar] [CrossRef]
- Pietta, P.; Marshall, G.R. Amide protection and amide supports in solid-phase peptide synthesis. J. Chem. Soc. Chem. Comm. 1970, 11, 650–651. [Google Scholar]
- Carpino, L.A.; Han, G.Y. 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J. Am. Chem. Soc. 1970, 92, 5748–5749. [Google Scholar] [CrossRef]
- Wang, S.-S. P-alkoxybenzyl alcohol resin and p-alkoxybenzyloxycarbonylhydrazide resin for solid phase synthesis of protected peptide fragments. J. Am. Chem. Soc. 1973, 95, 1328–1333. [Google Scholar]
- Barany, G.; Merrifield, R. A new amino protecting group removable by reduction. Chemistry of the dithiasuccinoyl (dts) function. J. Am. Chem. Soc. 1977, 99, 7363–7365. [Google Scholar] [CrossRef]
- Rink, H. Solid-phase synthesis of protected peptide fragments using a trialkoxy-diphenyl-methylester resin. Tetrahedron Lett. 1987, 28, 3787–3790. [Google Scholar] [CrossRef]
- Sieber, P. A new acid-labile anchor group for the solid-phase synthesis of c-terminal peptide amides by the fmoc method. Tetrahedron Lett. 1987, 28, 2107–2110. [Google Scholar] [CrossRef]
- Alewood, P.; Alewood, D.; Miranda, L.; Love, S.; Meutermans, W.; Wilson, D. Rapid in situ neutralization protocols for boc and fmoc solid-phase chemistries. Methods Enzymol. 1997, 289, 14–29. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–778. [Google Scholar]
- Liu, C.F.; Rao, C.; Tam, J.P. Orthogonal ligation of unprotected peptide segments through pseudoproline formation for the synthesis of hiv-1 protease analogs. J. Am. Chem. Soc. 1996, 118, 307–312. [Google Scholar] [CrossRef]
- Wöhr, T.; Wahl, F.; Nefzi, A.; Rohwedder, B.; Sato, T.; Sun, X.; Mutter, M. Pseudo-prolines as a solubilizing, structure-disrupting protection technique in peptide synthesis. J. Am. Chem. Soc. 1996, 118, 9218–9227. [Google Scholar] [CrossRef]
- Carpino, L.A.; Ghassemi, S.; Ionescu, D.; Ismail, M.; Sadat-Aalaee, D.; Truran, G.A.; Mansour, E.; Siwruk, G.A.; Eynon, J.S.; Morgan, B. Rapid, continuous solution-phase peptide synthesis: Application to peptides of pharmaceutical interest. Org. Process Res. Dev. 2003, 7, 28–37. [Google Scholar] [CrossRef]
- Nishiuchi, Y.; Inui, T.; Nishio, H.; Bódi, J.; Kimura, T.; Tsuji, F.I.; Sakakibara, S. Chemical synthesis of the precursor molecule of the aequorea green fluorescent protein, subsequent folding, and development of fluorescenc. P. Natl. A. Sci. USA 1998, 95, 13549–13554. [Google Scholar] [CrossRef]
- Vigneaud, V.; Ressler, C.; Swan, C.J.M.; Roberts, C.W.; Katsoyannis, P.G.; Gordon, S. The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J. Am. Chem. Soc. 1953, 75, 4879–4880. [Google Scholar] [CrossRef]
