
Continuous-Flow Synthesis of Deuterium-Labeled Antidiabetic Chalcones: Studies towards the Selective Deuteration of the Alkynone Core

 ,
,  and
and
Abstract
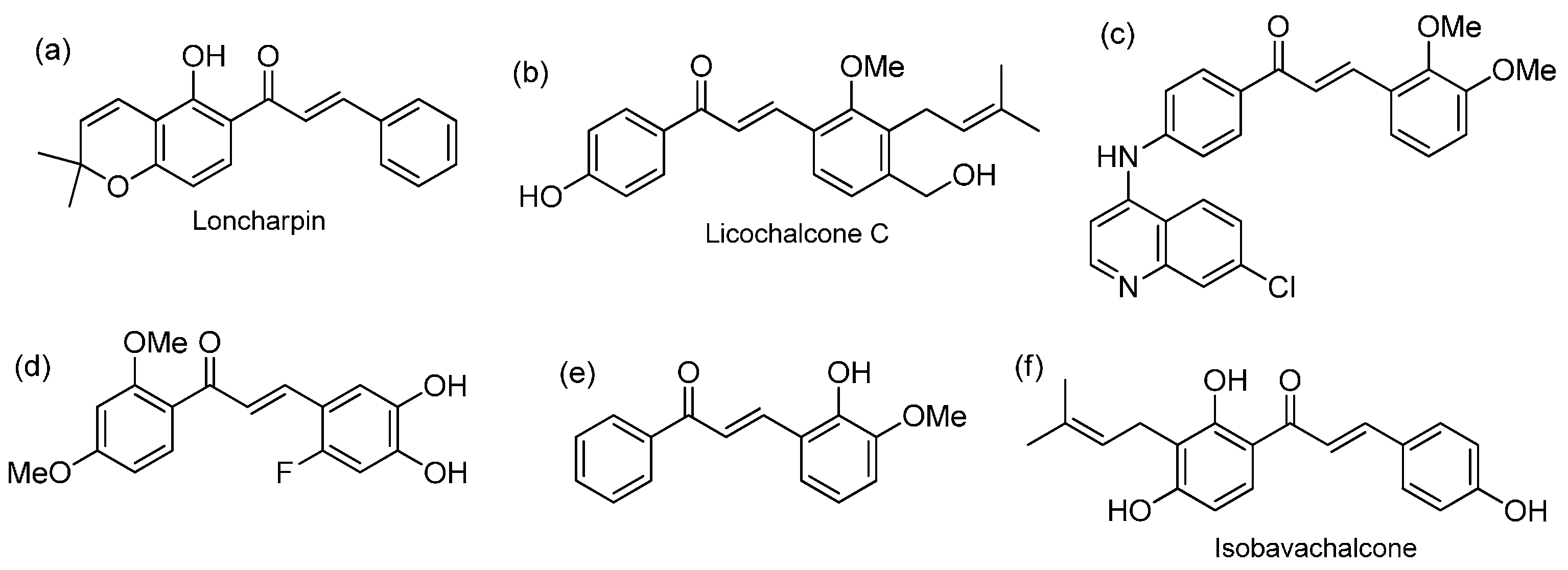
:1. Introduction
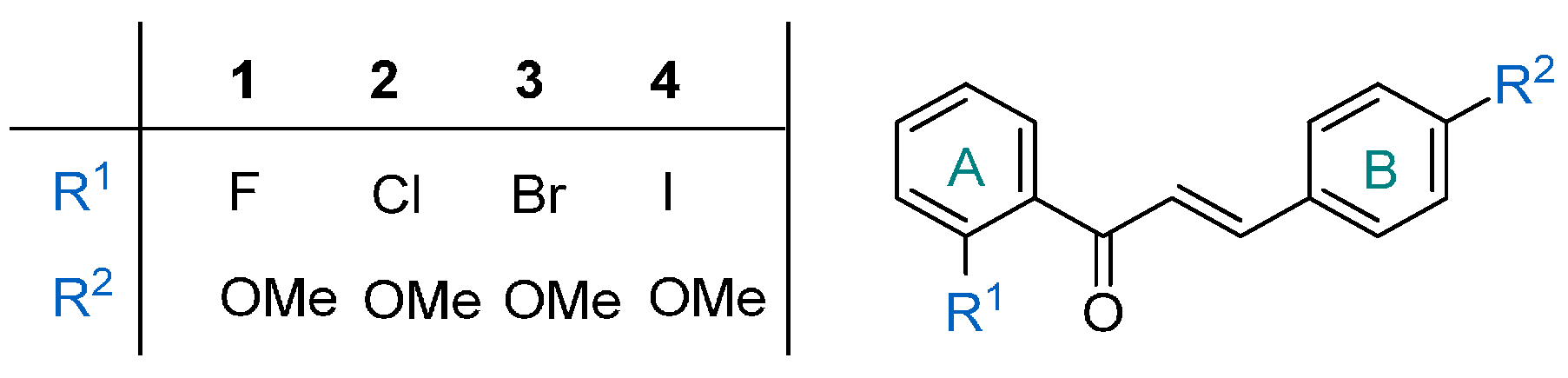
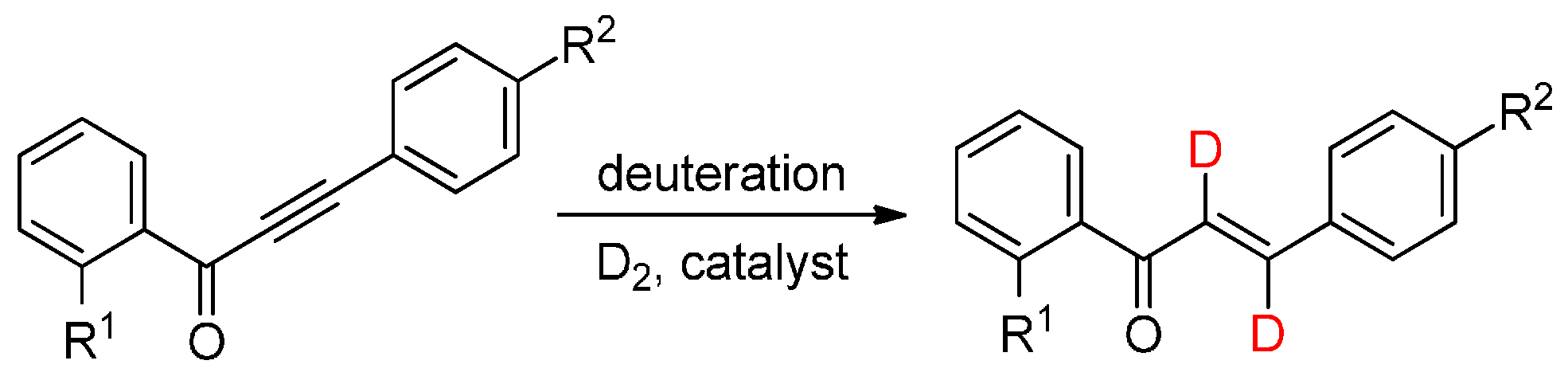
2. Results and Discussion
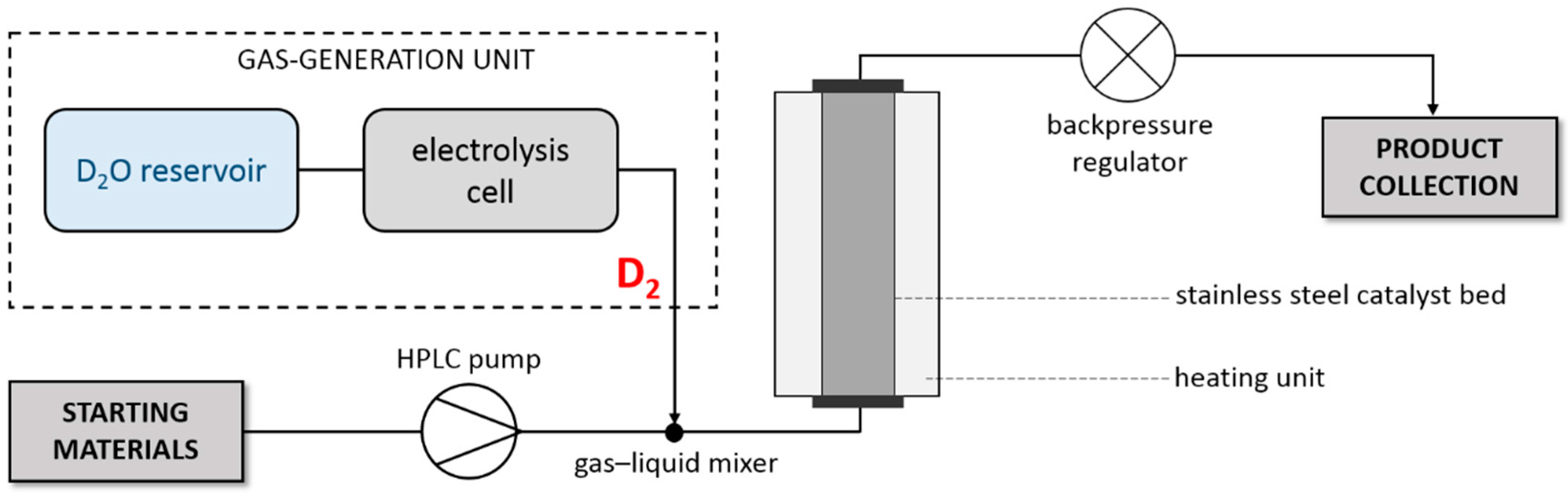
3. Materials and Methods
3.1. Materials
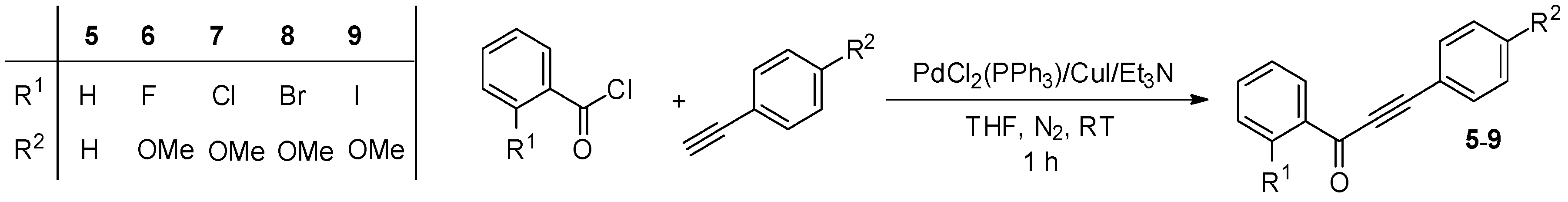
3.2. Synthesis of the Starting Materials
3.3. Continuous-Flow Deuterations
3.4. Product Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bukhari, S.N.A.; Jasamai, M.; Jantan, I. Synthesis and biological evaluation of chalcone derivatives (mini review). Mini-Rev. Med. Chem. 2012, 12, 1394–1403. [Google Scholar] [PubMed]
- Moore, B.S.; Hertweck, C.; Hopke, J.N.; Izumikawa, M.; Kalaitzis, J.A.; Nilsen, G.; O’Hare, T.; Piel, J.; Shipley, P.R.; Xiang, L.; et al. Plant-like biosynthetic pathways in bacteria: From benzoic acid to chalcone1. J. Nat. Prod. 2002, 65, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Barberán, F.A.; Clifford, M.N. Flavanones, chalcones and dihydrochalcones–nature, occurrence and dietary burden. J. Sci. Food Agric. 2000, 80, 1073–1080. [Google Scholar] [CrossRef]
- Nishimura, R.; Tabata, K.; Arakawa, M.; Ito, Y.; Kimura, Y.; Akihisa, T.; Nagai, H.; Sakuma, A.; Kohno, H.; Suzuki, T. Isobavachalcone, a chalcone constituent of angelica keiskei, induces apoptosis in neuroblastoma. Biol. Pharm. Bull. 2007, 30, 1878–1883. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Kelley, J.A.; Barchi, J.J.; Sanchez, T.; Dayam, R.; Pommier, Y.; Neamati, N. Mining the NCI antiviral compounds for HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2006, 14, 3785–3792. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, H.; Tanimoto, K.; Tamura, Y.; Mizutani, K.; Kinoshita, T. Mode of antibacterial action of retrochalcones from glycyrrhiza inflata. Phytochemistry 1998, 48, 125–129. [Google Scholar] [CrossRef]
