
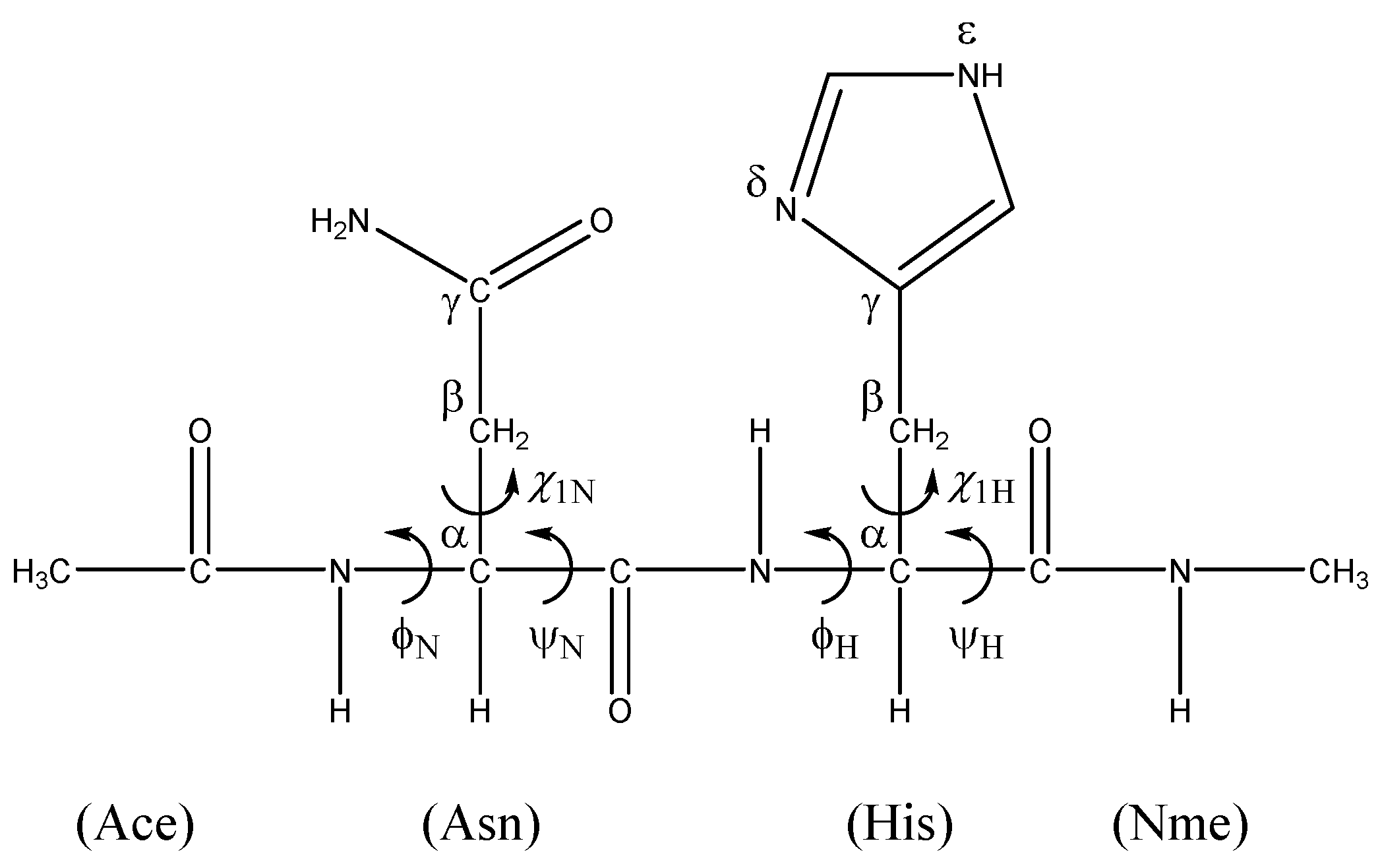
A Computational Study of the Mechanism of Succinimide Formation in the Asn–His Sequence: Intramolecular Catalysis by the His Side Chain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
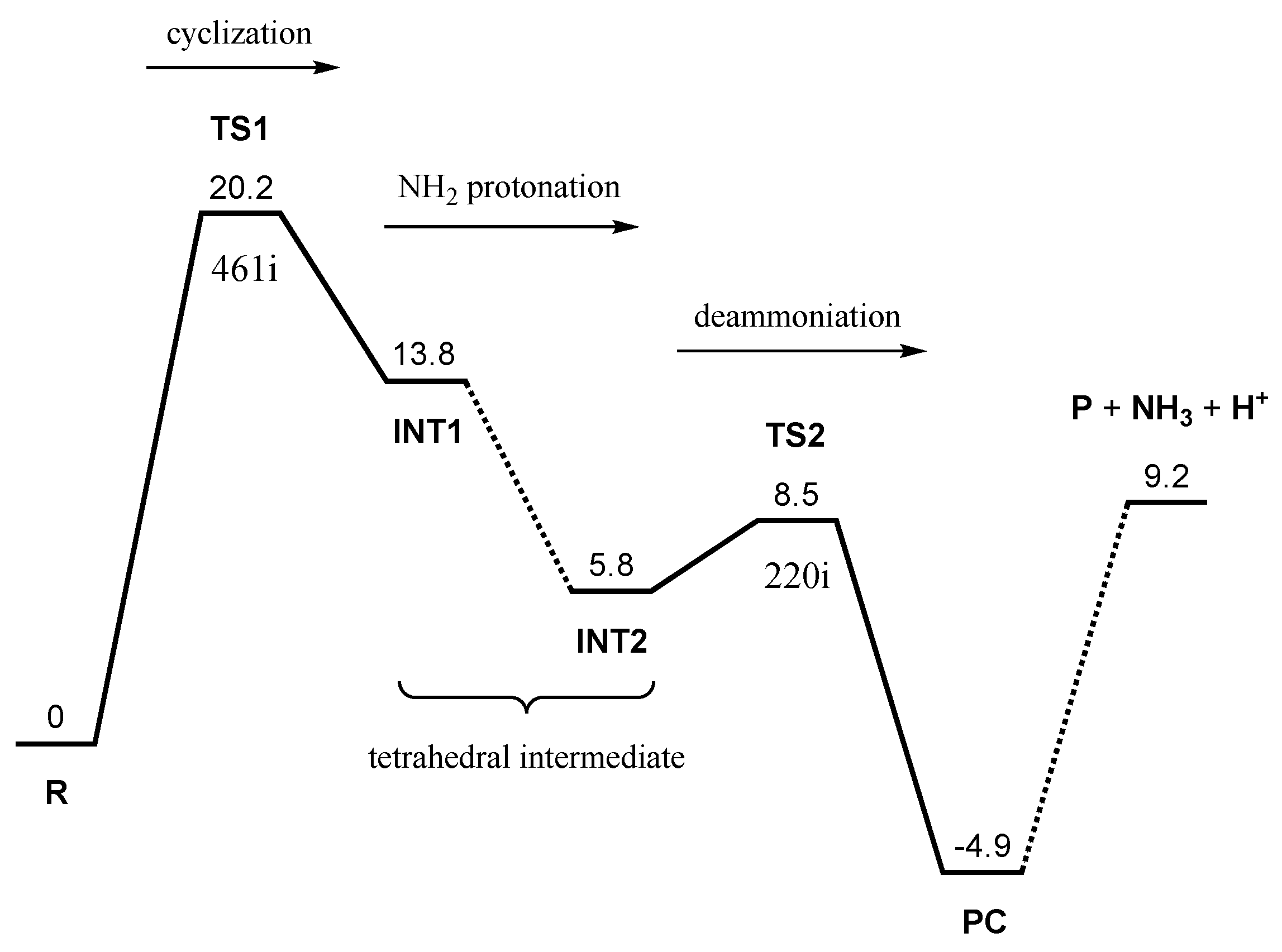
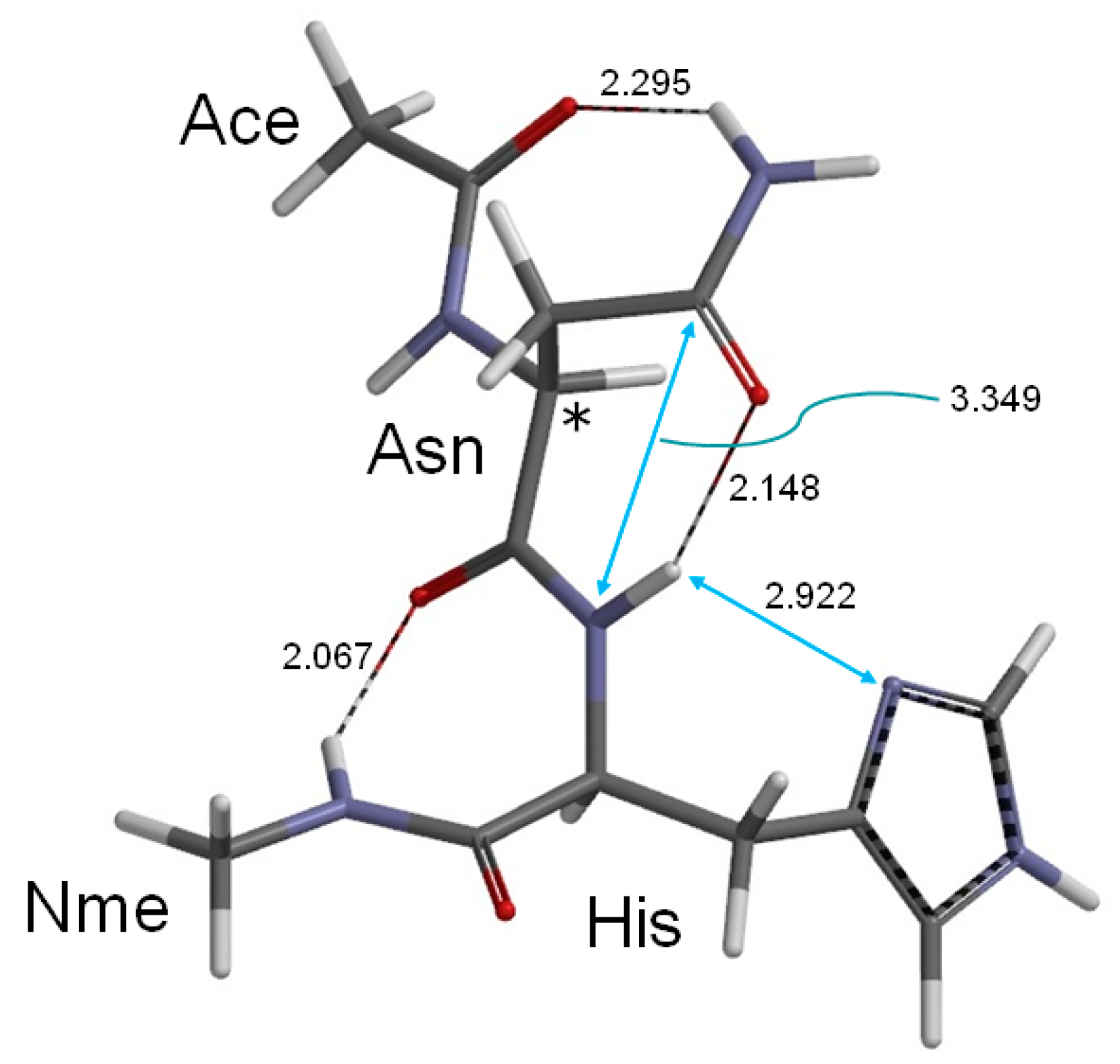
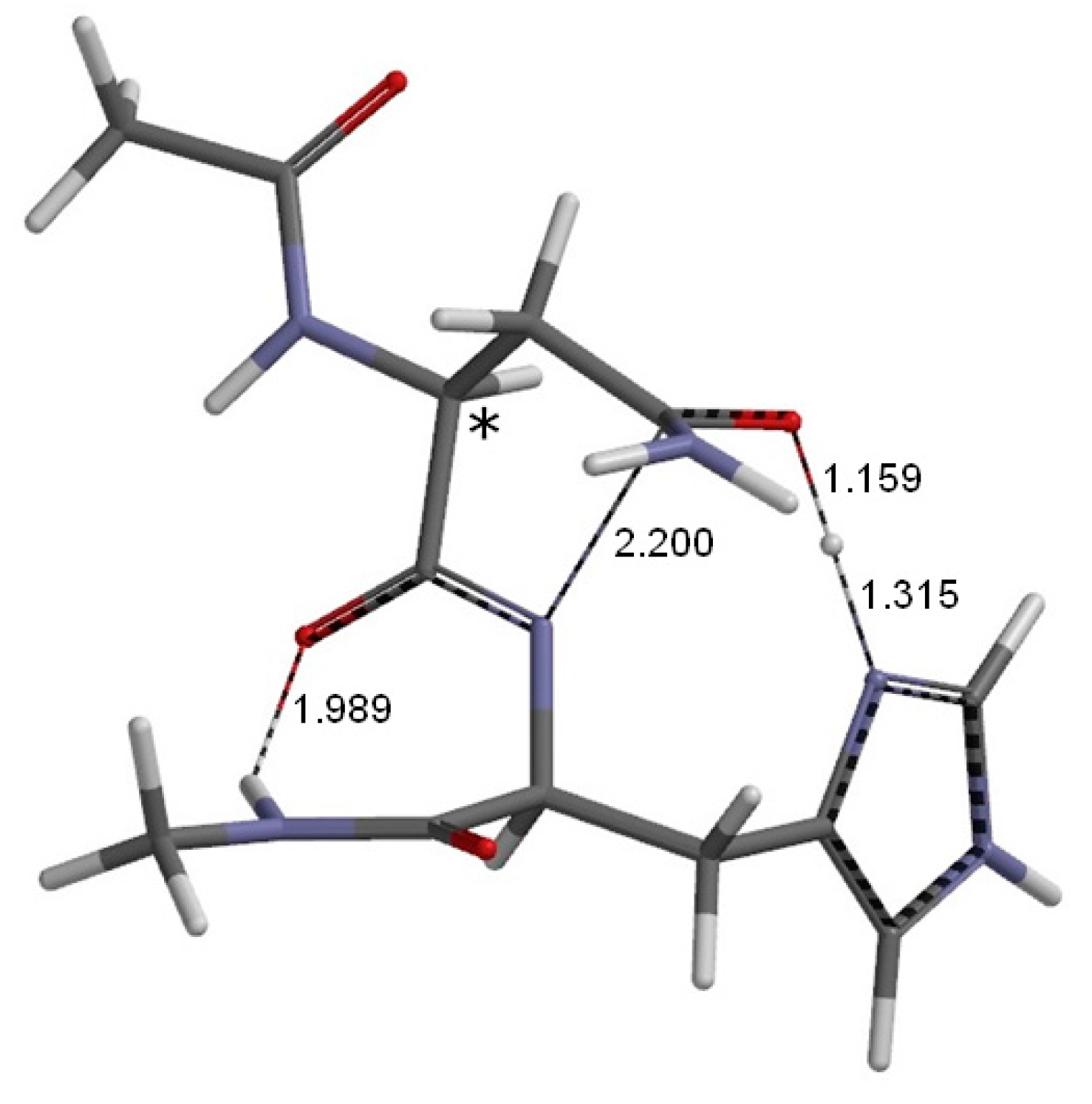
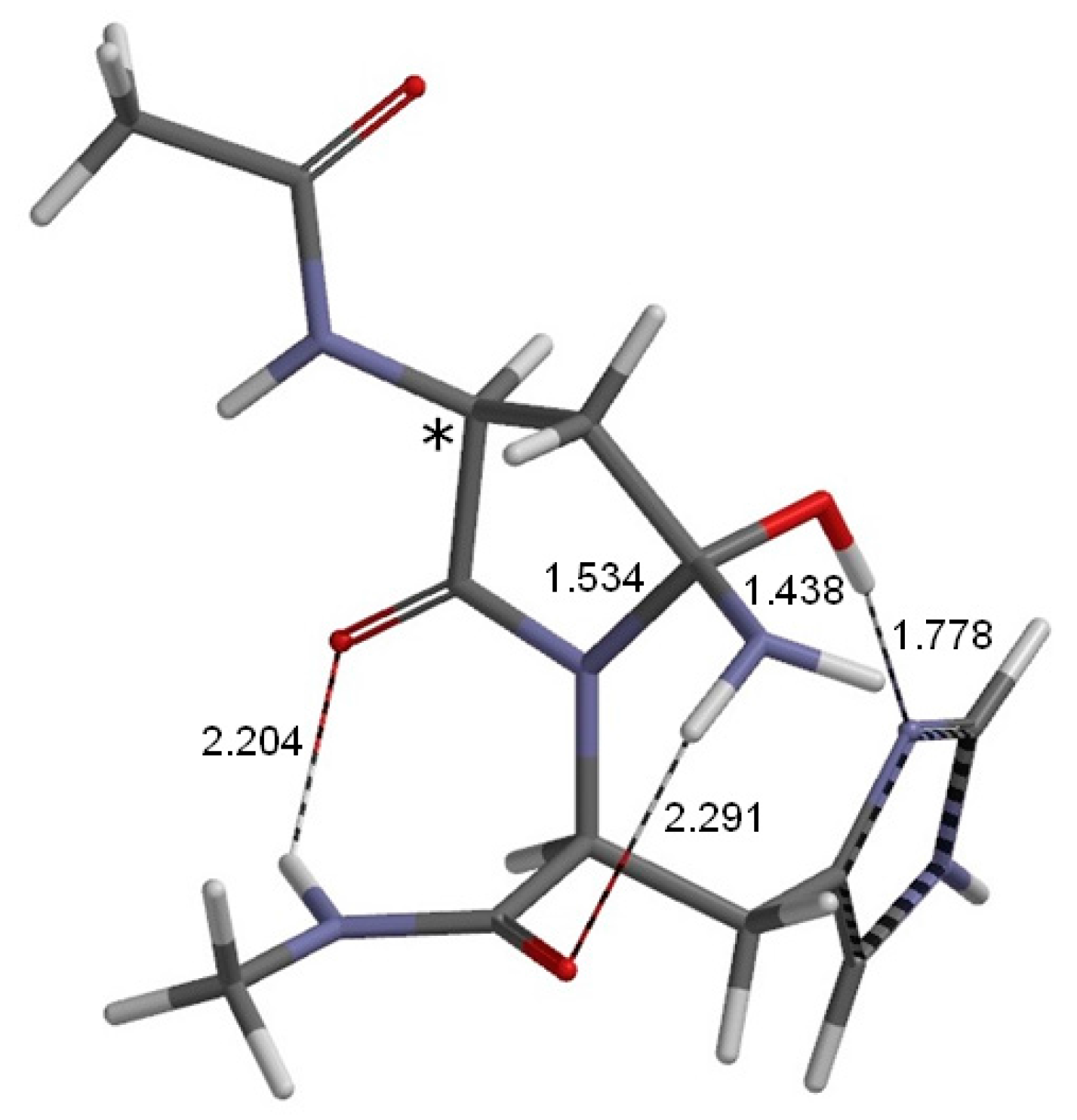
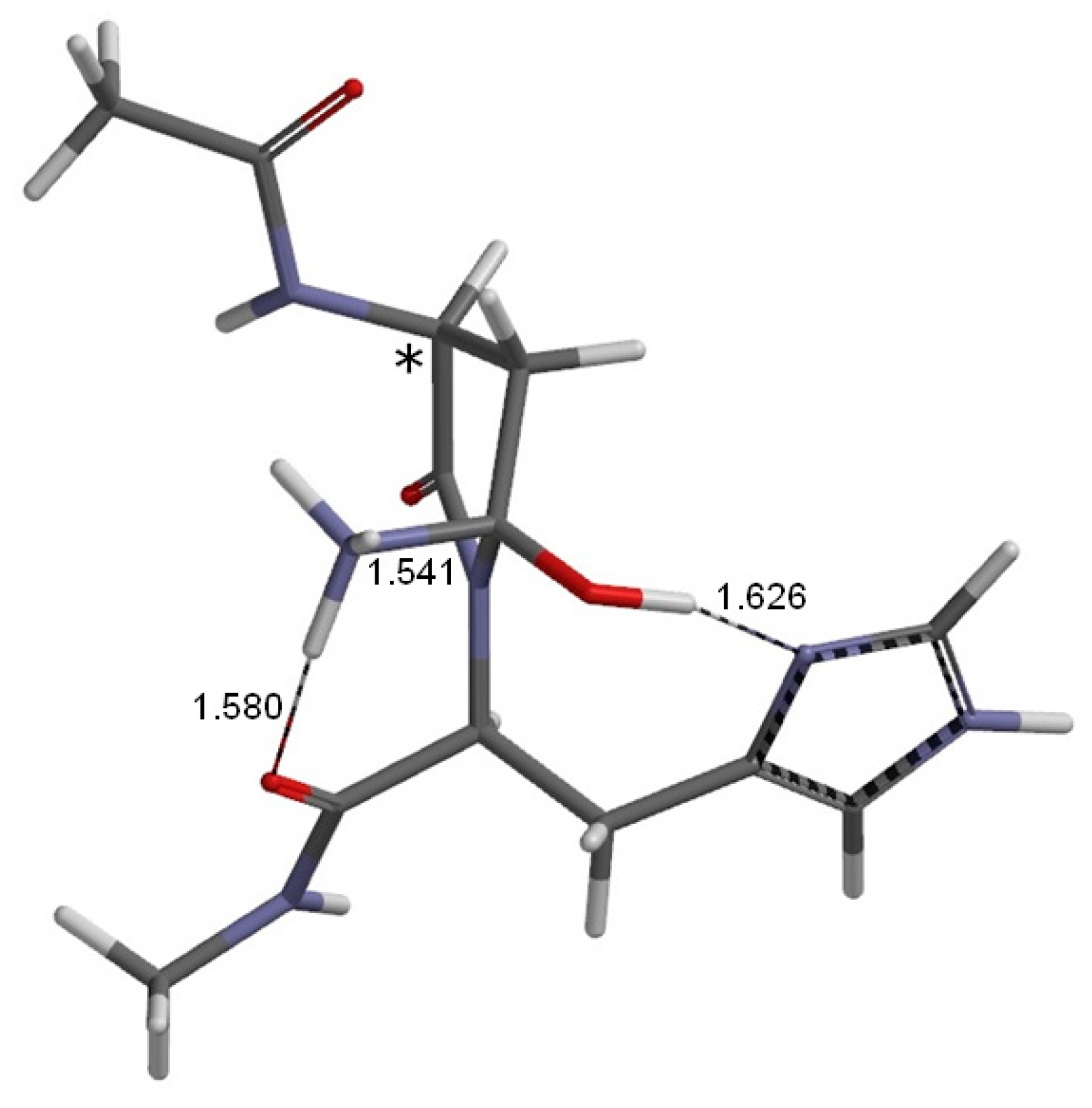
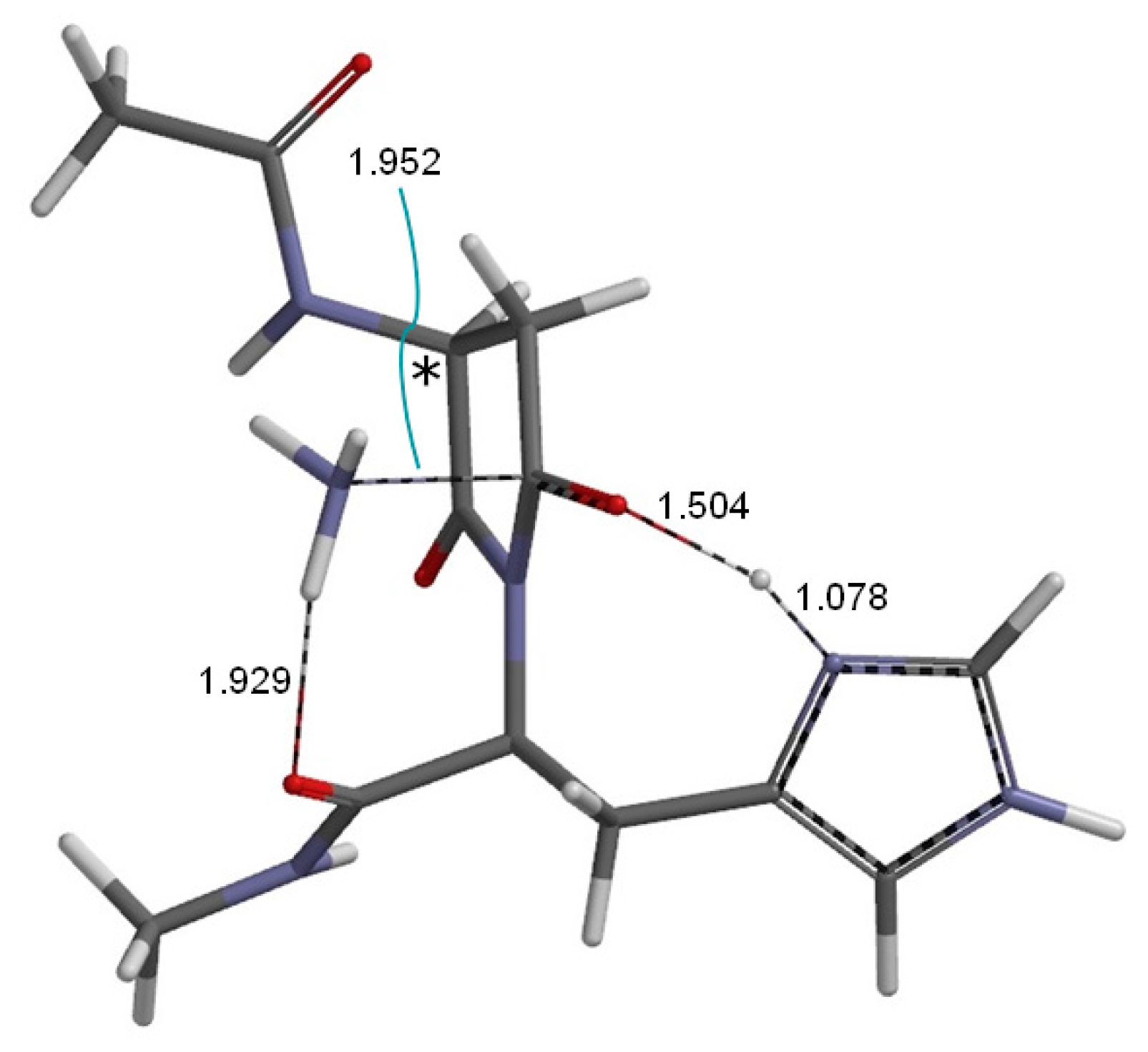
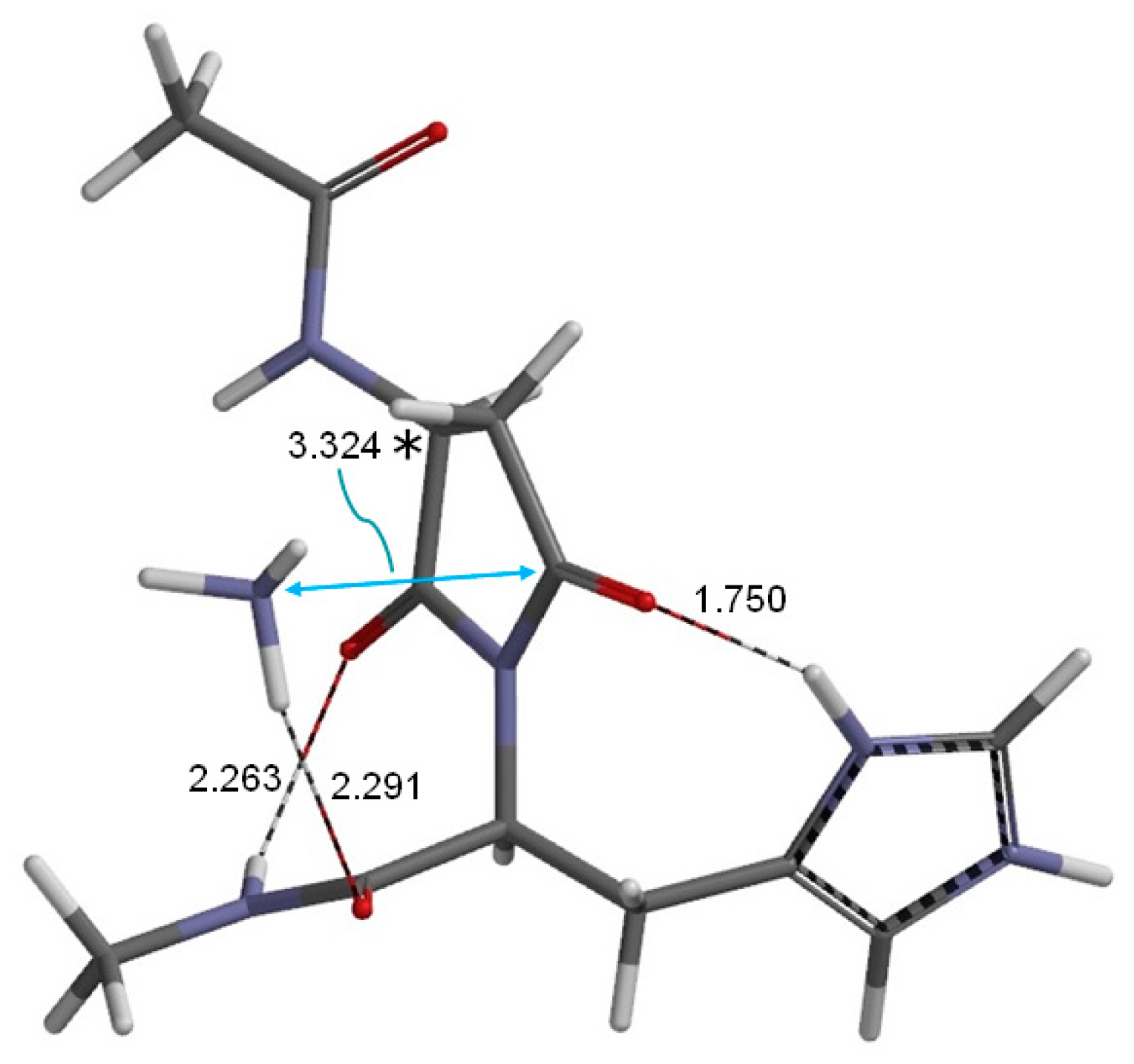
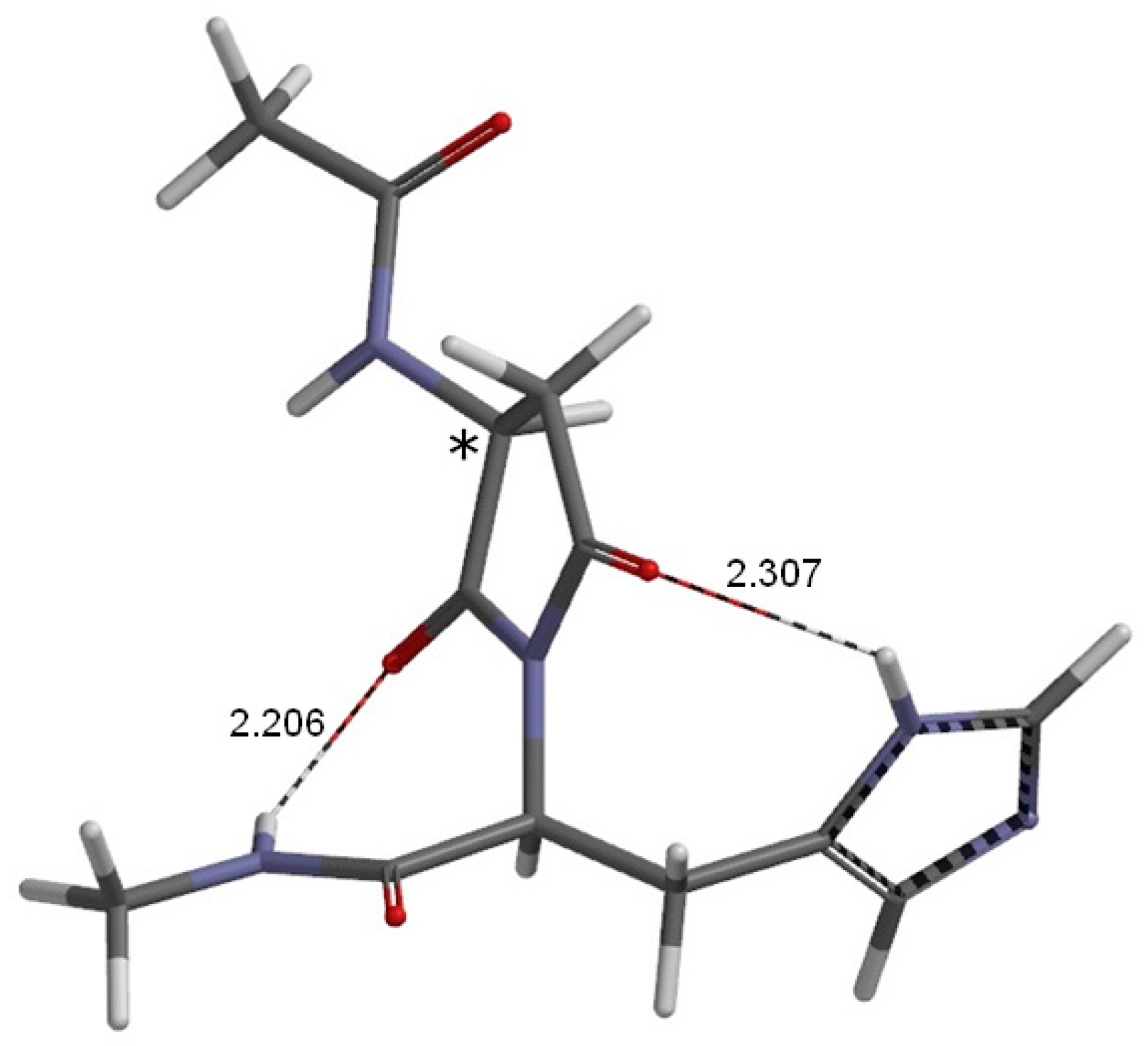
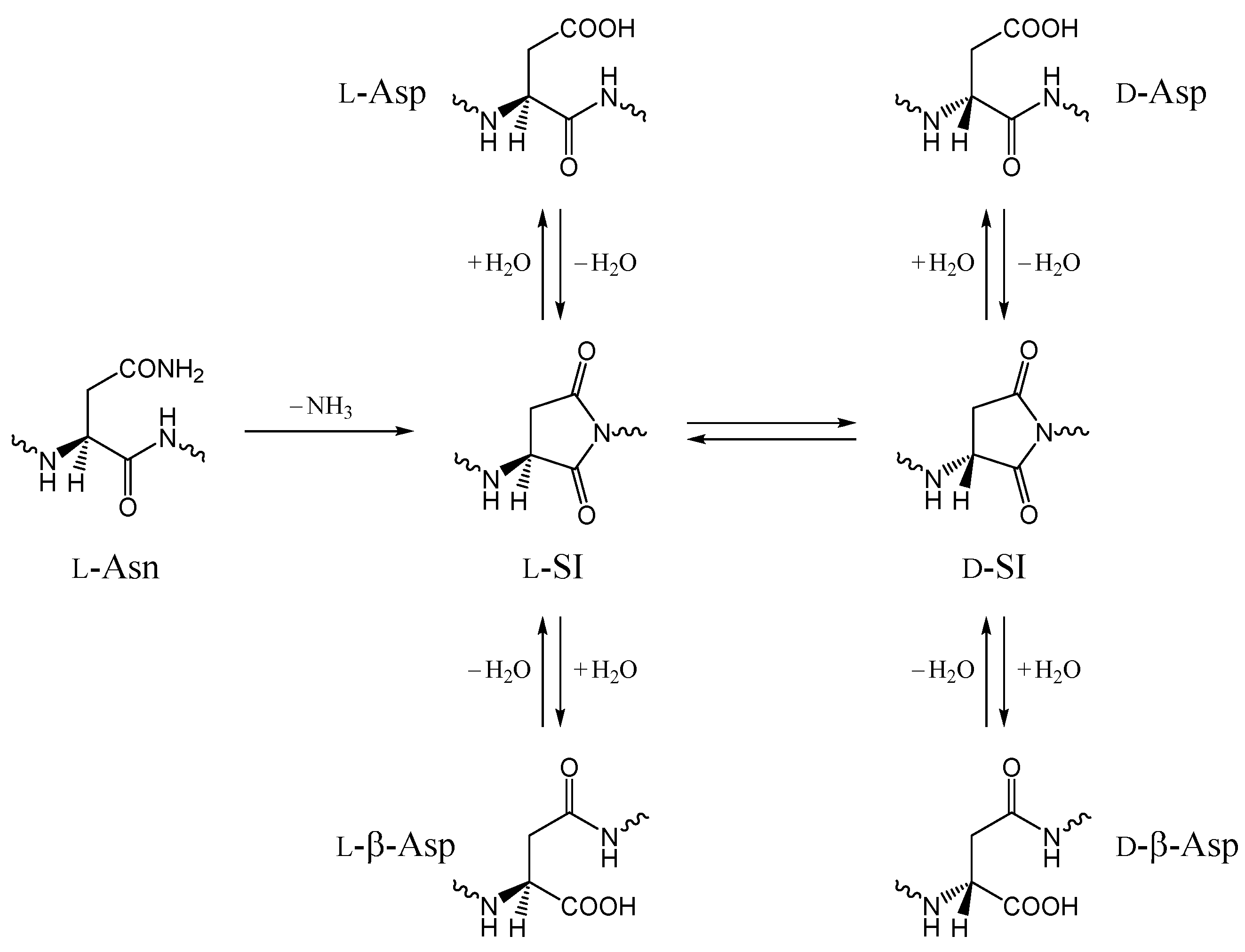
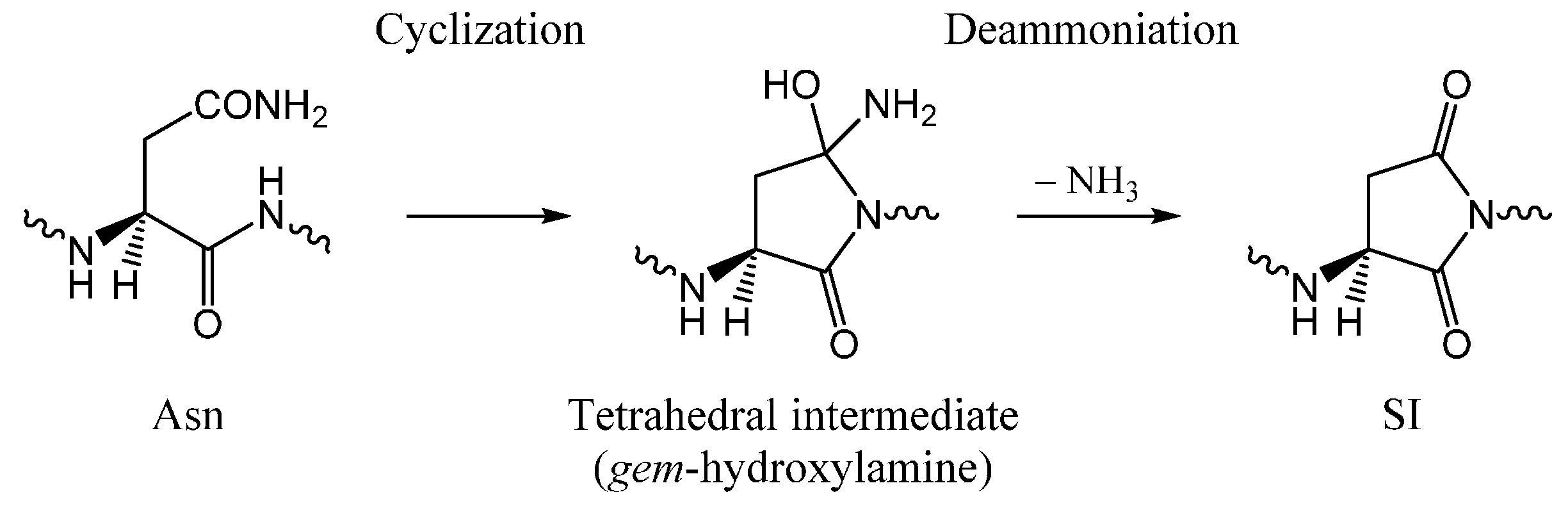
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
Abbreviations
| DFT | density functional theory |
| INT | intermediate |
| IRC | intrinsic reaction coordinate |
| P | product |
| PC | product complex |
| R | reactant |
| SI | succinimide |
| TS | transition state |
| ZPE | zero-point energy |
References
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [PubMed]
- Capasso, S.; Mazzarella, L.; Sica, F.; Zagari, A. Deamidation via cyclic imide in asparaginyl peptides. Pept. Res. 1989, 2, 195–200. [Google Scholar] [PubMed]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm. Res. 1990, 7, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Borchardt, R.T. Chemical pathways of peptide degradation. III. Effect of primary sequence on the pathways of deamidation of asparaginyl residues in hexapeptides. Pharm. Res. 1990, 7, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.T.; Urry, D.W. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in proteins. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Tyler-Cross, R.; Schirch, V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J. Biol. Chem. 1991, 266, 22549–22556. [Google Scholar] [PubMed]
- Clarke, S.; Stephenson, R.C.; Lowenson, J.D. Lability of asparagine and aspartic acid residues in proteins and peptides: Spontaneous deamidation and isomerization reactions. In Stability of Protein Pharmaceuticals, Part A: Chemical and Physical Pathways of Protein Degradation; Ahern, T.J., Manning, M.C., Eds.; Plenum Press: New York, NY, USA, 1992; pp. 1–29. [Google Scholar]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; Greenberg, B.D. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [PubMed]
- Capasso, S.; Mazzarella, L.; Sica, F.; Zagari, A.; Salvadori, S. Kinetics and mechanism of succinimide ring formation in the deamidation process of asparagine residues. J. Chem. Soc. Perkin Trans. 1993, 2, 679–682. [Google Scholar] [CrossRef]
- Capasso, S.; Kirby, A.J.; Salvadori, S.; Sica, F.; Zagari, A. Kinetics and mechanism of the reversible isomerization of aspartic acid residues in tetrapeptides. J. Chem. Soc. Perkin Trans. 1995, 2, 437–442. [Google Scholar] [CrossRef]
- Riha, W.E., III; Izzo, H.V.; Zhang, J.; Ho, C.T. Nonenzymatic deamidation of food proteins. Crit. Rev. Food Sci. Nutr. 1996, 36, 225–255. [Google Scholar] [CrossRef] [PubMed]