- du Vigneaud, V.; Ressler, C.; Swan, J.M.; Roberts, C.W.; Katsoyannis, P.G. The synthesis of oxytocin1. J. Am. Chem. Soc. 1954, 76, 3115–3121. [Google Scholar]
- Carpino, L.A. The 9-fluorenylmethyloxycarbonyl family of base-sensitive amino-protecting groups. Accounts Chem. Res. 1987, 20, 401–407. [Google Scholar] [CrossRef]
- Chen, S.; Janda, K.D. Total synthesis of naturally occurring prostaglandin f2α on a non-cross-linked polystyrene support. Tetrahedron Lett. 1998, 39, 3943–3946. [Google Scholar] [CrossRef]
- Gutte, B.; Merrifield, R. The synthesis of ribonuclease a. J. Biol. Chem. 1971, 246, 1922–1941. [Google Scholar]
- Palasek, S.A.; Cox, Z.J.; Collins, J.M. Limiting racemization and aspartimide formation in microwave-enhanced fmoc solid phase peptide synthesis. Pept. Sci. 2006, 13, 143–148. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Hussein, W.M.; Liu, T.-Y.; Toth, I. Microwave-assisted synthesis of difficult sequence-containing peptide using the isopeptide method. Org. Biomol. Chem. 2013, 11, 2370–2376. [Google Scholar] [CrossRef]
- Kemp, D.; Kerkman, D.J. Models that demonstrate peptide bond formation by prior thiol capture--II capture by organomercury derivatives. Tetrahedron Lett. 1981, 22, 185–186. [Google Scholar] [CrossRef]
- Fotouhi, N.; Galakatos, N.G.; Kemp, D. Peptide synthesis by prior thiol capture. 6. Rates of the disulfide-bond-forming capture reaction and demonstration of the overall strategy by synthesis of the c-terminal 29-peptide sequence of bpti. J. Org. Chem. 1989, 54, 2803–2817. [Google Scholar] [CrossRef]
- Schnölzer, M.; Kent, S. Constructing proteins by dovetailing unprotected synthetic peptides: Backbone-engineered hiv protease. Science 1992, 256, 221. [Google Scholar]
- Dirksen, A.; Meijer, E.; Adriaens, W.; Hackeng, T.M. Strategy for the synthesis of multivalent peptide-based nonsymmetric dendrimers by native chemical ligation. Chem. Commun. 2006, 15, 1667–1669. [Google Scholar]
- Dawson, P.E.; Kent, S.B.H. Synthesis of native proteins by chemical ligation 1. Annu. Rev. Biochem. 2000, 69, 923–960. [Google Scholar] [CrossRef]
- Camarero, J.A.; Shekhtman, A.; Campbell, E.A.; Chlenov, M.; Gruber, T.M.; Bryant, D.A.; Darst, S.A.; Cowburn, D.; Muir, T.W. Autoregulation of a bacterial σ factor explored by using segmental isotopic labeling and nmr. P. Natl. A. Sci. USA 2002, 99, 8536–8541. [Google Scholar] [CrossRef]
- Cowburn, D.; Muir, T.W. Segmental isotopic labeling using expressed protein ligation. Methods Enzymol. 2001, 339, 41–54. [Google Scholar] [CrossRef]
- Ficht, S.; Dose, C.; Seitz, O. As fast and selective as enzymatic ligations: Unpaired nucleobases increase the selectivity of DNA-controlled native chemical pna ligation. ChemBioChem 2005, 6, 2098–2103. [Google Scholar] [CrossRef]