- Nakamura, C.; Kawasaki, N.; Miyataka, H.; Jayachandran, E.; Kim, I.H.; Kirk, K.L.; Taguchi, T.; Takeuchi, Y.; Hori, H.; Satoh, T. Synthesis and biological activities of fluorinated chalcone derivatives. Bioorg. Med. Chem. 2002, 10, 699–706. [Google Scholar] [CrossRef]
- Fontenele, J.B.; Leal, L.K.A.M.; Ferreira, M.A.D.; Silveira, E.R.; Viana, G.S.B. Antiplatelet effect of lonchocarpin and derricin isolated from lonchocarpus sericeus. Pharm. Biol. 2005, 43, 726–731. [Google Scholar] [CrossRef]
- Sharma, M.; Chaturvedi, V.; Manju, Y.K.; Bhatnagar, S.; Srivastava, K.; Puri, S.K.; Chauhan, P.M.S. Substituted quinolinyl chalcones and quinolinyl pyrimidines as a new class of anti-infective agents. Eur. J. Med. Chem. 2009, 44, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.R.; Prasad, S.; Sung, B.; Aggarwal, B.B. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int. Immunopharmacol. 2011, 11, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Batovska, D.I.; Todorova, I.T. Trends in utilization of the pharmacological potential of chalcones. Curr. Clin. Pharmacol. 2010, 5, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Meng, C.Q.; Sikorski, J.A. Recent advances in therapeutic chalcones. Expert. Opin. Ther. Pat. 2004, 14, 1669–1691. [Google Scholar] [CrossRef]
- Simmons, D. Prevention of gestational diabetes mellitus: Where are we now? Diabetes Obes. Metab. 2015, 17, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. The current drug treatment landscape for diabetes and perspectives for the future. Clin. Pharmacol. Ther. 2015, 98, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Florez, J.C. Therapeutic challenges in diabetes prevention: We have not found the “exercise pill”. Clin. Pharmacol. Ther. 2015, 98, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Seuring, T.; Archangelidi, O.; Suhrcke, M. The economic costs of type 2 diabetes: A global systematic review. Pharmacoeconomics 2015, 33, 811–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, P.; Srivastava, S.P.; Srivastava, R.; Rawat, A.K.; Srivastava, A.K.; Pratap, R. Synthesis and antidyslipidemic activity of chalcone fibrates. Bioorg. Med. Chem. Lett. 2011, 21, 3475–3478. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Zou, H.-J.; Wu, A.-H.; Ye, Y.-L.; Shen, J.-H. Structure-based drug design of a novel family of chalcones as PPARα agonists: Virtual screening, synthesis, and biological activities in vitro. Acta Pharmacol. Sin. 2007, 28, 2040–2052. [Google Scholar] [CrossRef] [PubMed]
- Enoki, T.; Ohnogi, H.; Nagamine, K.; Kudo, Y.; Sugiyama, K.; Tanabe, M.; Kobayashi, E.; Sagawa, H.; Kato, I. Antidiabetic activities of chalcones isolated from a japanese herb, angelica keiskei. J. Agric. Food Chem. 2007, 55, 6013–6017. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-T.; Hsieh, T.-J.; El-Shazly, M.; Chuang, D.-W.; Tsai, Y.-H.; Yen, C.-T.; Wu, S.-F.; Wu, Y.-C.; Chang, F.-R. Synthesis of chalcone derivatives as potential anti-diabetic agents. Bioorg. Med. Chem. Lett. 2012, 22, 3912–3915. [Google Scholar] [CrossRef] [PubMed]
- Hiroaki, H. Recent applications of isotopic labeling for protein nmr in drug discovery. Expert. Opin. Drug Discov. 2013, 8, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Rey, M.; Burns, K.M.; Schriemer, D.C. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry—A review. Anal. Chim. Acta 2012, 721, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, K.; Impens, F.; Ghesquiere, B.; Van Damme, P.; Lambrechts, A.; Vandekerckhove, J. Stable isotopic labeling in proteomics. Proteomics 2008, 8, 4873–4885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Jones, G.C.; Munoz, M.P. Isotopic labelling in the study of organic and organometallic mechanism and structure: An account. J. Label. Compd. Radiopharm. 2007, 50, 1072–1087. [Google Scholar] [CrossRef]
- Gant, T.G. Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 2014, 57, 3595–3611. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Sleno, L.; A Volmer, D. Isotopic labeling of metabolites in drug discovery applications. Curr. Drug Metab. 