- Capasso, S. Thermodynamic parameters of the reversible isomerization of aspartic residues via a succinimide derivative. Thermochim. Acta 1996, 286, 41–50. [Google Scholar] [CrossRef]
- Powell, M.F. A compendium and hydropathy/flexibility analysis of common reactive sites in proteins: Reactivity at Asn, Asp, Gln, and Met motifs in neutral pH solution. In Formulation, Characterization, and Stability of Protein Drugs; Pearlman, R., Wang, Y.J., Eds.; Springer Science + Business Media: New York, NY, USA, 1996; pp. 1–140. [Google Scholar]
- Fujii, N.; Takemoto, L.J.; Momose, Y.; Matsumoto, S.; Hiroki, K.; Akaboshi, M. Formation of four isomers at the Asp-151 residue of aged human αA-crystallin by natural aging. Biochem. Biophys. Res. Commun. 1999, 265, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Capasso, S. Estimation of deamidation rate of asparagine side chains. J. Peptide Res. 2000, 55, 224–229. [Google Scholar] [CrossRef]
- Goolcharran, C.; Stauffer, L.L.; Cleland, J.L.; Borchardt, R.T. The effects of a histidine residue on the C-terminal side of an asparaginyl residue on the rate of deamidation using model peptides. J. Pharm. Sci. 2000, 89, 818–825. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B. Molecular Clocks. Proc. Natl. Acad. Sci. USA 2001, 98, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B.; Merrifield, R.B. Mass spectrometric evaluation of synthetic peptides as