- Lovrinovic, M.; Niemeyer, C.M. Microtiter plate-based screening for the optimization of DNA–protein conjugate synthesis by means of expressed protein ligation. ChemBioChem 2006, 8, 61–67. [Google Scholar] [CrossRef]
- Paramonov, S.E.; Gauba, V.; Hartgerink, J.D. Synthesis of collagen-like peptide polymers by native chemical ligation. Macromolecules 2005, 38, 7555–7561. [Google Scholar] [CrossRef]
- Zhong, W.; Skwarczynski, M.; Fujita, Y.; Simerska, P.; Good, M.F.; Toth, I. Design and synthesis of lipopeptide–carbohydrate assembled multivalent vaccine candidates using native chemical ligation. Aust. J. Chem. 2009, 62, 993–999. [Google Scholar] [CrossRef]
- Blanco-Canosa, J.B.; Dawson, P.E. An efficient fmoc-spps approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew. Chem. Int. Edit. 2008, 47, 6851–6855. [Google Scholar] [CrossRef]
- Bu, X.; Xie, G.; Law, C.W.; Guo, Z. An improved deblocking agent for direct fmoc solid-phase synthesis of peptide thioesters. Tetrahedron Lett. 2002, 43, 2419–2422. [Google Scholar] [CrossRef]
- Ollivier, N.; Behr, J.B.; El-Mahdi, O.; Blanpain, A.; Melnyk, O. Fmoc solid-phase synthesis of peptide thioesters using an intramolecular n, s-acyl shift. Org. Lett. 2005, 7, 2647–2650. [Google Scholar] [CrossRef]
- Alsina, J.; Yokum, T.S.; Albericio, F.; Barany, G. Backbone amide linker (bal) strategy for n α-9-fluorenylmethoxycarbonyl (fmoc) solid-phase synthesis of unprotected peptide p-nitroanilides and thioesters1. J. Org. Chem. 1999, 64, 8761–8769. [Google Scholar] [CrossRef]
- Pentelute, B.L.; Gates, Z.P.; Tereshko, V.; Dashnau, J.L.; Vanderkooi, J.M.; Kossiakoff, A.A.; Kent, S.B.H. X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J. Am. Chem. Soc. 2008, 130, 9695–9701. [Google Scholar] [CrossRef]
- Pentelute, B.L.; Gates, Z.P.; Dashnau, J.L.; Vanderkooi, J.M.; Kent, S.B.H. Mirror image forms of snow flea antifreeze protein prepared by total chemical synthesis have identical antifreeze activities. J. Am. Chem. Soc. 2008, 130, 9702–9707. [Google Scholar] [CrossRef]
- Brik, A.; Keinan, E.; Dawson, P.E. Protein synthesis by solid-phase chemical ligation using a safety catch linker. J. Org. Chem. 2000, 65, 3829–3835. [Google Scholar] [CrossRef]
- Bang, D.; Pentelute, B.L.; Kent, S.B. Kinetically controlled ligation for the convergent chemical synthesis of proteins. Angew. Chem. Int. Ed. 2006, 45, 3985–3988. [Google Scholar] [CrossRef]
- Leleu, S.; Penhoat, M.; Bouet, A.; Dupas, G.; Papamicaël, C.; Marsais, F.; Levacher, V. Amine capture strategy for peptide bond formation by means of quinolinium thioester salts. J. Am. Chem. Soc. 2005, 127, 15668–15669. [Google Scholar] [CrossRef]