2012, 13, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Krumbiegel, P. Large deuterium isotope effects and their use: A historical review. Isot. Environ. Health Stud. 2011, 47, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Robins, R.; Billault, I.; Duan, J.-R.; Guiet, S.; Pionnier, S.; Zhang, B.-L. Measurement of 2H distribution in natural products by quantitative 2H-NMR: An approach to understanding metabolism and enzyme mechanism? Phytochem. Rev. 2003, 2, 87–102. [Google Scholar] [CrossRef]
- Kharasch, E.D.; Bedynek, P.S.; Park, S.; Whittington, D.; Walker, A.; Hoffer, C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics: I. Evidence against CYP3A mediation of methadone clearance. Clin. Pharmacol. Ther. 2008, 84, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E.; Raghavan, A.S.; Hess, B.A., Jr.; Smentek, L. Thermal 1,5 hydrogen sigmatropic shifts in cis,cis-1,3-cyclononadienes probed by gas-phase kinetic studies and density functional theory calculations. J. Am. Chem. Soc. 2006, 128, 14854–14862. [Google Scholar] [CrossRef] [PubMed]
- Stringer, R.A.; Williams, G.; Picard, F.; Sohal, B.; Kretz, O.; McKenna, J.; Krauser, J.A. Application of a deuterium replacement strategy to modulate the pharmacokinetics of 7-(3,5-dimethyl-1H-1,2,4-triazol-1-yl)-3-(4-methoxy-2-methylphenyl)-2,6-dimethylpyrazolo[5,1-b]oxazole, a novel CRF1 antagonist. Drug Metab. Dispos. 2014, 42, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Braman, V.; Graham, P.; Cheng, C.; Turnquist, D.; Harnett, M.; Sabounjian, L.; Shipley, J. A randomized phase I evaluation of CTP-499, a novel deuterium-containing drug candidate for diabetic nephropathy. Clin. Pharmacol. Drug Dev. 2013, 2, 53–66. [Google Scholar] [CrossRef]
- Nelson, S.D.; Trager, W.F. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab. Dispos. 2003, 31, 1481–1497. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-T.; Ötvös, S.B.; Wu, Y.-C.; Mándity, I.M.; Chang, F.-R.; Fülöp, F. Highly selective continuous-flow synthesis of potentially bioactive deuterated chalcone derivatives. ChemPlusChem 2015, 80, 859–864. [Google Scholar] [CrossRef]
