
Spectroscopic and Kinetic Characterization of Peroxidase-Like π-Cation Radical Pinch-Porphyrin-Iron(III) Reaction Intermediate Models of Peroxidase Enzymes

 , ,
, ,
Abstract
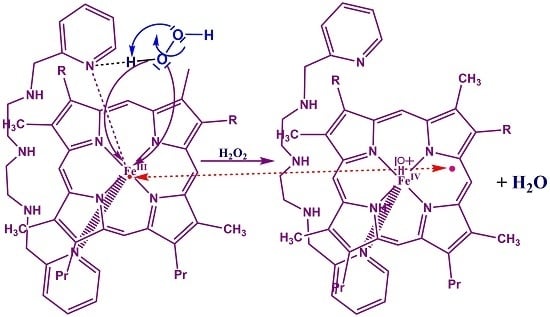
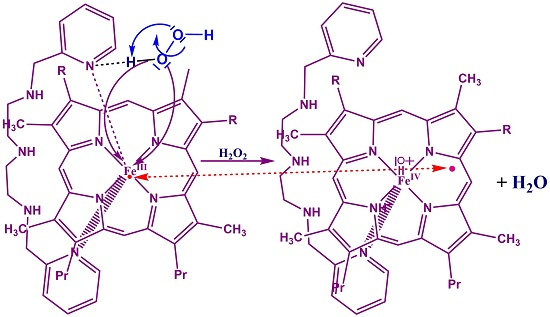