- Perler, F.B.; Adam, E. Protein splicing and its applications. Curr. Opin. Biotech. 2000, 11, 377–383. [Google Scholar] [CrossRef]
- Muir, T.W.; Sondhi, D.; Cole, P.A. Expressed protein ligation: A general method for protein engineering. P. Natl. A. Sci. USA 1998, 95, 6705–6710. [Google Scholar] [CrossRef]
- Nilsson, B.L.; Hondal, R.J.; Soellner, M.B.; Raines, R.T. Protein assembly by orthogonal chemical ligation methods. J. Am. Chem. Soc. 2003, 125, 5268–5269. [Google Scholar] [CrossRef]
- Saleh, L.; Perler, F.B. Protein splicing in cis and in trans. Chem. Rec. 2006, 6, 183–193. [Google Scholar] [CrossRef]
- De Rosa, L.; Cortajarena, A.L.; Romanelli, A.; Regan, L.; D'Andrea, L.D. Site-specific protein double labeling by expressed protein ligation: Applications to repeat proteins. Org. Biomol. Chem. 2012, 10, 273–280. [Google Scholar] [CrossRef]
- Saxon, E.; Armstrong, J.I.; Bertozzi, C.R. A “traceless” staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2000, 2, 2141–2143. [Google Scholar] [CrossRef]
- Soellner, M.B.; Nilsson, B.L.; Raines, R.T. Staudinger ligation of α-azido acids retains stereochemistry. J. Org. Chem. 2002, 67, 4993–4996. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell surface engineering by a modified staudinger reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef]
- Szymański, W.; Wu, B.; Poloni, C.; Janssen, D.B.; Feringa, B.L. Azobenzene photoswitches for staudinger–bertozzi ligation. Angew. Chem. Int. Ed. 2013, 52, 2068–2072. [Google Scholar] [CrossRef]
- Li, P.; Wang, L. One-pot synthesis of 1, 2, 3-triazoles from benzyl and alkyl halides, sodium azide and alkynes in water under transition-metal-catalyst free reaction conditions. Lett. Org. Chem. 2007, 4, 23–26. [Google Scholar] [CrossRef]
- Manetsch, R.; Krasinski, A.; Radic, Z.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In situ click chemistry: Enzyme inhibitors made to their own specifications. J. Am. Chem. Soc. 2004, 126, 12809–12818. [Google Scholar] [CrossRef]
- Meutermans, W.D.F.; Golding, S.W.; Bourne, G.T.; Miranda, L.P.; Dooley, M.J.; Alewood, P.F.; Smythe, M.L. Synthesis of difficult cyclic peptides by inclusion of a novel photolabile auxiliary in a ring contraction strategy. J. Am. Chem. Soc. 1999, 121, 9790–9796. [Google Scholar] [CrossRef]
- Sohma, Y.; Taniguchi, A.; Yoshiya, T.; Chiyomori, Y.; Fukao, F.; Nakamura, S.; Skwarczynski, M.; Okada, T.; Ikeda, K.; Hayashi, Y. ‘Click peptide’: A novel ‘o-acyl isopeptide method’for peptide synthesis and chemical biology-oriented synthesis of amyloid β peptide analogues. J. Pept. Sci. 2006, 12, 823–828. [Google Scholar] [CrossRef]