primary structure models for peptide and protein deamidation. J. Pept. Res. 2001, 57, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Lindner, H.; Helliger, W. Age-dependent deamidation of asparagine residues in proteins. Exp. Gerontol. 2001, 36, 1551–1563. [Google Scholar] [CrossRef]
- Fujii, N.; Matsumoto, S.; Hiroki, K.; Takemoto, L. Inversion and isomerization of Asp-58 residue in human αA-crystallin from normal aged lenses and cataractous lenses. Biochim. Biophys. Acta 2001, 1549, 179–187. [Google Scholar] [CrossRef]
- Ritz-Timme, S.; Collins, M.J. Racemization of aspartic acid in human proteins. Aging Res. Rev. 2002, 1, 43–59. [Google Scholar] [CrossRef]
- Reissner, K.J.; Aswad, D.W. Deamidation and isoaspartate formation in proteins: Unwanted alterations or surreptitious signals? Cell. Mol. Life Sci. 2003, 60, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, Z.W.; Robinson, B.R.; Robinson, A.L.; Robinson, J.A.; Robinson, M.L.; Robinson, A.B. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl peptapeptides. J. Pept. Res. 2004, 63, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Molecular Clocks: Deamidation of Asparaginyl and Glutaminyl Residues in Peptides and Proteins; Althouse Press: Cave Junction, OR, USA, 2004. [Google Scholar]