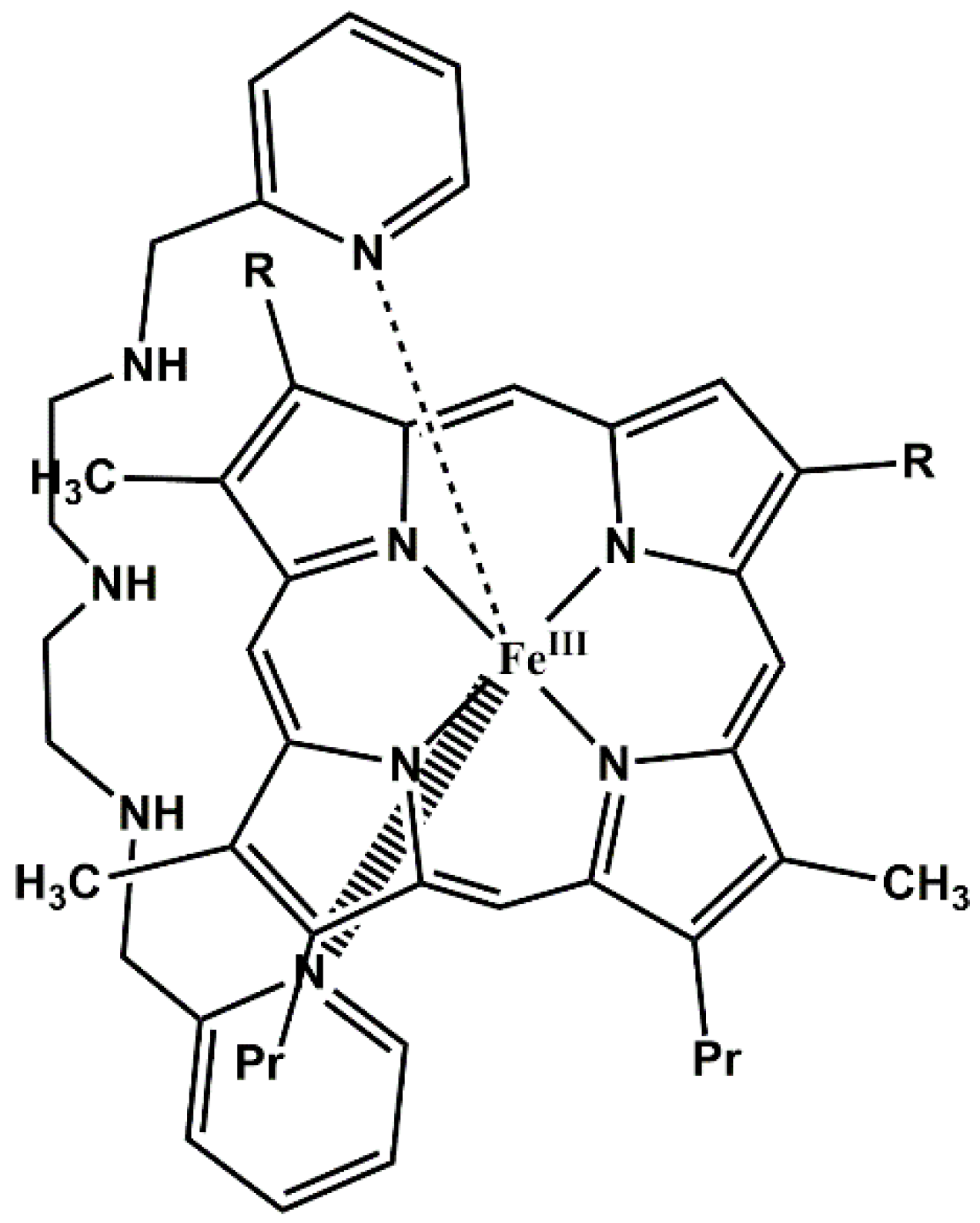
:
1. Introduction
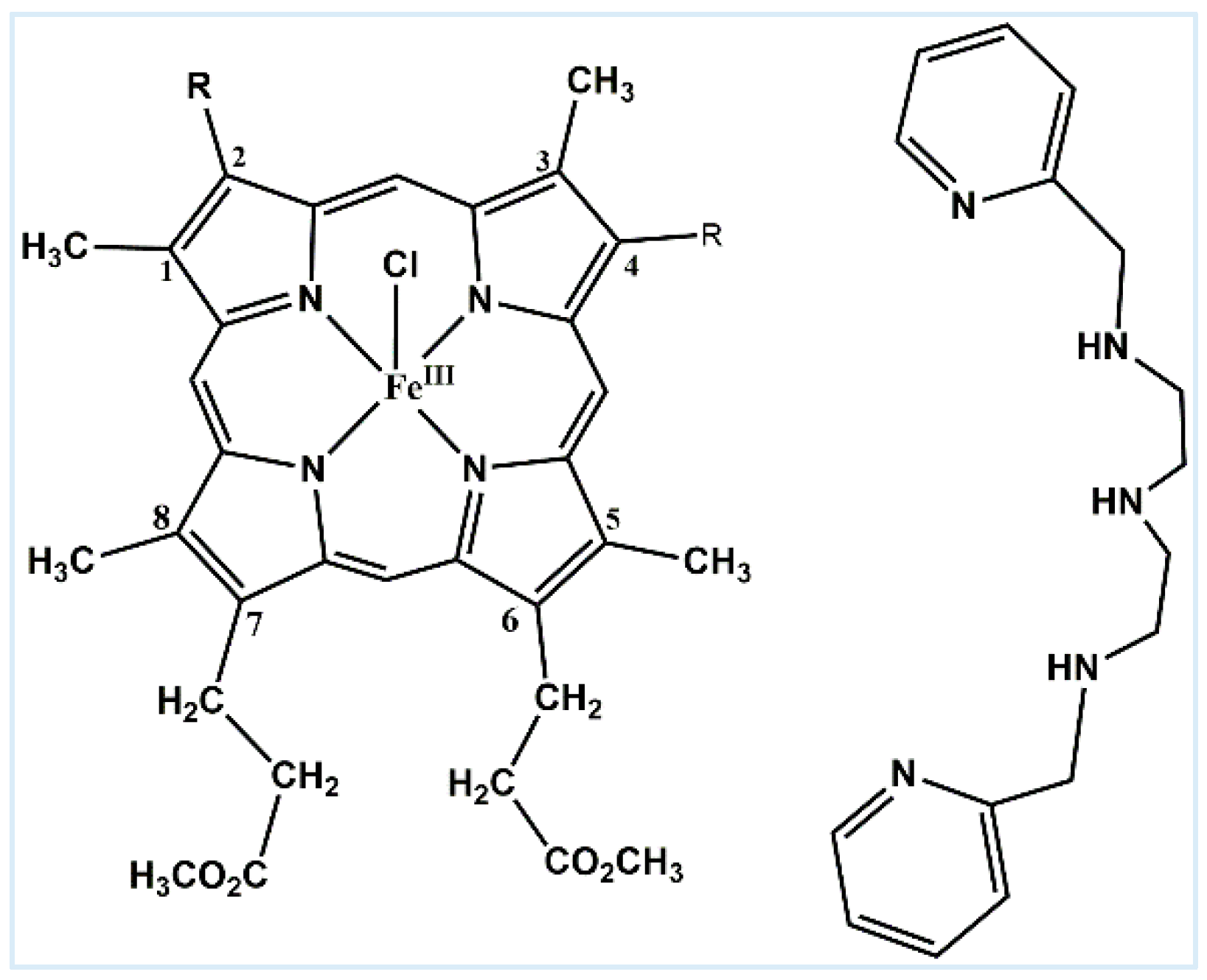
2. Results and Discussion
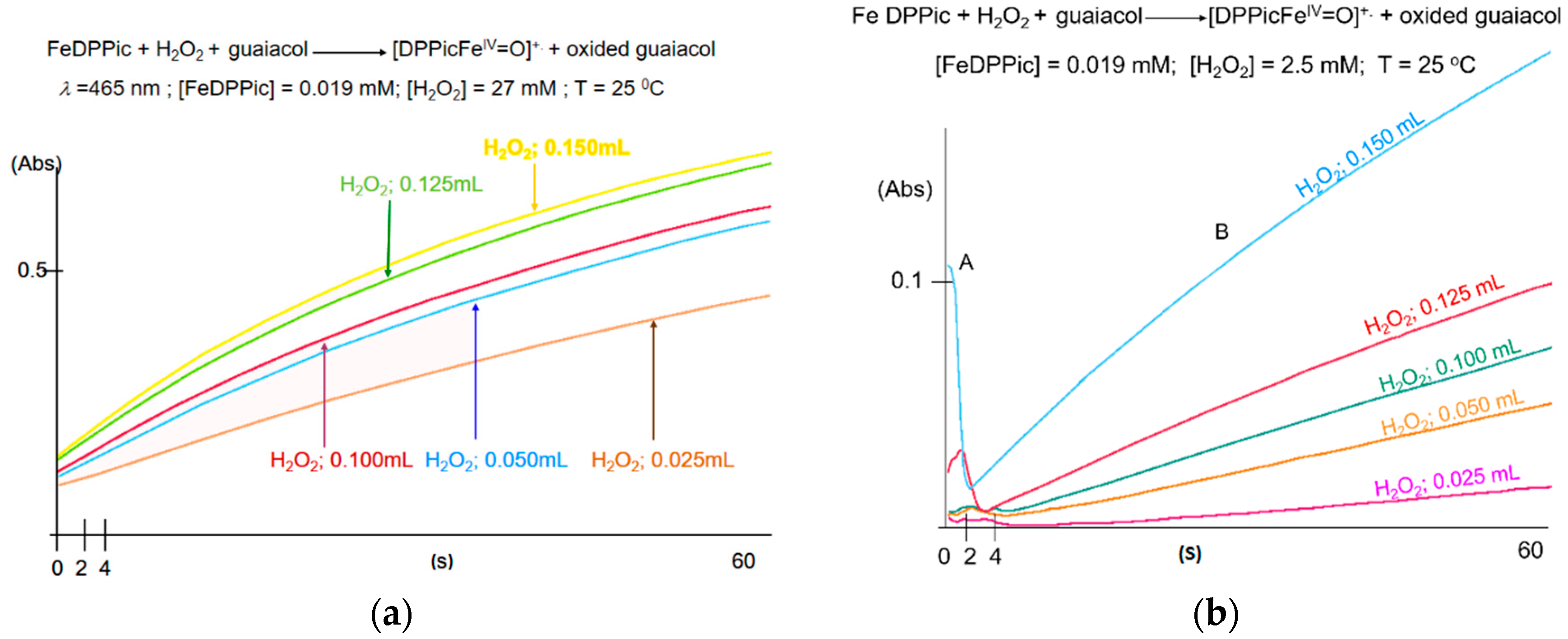