- Microreactors in Organic Chemistry and Catalysis; Wirth, T. (Ed.) John Wiley & Sons: Hoboken, NJ, USA, 2013.
- Ley, S.V.; Fitzpatrick, D.E.; Ingham, R.J.; Myers, R.M. Organic synthesis: March of the machines. Angew. Chem. Int. Ed. 2015, 54, 3449–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mándity, I.M.; Ötvös, S.B.; Fülöp, F. Strategic application of residence-time control in continuous-flow reactors. ChemistryOpen 2015, 4, 212–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ötvös, S.B.; Fülöp, F. Flow chemistry as a versatile tool for the synthesis of triazoles. Catal. Sci. Technol. 2015, 5, 4926–4941. [Google Scholar] [CrossRef]
- Vaccaro, L.; Lanari, D.; Marrocchi, A.; Strappaveccia, G. Flow approaches towards sustainability. Green Chem. 2014, 16, 3680–3704. [Google Scholar] [CrossRef]
- McQuade, D.T.; Seeberger, P.H. Applying flow chemistry: Methods, materials, and multistep synthesis. J. Org. Chem. 2013, 78, 6384–6389. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.-I.; Takahashi, Y.; Nagaki, A. Flash chemistry: Flow chemistry that cannot be done in batch. Chem. Commun. 2013, 49, 9896–9904. [Google Scholar] [CrossRef] [PubMed]
- Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. Novel process windows for enabling, accelerating, and uplifting flow chemistry. ChemSusChem 2013, 6, 746–789. [Google Scholar] [CrossRef] [PubMed]
- Hartman, R.L.; McMullen, J.P.; Jensen, K.F. Deciding whether to go with the flow: Evaluating the merits of flow reactors for synthesis. Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [Google Scholar] [CrossRef] [PubMed]
- Wegner, J.; Ceylan, S.; Kirschning, A. Ten key issues in modern flow chemistry. Chem. Commun. 2011, 47, 4583–4592. [Google Scholar] [CrossRef] [PubMed]
- Irfan, M.; Glasnov, T.N.; Kappe, C.O. Heterogeneous catalytic hydrogenation reactions in continuous-flow reactors. ChemSusChem 2011, 4, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Inorganic Isotopic Syntheses; Herber, R.H. (Ed.) Benjamin Inc.: New York, NY, USA, 1962.