- Fujii, N. d-Amino acid in elderly tissues. Biol. Pharm. Bull. 2005, 28, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Houchin, M.L.; Heppert, K.; Topp, E.M. Deamidation, acylation and proteolysis of a model peptide in PLGA films. J. Control. Release 2006, 112, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.A.; Borchardt, R.T. Formulation considerations for proteins susceptible to asparagine deamidation and aspartate isomerization. J. Pharm. Sci. 2006, 95, 2321–2336. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.A.; Borchardt, R.T.; Eigenbrot, C.; Shia, S.; Wang, Y.J.; Shire, S.J.; Liu, J.L. Aspartate isomerization in the complementarity-determining regions of two closely related monoclonal antibodies. Biochemistry 2007, 46, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Houchin, M.L.; Topp, E.M. Chemical degradation of peptides and proteins in PLGA: A review of reactions and mechanisms. J. Pharm. Sci. 2008, 97, 2395–2404. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, Y.; Konoha, K.; Kawahara, M.; Nakagomi, K. Quantification of structural alterations of l-Asp and l-Asn residues in peptides related to neuronal diseases by reversed-phase high-performance liquid chromatography. Chem. Biodiv. 2010, 7, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Hooi, M.Y.S.; Truscott, R.J.W. Racemisation and human cataract. d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. AGE 2011, 33, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Kawaguchi, T.; Sasaki, H.; Fujii, N. Simultaneous stereoinversion and isomerization at the Asp-4 residue in βB2-crystallin from the aged human eye lenses. Biochemistry 2011, 50, 8628–8635. [Google Scholar] [CrossRef] [PubMed]