- Sohma, Y.; Sasaki, M.; Hayashi, Y.; Kimura, T.; Kiso, Y. Novel and efficient synthesis of difficult sequence-containing peptides through o–n intramolecular acyl migration reaction of o-acyl isopeptides. Chem. Commun. 2004, 124–125. [Google Scholar] [CrossRef]
- Fang, G.M.; Li, Y.M.; Shen, F.; Huang, Y.C.; Li, J.B.; Lin, Y.; Cui, H.K.; Liu, L. Protein chemical synthesis by ligation of peptide hydrazides. Angew. Chem. Int. Ed. 2011, 50, 7645–7649. [Google Scholar]
- Fang, G.M.; Wang, J.X.; Liu, L. Convergent chemical synthesis of proteins by ligation of peptide hydrazides. Angew. Chem. Int. Ed. 2012, 51, 10347–10350. [Google Scholar] [CrossRef]
- Pattabiraman, V.R.; Ogunkoya, A.O.; Bode, J.W. Chemical protein synthesis by chemoselective α-ketoacid–hydroxylamine (kaha) ligations with 5-oxaproline. Angew. Chem. Int. Ed. 2012, 51, 5114–5118. [Google Scholar] [CrossRef]
- Ogunkoya, A.O.; Pattabiraman, V.R.; Bode, J.W. Sequential α-ketoacid-hydroxylamine (kaha) ligations: Synthesis of c-terminal variants of the modifier protein ufm1. Angew. Chem. Int. Ed. 2012, 51, 9693–9697. [Google Scholar] [CrossRef]
- Bode, J.W.; Fox, R.M.; Baucom, K.D. Chemoselective amide ligations by decarboxylative condensations of n-alkylhydroxylamines and α-ketoacids. Angew. Chem. Int. Ed. 2006, 45, 1248–1252. [Google Scholar] [CrossRef]
- Carrillo, N.; Davalos, E.A.; Russak, J.A.; Bode, J.W. Iterative, aqueous synthesis of β3-oligopeptides without coupling reagents. J. Am. Chem. Soc. 2006, 128, 1452–1453. [Google Scholar] [CrossRef]
- Hughes, B. 2009 fda drug approvals. Nat. Rev. Drug. Discov. 2010, 9, 89–92. [Google Scholar] [CrossRef]
- Mullard, A. 2010 fda drug approvals. Nat. Rev. Drug. Discov. 2011, 10, 82–85. [Google Scholar] [CrossRef]
- Mullard, A. 2011 fda drug approvals. Nat. Rev. Drug. Discov. 2012, 11, 91–94. [Google Scholar] [CrossRef]
- Mullard, A. 2012 fda drug approvals. Nat. Rev. Drug. Discov. 2013, 12, 87–90. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Ayoub, M.; Scheidegger, D. Peptide drugs, overcoming the challenges, a growing business. Chim. Oggi. 2006, 24, 46–48. [Google Scholar]
- Takahashi, H.; Okamoto, M.; Shimodaira, S.; Tsujitani, S.-I.; Nagaya, M.; Ishidao, T.; Kishimoto, J.; Yonemitsu, Y. Impact of dendritic cell vaccines pulsed with wilms’ tumour-1 peptide antigen on the survival of patients with advanced non-small cell lung cancers. Eur. J. Cancer 2012, 49, 852–859. [Google Scholar]
- Sosinowski, T.; Eisenbarth, G.S. Type 1 diabetes: Primary antigen/peptide/register/trimolecular complex. Immunol. Res. 2012, 55, 270–276. [Google Scholar] [CrossRef]