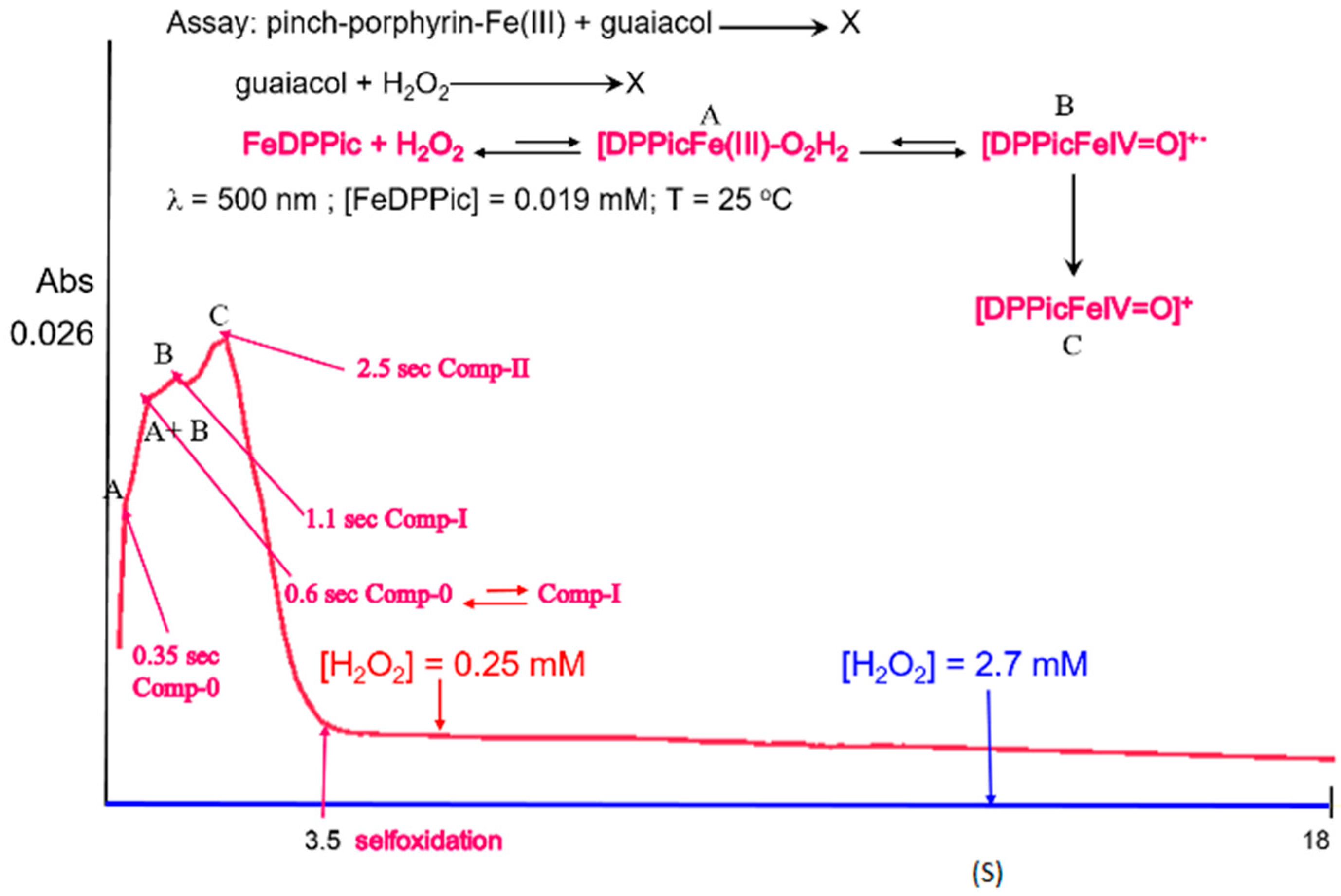
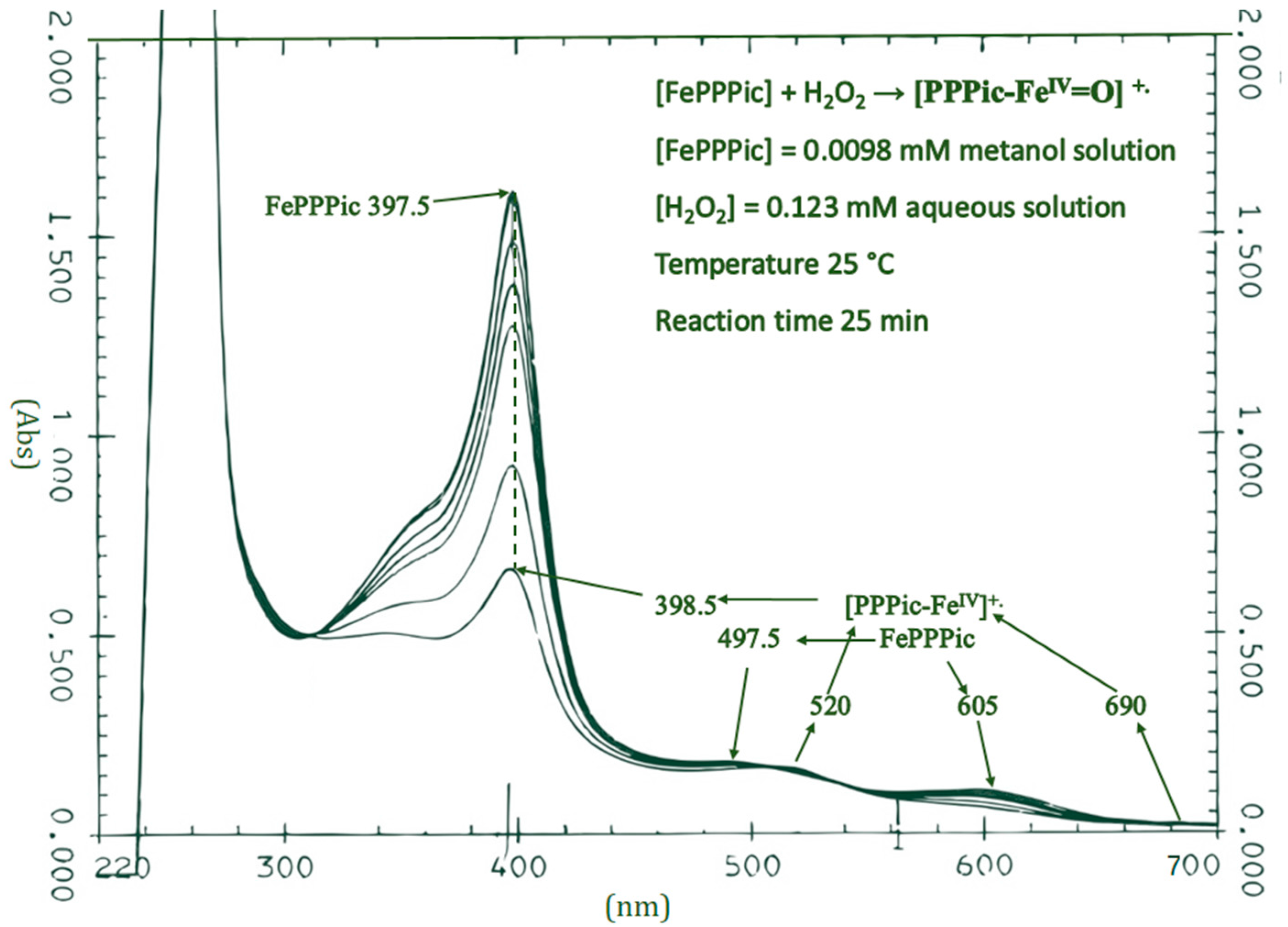
3.1. UV-Vis
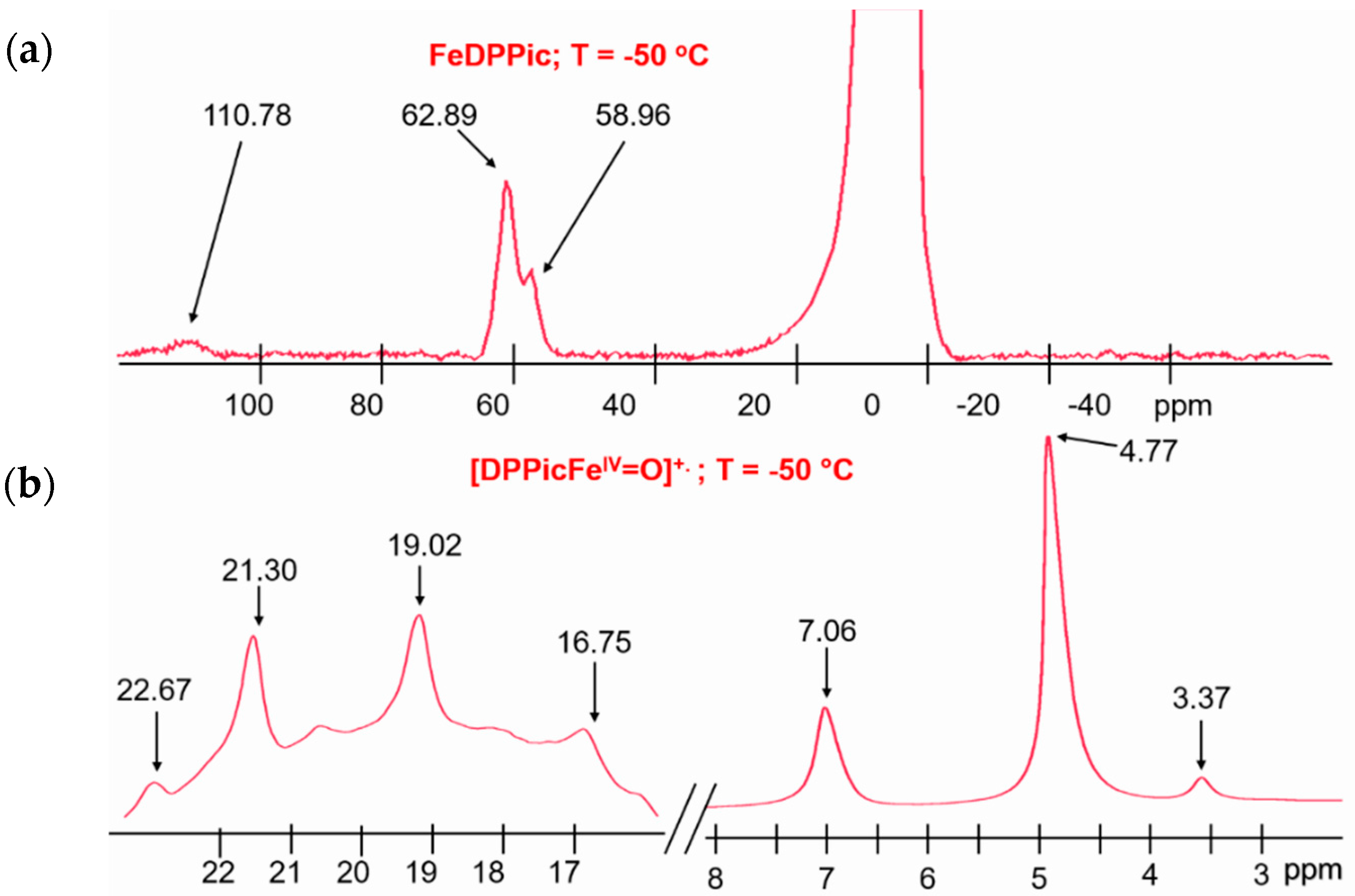