- Sreedhara, A.; Cordoba, A.; Zhu, Q.; Kwong, J.; Liu, J. Characterization of the isomerization products of aspartate residues at two different sites in a monoclonal antibody. Pharm. Res. 2012, 29, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Aki, K.; Fujii, N.; Fujii, N. Kinetics of isomerization and inversion of aspartate 58 of α-crystalline peptide mimics under physiological conditions. PLoS ONE 2013, 8, e58515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, B.D.; Tran, B.; Moore, J.M.R.; Sharma, V.K.; Kosky, A. Specific catalysis of asparaginyl deamidation by carboxylic acids: Kinetic, thermodynamic, and quantitative structure-property relationship analyses. Mol. Pharm. 2014, 11, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K. Isomerization of aspartyl residues in crystallins and its influence upon cataract. Biochim. Biophys. Acta 2016, 1860, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Fujii, N. Isomerization of Asp residues plays an important role in α-crystallin dissociation. FEBS J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Radkiewicz, J.L.; Zipse, H.; Clarke, S.; Houk, K.N. Accelerated racemization of aspartic acid and asparagine residues via succinimide intermediates: An ab initio theoretical exploration of mechanism. J. Am. Chem. Soc. 1996, 118, 9148–9155. [Google Scholar] [CrossRef]