- Chen, H.H.; Glockner, J.F.; Schirger, J.A.; Cataliotti, A.; Redfield, M.M.; Burnett, J.C. Novel protein therapeutics for systolic heart failure: chronic subcutaneous B-type natruiuretic peptide. J. Am. Coll. Cardiol. 2012, 60, 2305–2312. [Google Scholar] [CrossRef]
- Dawgul, M.; Baranska-Rybak, W.; Kamysz, E.; Karafova, A.; Nowicki, R.; Kamysz, W. Activity of short lipopeptides and conventional antimicrobials against planktonic cells and biofilms formed by clinical strains of staphylococcus aureus. Future 2012, 4, 1541–1551. [Google Scholar]
- Coles, D.J.; Simerska, P.; Fujita, Y.; Toth, I. The influence of incorporating lipids or liposaccharides on the particle size of peptide therapeutics. Biopolymers Pept. Sci. 2011, 96, 172–176. [Google Scholar] [CrossRef]
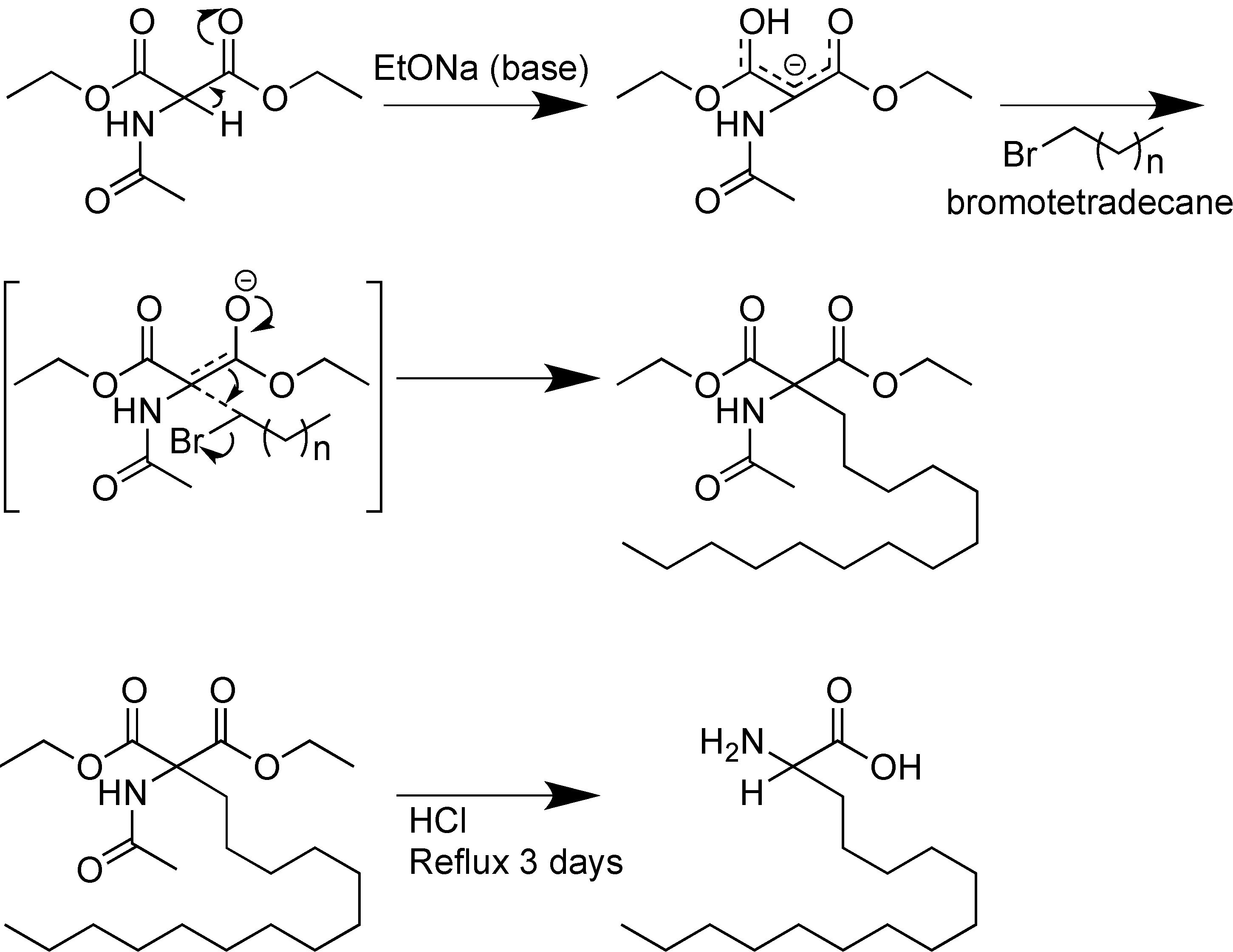
- Gibbons, W.A.; Hughes, R.A.; Charalambous, M.; Christodoulou, M.; Szeto, A.; Aulabaugh, A.E.; Mascagni, P.; Toth, I. Lipidic peptides, i. Synthesis, resolution and structural elucidation of lipidic amino acids and their homo-and hetero-oligomers. Liebigs. Ann. Chem. 2006, 1990, 1175–1183. [Google Scholar]
- Kellam, B.; Drouillat, B.; Dekany, G.; Starr, M.S.; Toth, I. Synthesis and in vitro evaluation of lipoamino acid and carbohydrate-modified enkephalins as potential antinociceptive agents. Int. J. Pharm. 1998, 161, 55–64. [Google Scholar] [CrossRef]
- Ross, B.P.; Falconer, R.A.; Toth, I. N-1-(4, 4-dimethyl-2, 6-dioxocyclohex-1-ylidene) ethyl (n-dde) lipoamino acids. Molbank 2008, 2008, M566. [Google Scholar] [CrossRef]
- Goodwin, D.; Simerska, P.; Toth, I. Peptides as therapeutics with enhanced bioactivity. Curr. Med. Chem. 2012, 19, 4451–4461. [Google Scholar] [CrossRef]
- Simerska, P.; Moyle, P.M.; Toth, I. Modern lipid-, carbohydrate-, and peptide-based delivery systems for peptide, vaccine, and gene products. Med. Res. Rev. 2009, 31, 520–547. [Google Scholar] [CrossRef]
- Varamini, P.; Mansfeld, F.M.; Blanchfield, J.T.; Wyse, B.D.; Smith, M.T.; Toth, I. Lipo-endomorphin-1 derivatives with systemic activity against neuropathic pain without producing constipation. PLOS One 2012, 7, e41909. [Google Scholar]
- Simerska, P.; Abdel-Aal, A.-B.M.; Fujita, Y.; Moyle, P.M.; McGeary, R.P.; Batzloff, M.R.; Olive, C.; Good, M.F.; Toth, I. Development of a liposaccharide-based delivery system and its application to the design of group a streptococcal vaccines. J. Med. Chem. 2008, 51, 1447–1452. [Google Scholar] [CrossRef]
- Simerska, P.; Abdel-Aal, A.B.M.; Fujita, Y.; Batzloff, M.R.; Good, M.F.; Toth, I. Synthesis and in vivo studies of carbohydrate-based vaccines against group a streptococcus. Biopolymers Pept. Sci. 2008, 90, 611–616. [Google Scholar] [CrossRef]