- Sawama, Y.; Monguchi, Y.; Sajiki, H. Efficient H–D exchange reactions using heterogeneous platinum-group metal on carbon–H2–D2O system. Synlett 2012, 23, 959–972. [Google Scholar] [CrossRef]
- Modutlwa, N.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Synthesis of deuterium-labelled drugs by hydrogen–deuterium (H–D) exchange using heterogeneous catalysis. J. Label. Compd. Radiopharm. 2010, 53, 686–692. [Google Scholar] [CrossRef]
- Maegawa, T.; Fujiwara, Y.; Inagaki, Y.; Esaki, H.; Monguchi, Y.; Sajiki, H. Mild and efficient H/D exchange of alkanes based on C‒H activation catalyzed by rhodium on charcoal. Angew. Chem. Int. Ed. 2008, 47, 5394–5397. [Google Scholar] [CrossRef] [PubMed]
- Kurita, T.; Aoki, F.; Mizumoto, T.; Maejima, T.; Esaki, H.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Facile and convenient method of deuterium gas generation using a Pd/C-catalyzed H2–D2 exchange reaction and its application to synthesis of deuterium-labeled compounds. Chem. Eur. J. 2008, 14, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, T.; Fujiwara, Y.; Inagaki, Y.; Monguchi, Y.; Sajiki, H. A convenient and effective method for the regioselective deuteration of alcohols. Adv. Synth. Catal. 2008, 350, 2215–2218. [Google Scholar] [CrossRef]
- Esaki, H.; Ito, N.; Sakai, S.; Maegawa, T.; Monguchi, Y.; Sajiki, H. General method of obtaining deuterium-labeled heterocyclic compounds using neutral D2O with heterogeneous Pd/C. Tetrahedron 2006, 62, 10954–10961. [Google Scholar] [CrossRef]
- Mutsumi, T.; Iwata, H.; Maruhashi, K.; Monguchi, Y.; Sajiki, H. Halogen–deuterium exchange reaction mediated by tributyltin hydride using THF-d8 as the deuterium source. Tetrahedron 2011, 67, 1158–1165. [Google Scholar] [CrossRef]
- Di Giuseppe, A.; Castarlenas, R.; Pérez-Torrente, J.J.; Lahoz, F.J.; Polo, V.; Oro, L.A. Mild and selective H/D exchange at the β-position of aromatic α-olefins by N-heterocyclic carbene–hydride–rhodium catalysts. Angew. Chem. Int. Ed. 2011, 50, 3938–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockley, W.J.S.; Hesk, D. Rhodium- and ruthenium-catalysed hydrogen isotope exchange. J. Label. Compd. Radiopharm. 2010, 53, 704–715. [Google Scholar] [CrossRef]
- Derdau, V.; Atzrodt, J.; Zimmermann, J.; Kroll, C.; Brückner, F. Hydrogen–deuterium exchange reactions of aromatic compounds and heterocycles by NaBD4-activated rhodium, platinum and palladium catalysts. Chem. Eur. J. 2009, 15, 10397–10404. [Google Scholar] [CrossRef] [PubMed]