- Takahashi, O.; Kobayashi, K.; Oda, A. Modeling the enolization of succinimide derivatives, a key step of racemization of aspartic acid residues: Importance of a two-H2O mechanism. Chem. Biodiv. 2010, 7, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O. Two-water-assisted racemization of the succinimide intermediate formed in proteins: A computational model study. Health 2013, 5, 2018–2021. [Google Scholar] [CrossRef]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Reaction mechanism of deamidation of asparaginyl residues in peptides: Effect of solvent molecules. J. Phys. Chem. A 2006, 110, 8354–8365. [Google Scholar] [CrossRef] [PubMed]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Deamidation of asparagine residues: Direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A 2009, 113, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Manabe, N.; Kirikoshi, R.; Takahashi, O. Glycolic acid-catalyzed deamidation of asparagine residues in degrading PLGA matrices: A computational study. Int. J. Mol. Sci. 2015, 16, 7261–7272. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Acetic acid can catalyze succinimide formation from aspartic acid residues by a concerted bond reorganization mechanism: A computational study. Int. J. Mol. Sci. 2015, 16, 1613–1626. [Google Scholar] [CrossRef] [PubMed]
- Halim, M.A.; Almatarneh, M.H.; Poirier, R.A. Mechanistic study of the deamidation reaction of glutamine: A computational approach. J. Phys. Chem. B 2014, 118, 2316–2330. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Gokel, G.W. Dean’s Handbook of Organic Chemistry, 2nd ed.; McGraw-Hill: New York, NY, USA, 2004; Section 8; p. 9. [Google Scholar]