- Zhong, W.; Skwarczynski, M.; Simerska, P.; Good, M.F.; Toth, I. Development of highly pure α-helical lipoglycopeptides as self-adjuvanting vaccines. Tetrahedron 2009, 65, 3459–3464. [Google Scholar] [CrossRef]
- Abdel-Aal, A.-B.M.; Batzloff, M.R.; Fujita, Y.; Barozzi, N.; Faria, A.; Simerska, P.; Moyle, P.M.; Good, M.F.; Toth, I. Structure–activity relationship of a series of synthetic lipopeptide self-adjuvanting group a streptococcal vaccine candidates. J. Med. Chem. 2007, 51, 167–172. [Google Scholar]
- Zaman, M.; Abdel-Aal, A.-B.M.; Fujita, Y.; Ziora, Z.M.; Batzloff, M.R.; Good, M.F.; Toth, I. Structure-activity relationship for the development of a self-adjuvanting mucosally active lipopeptide vaccine against streptococcus pyogenes. J. Med. Chem. 2012, 55, 8515–8523. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Dougall, A.M.; Khoshnejad, M.; Chandrudu, S.; Pearson, M.S.; Loukas, A.; Toth, I. Peptide-based subunit vaccine against hookworm infection. Plos One 2012, 7, e46870. [Google Scholar]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Shi, L.; Sings, H.; Bryan, J.; Wang, B.; Wang, Y.; Mach, H.; Kosinski, M.; Washabaugh, M.; Sitrin, R.; Barr, E. Gardasil®: Prophylactic human papillomavirus vaccine development–from bench top to bed-side. Clin. Pharmacol. Ther. 2007, 81, 259–264. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Verdine, G.L.; Hilinski, G. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012, 503, 3–33. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Sci. Signal. 2004, 305, 1466–1470. [Google Scholar]
- Rojas-Cervellera, V.c.; Giralt, E.; Rovira, C. Staple motifs, initial steps in the formation of thiolate-protected gold nanoparticles: How do they form? Inorg. Chem. 2012, 51, 11422–11429. [Google Scholar] [CrossRef]
- Green, B.R.; Klein, B.D.; Lee, H.-K.; Smith, M.D.; Steve White, H.; Bulaj, G. Cyclic analogs of galanin and neuropeptide Y by hydrocarbon stapling. Bioorg. Med. Chem. 2012, 21, 303–310. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chandrudu, S.; Simerska, P.; Toth, I. Chemical Methods for Peptide and Protein Production. Molecules 2013, 18, 4373-4388. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18044373
Chandrudu S, Simerska P, Toth I. Chemical Methods for Peptide and Protein Production. Molecules. 2013; 18(4):4373-4388. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18044373
Chicago/Turabian StyleChandrudu, Saranya, Pavla Simerska, and Istvan Toth. 2013. "Chemical Methods for Peptide and Protein Production" Molecules 18, no. 4: 4373-4388. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18044373