- Derdau, V. Deuterated ammonium formate as deuterium source in a mild catalytic deuterium transfer reaction of pyridines, pyrazines and isoquinolines. Tetrahedron Lett. 2004, 45, 8889–8893. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Mándity, I.M.; Fülöp, F. Highly selective deuteration of pharmaceutically relevant nitrogen-containing heterocycles: A flow chemistry approach. Mol. Divers. 2011, 15, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Mándity, I.M.; Martinek, T.A.; Darvas, F.; Fülöp, F. A simple, efficient, and selective deuteration via a flow chemistry approach. Tetrahedron Lett. 2009, 50, 4372–4374. [Google Scholar] [CrossRef]
- Jones, R.V.; Godorhazy, L.; Varga, N.; Szalay, D.; Urge, L.; Darvas, F. Continuous-flow high pressure hydrogenation reactor for optimization and high-throughput synthesis. J. Comb. Chem. 2006, 8, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.; Kappe, C.O. Heterogeneous hydrogenation reactions using a continuous flow high pressure device. J. Comb. Chem. 2005, 7, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Habraken, E.; Haspeslagh, P.; Vliegen, M.; Noël, T. Iridium(I)-catalyzed ortho-directed hydrogen isotope exchange in continuous-flow reactors. J. Flow. Chem. 2015, 5, 2–5. [Google Scholar] [CrossRef]
- Noël, T.; Su, Y.; Hessel, V. Beyond organometallic flow chemistry: The principles behind the use of continuous-flow reactors for synthesis. Top. Organomet. Chem. 2015. [Google Scholar] [CrossRef]
- Chuang, D.-W.; El-Shazly, M.; Balaji, D.B.; Chung, Y.-M.; Chang, F.-R.; Wu, Y.-C. Synthesis of flavones and γ-benzopyranones using mild sonogashira coupling and 18-crown-6 ether mediated 6-endo cyclization. Eur. J. Org. Chem. 2012, 4533–4540. [Google Scholar] [CrossRef]
- Cox, R.J.; Ritson, D.J.; Dane, T.A.; Berge, J.; Charmant, J.P.H.; Kantacha, A. Room temperature palladium catalysed coupling of acyl chlorides with terminal alkynes. Chem. Commun. 2005, 1037–1039. [Google Scholar] [CrossRef] [PubMed]
- Karpov, A.S.; Müller, T.J.J. Straightforward novel one-pot enaminone and pyrimidine syntheses by coupling-addition-cyclocondensation sequences. Synthesis 2003, 2815–2826. [Google Scholar] [CrossRef]
- Siegel, S. Heterogeneous catalytic hydrogenation of C=C and C≡C. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon Press: New York, NY, USA, 1991; Volume 8, pp. 417–442. [Google Scholar]
- Lee, Y.; Motoyama, Y.; Tsuji, K.; Yoon, S.-H.; Mochida, I.; Nagashima, H. (Z)-selective partial hydrogenation of internal alkynes by using palladium nanoparticles supported on nitrogen-doped carbon nanofiber. ChemCatChem 2012, 4, 778–781. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Shioiri, T. Convenient stereoselective synthesis of (Z)-chalcone derivatives from 1,3-diaryl-2-propynyl silyl ethers. Tetrahedron Lett. 2006, 47, 4943–4945. [Google Scholar] [CrossRef]
- Nicodem, D.E.; de M.G. Matos, J.A. Photoisomerization of chalcone: Wavelength dependence. J. Photochem. 1981, 15, 193–202. [Google Scholar] [CrossRef]