3.2. 1H-NMR of Compound I
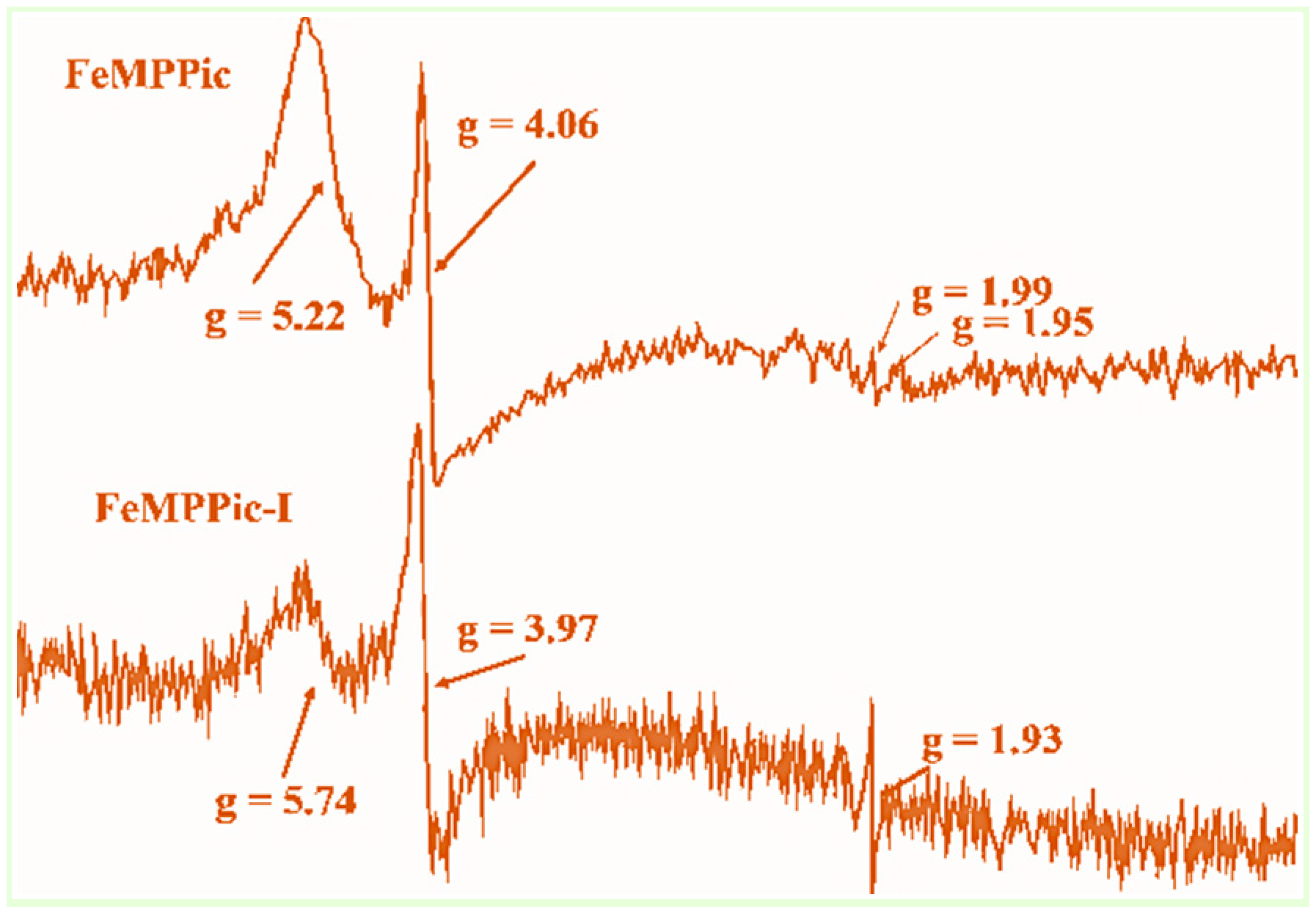
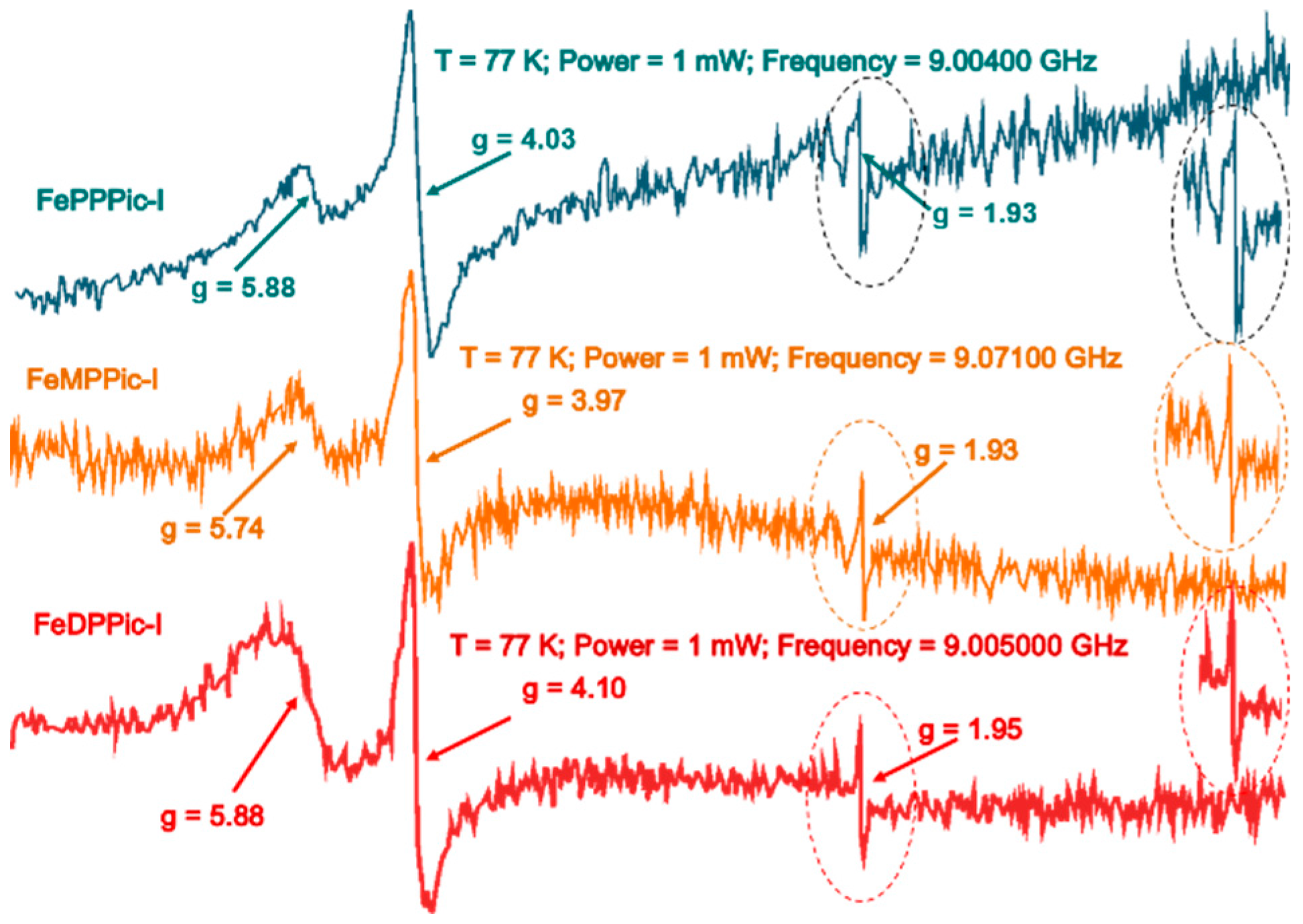