- Camaioni, D.M.; Schwerdtfeger, C.A. Comment on “Accurate experimental values for the free energies of hydration of H+, OH−, and H3O+”. J. Phys. Chem. A 2005, 109, 10795–10797. [Google Scholar] [CrossRef] [PubMed]
- Spartan’14, version 1.1.4 ed; software for molecular modeling; Wavefunction, Inc.: Irvine, CA, USA, 2014.
- Sample Availability: Samples of the compounds are not available from the authors.











© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, O.; Manabe, N.; Kirikoshi, R. A Computational Study of the Mechanism of Succinimide Formation in the Asn–His Sequence: Intramolecular Catalysis by the His Side Chain. Molecules 2016, 21, 327. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21030327
Takahashi O, Manabe N, Kirikoshi R. A Computational Study of the Mechanism of Succinimide Formation in the Asn–His Sequence: Intramolecular Catalysis by the His Side Chain. Molecules. 2016; 21(3):327. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21030327
Chicago/Turabian StyleTakahashi, Ohgi, Noriyoshi Manabe, and Ryota Kirikoshi. 2016. "A Computational Study of the Mechanism of Succinimide Formation in the Asn–His Sequence: Intramolecular Catalysis by the His Side Chain" Molecules 21, no. 3: 327. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21030327