- Oba, M.; Ohkuma, K.; Hitokawa, H.; Shirai, A.; Nishiyama, K. Convenient synthesis of deuterated glutamic acid, proline and leucine via catalytic deuteration of unsaturated pyroglutamate derivatives. J. Label. Compd. Radiopharm. 2006, 49, 229–235. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds 5‒9 are available from the authors in mg quantities.


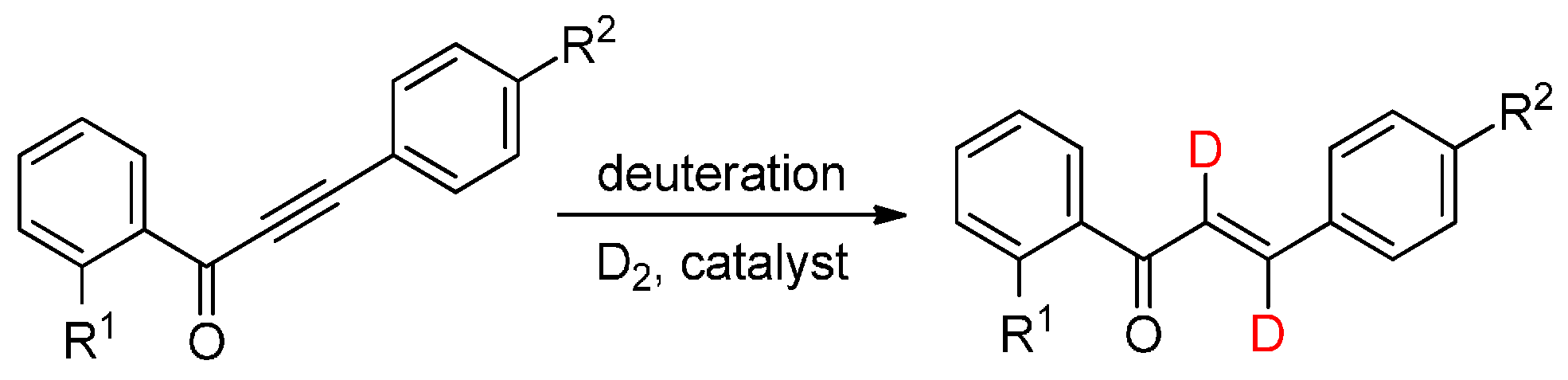




| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 5a c | 5b | 5c | |||||
| 1 | Lindlar catalyst | 10 | 25 | 47 | 92 | 8 | 0 |
| 2 | Lindlar catalyst | 20 | 25 | 57 | 86 | 14 | 0 |
| 3 | Lindlar catalyst | 40 | 25 | 63 | 77 | 23 | 0 |
| 4 | Lindlar catalyst | 80 | 25 | 82 | 73 | 23 | 4 |

| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 6a c | 6b | 6c | |||||
| 1 | Lindlar catalyst | 10 | 25 | 23 | 90 | 10 | 0 |
| 2 | Lindlar catalyst | 20 | 25 | 40 | 86 | 14 | 0 |
| 3 | Lindlar catalyst | 40 | 25 | 57 | 81 | 19 | 0 |
| 4 | Lindlar catalyst | 80 | 25 | 63 | 80 | 20 | 0 |

| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 7a c | 7b | 7c | |||||
| 1 | Lindlar catalyst | 20 | 25 | 33 | 89 | 11 | 0 |
| 2 | Lindlar catalyst | 40 | 25 | 43 | 88 | 12 | 0 |
| 3 | Lindlar catalyst | 80 | 25 | 47 | 79 | 21 | 0 |
| 4 | 5% Pd/BaSO4 | 20 | 25 | 75 | 62 | 38 | 0 |
| 5 | 5% Pd/BaSO4 | 40 | 25 | 86 | 60 | 38 | 2 |

| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 8a c | 8b | 8c | |||||
| 1 | 5% Pd/BaSO4 | 40 | 50 | 50 | 69 | 31 | 0 |
| 2 | 5% Pd/BaSO4 | 40 | 70 | 53 | 56 | 44 | 0 |
| 3 | 5% Pd/BaSO4 | 60 | 25 | 66 | 77 | 23 | 0 |
| 4 | 5% Pd/BaSO4 | 60 | 50 | 82 | 61 | 39 | 0 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 9a c | 9b | 9c | |||||
| 1 | 5% Pd/BaSO4 | 80 | 50 | 17 | 100 | 0 | 0 |
| 2 | 5% Pd/BaSO4 | 80 | 100 | 56 | 100 | 0 | 0 |
| 3 | 5% Pt/Al2O3 | 20 | 25 | 89 | 28 | 67 | 5 |
| 4 | 5% Pt/Al2O3 | 40 | 25 | 95 | 18 | 68 | 14 |
| 5 | 5% Pt/Al2O3 | 80 | 25 | 99 | 7 | 59 | 34 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ötvös, S.B.; Hsieh, C.-T.; Wu, Y.-C.; Li, J.-H.; Chang, F.-R.; Fülöp, F. Continuous-Flow Synthesis of Deuterium-Labeled Antidiabetic Chalcones: Studies towards the Selective Deuteration of the Alkynone Core. Molecules 2016, 21, 318. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21030318
Ötvös SB, Hsieh C-T, Wu Y-C, Li J-H, Chang F-R, Fülöp F. Continuous-Flow Synthesis of Deuterium-Labeled Antidiabetic Chalcones: Studies towards the Selective Deuteration of the Alkynone Core. Molecules. 2016; 21(3):318. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21030318
Chicago/Turabian StyleÖtvös, Sándor B., Chi-Ting Hsieh, Yang-Chang Wu, Jih-Heng Li, Fang-Rong Chang, and Ferenc Fülöp. 2016. "Continuous-Flow Synthesis of Deuterium-Labeled Antidiabetic Chalcones: Studies towards the Selective Deuteration of the Alkynone Core" Molecules 21, no. 3: 318. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21030318