3.3. ESR Spectroscopy
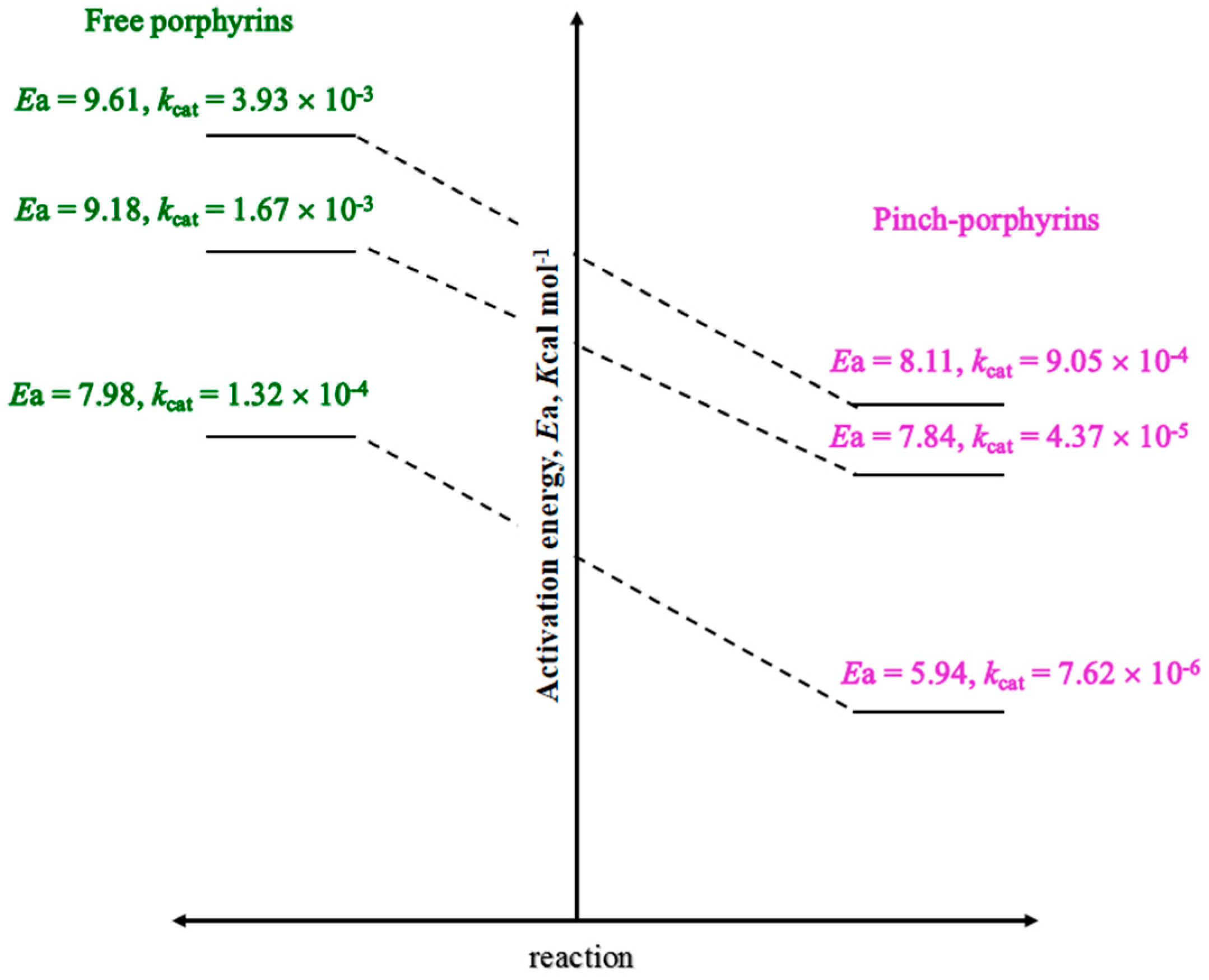
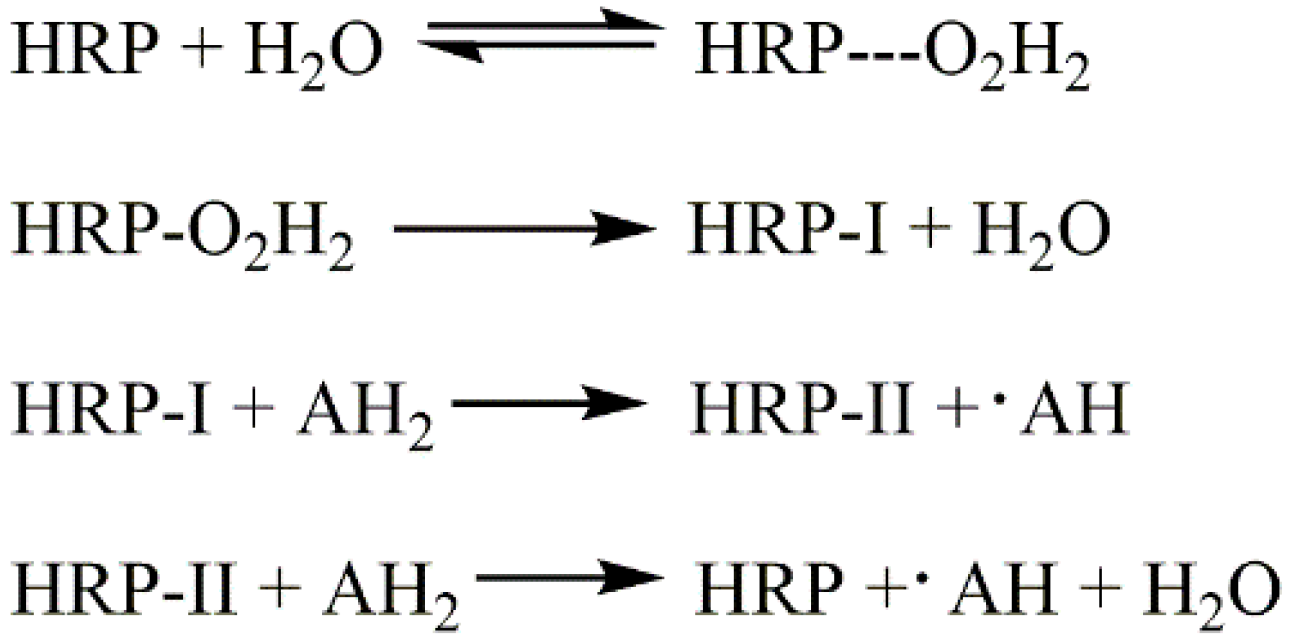
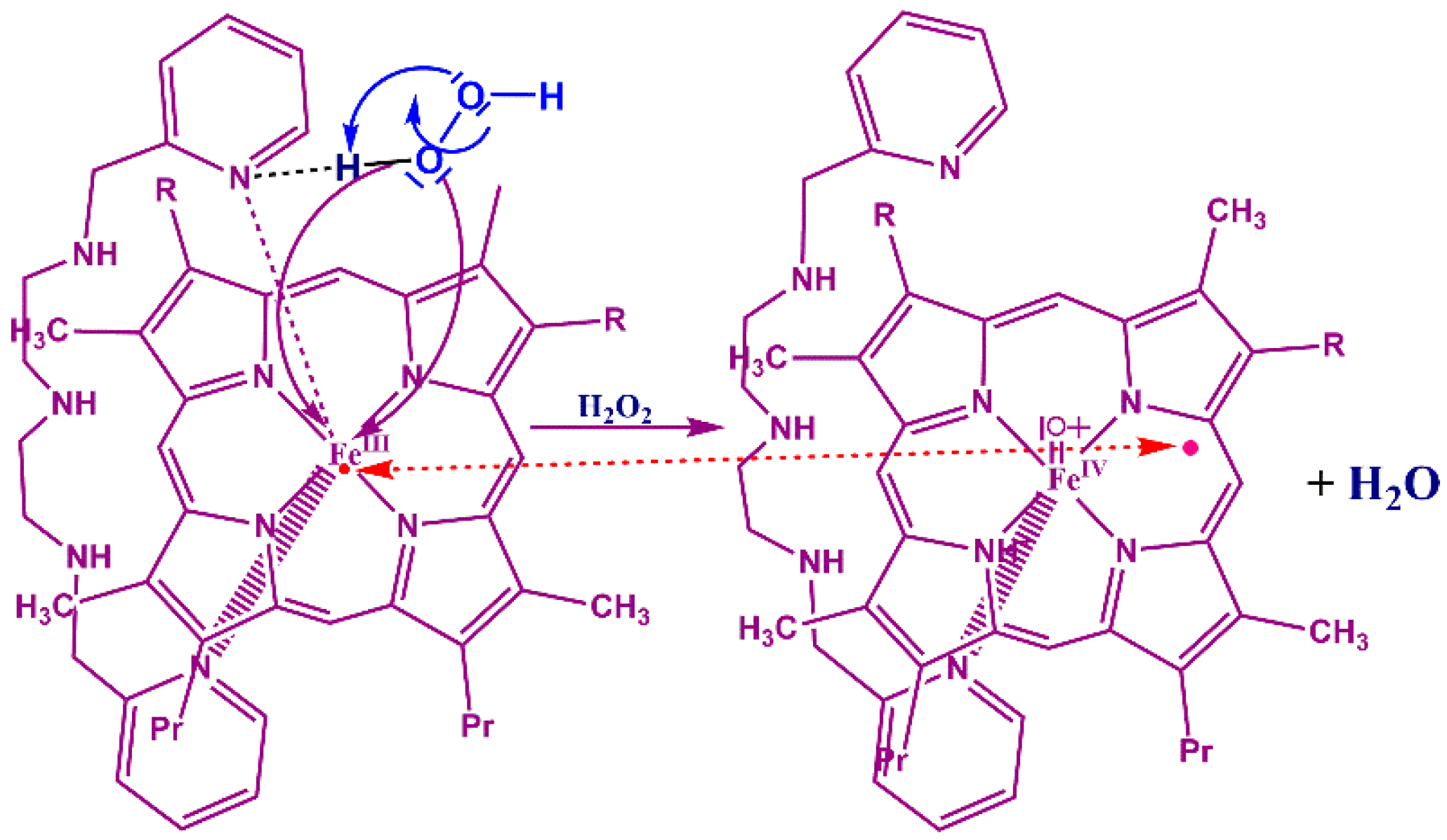
3.4. Reaction Mechanism
3. Material and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lehninger, A.L.; Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 5th ed.; Ahr, K., Ed.; W. H. Freeman and Company: New York, NY, USA, 2008. [Google Scholar]
- Hernández-Anzaldo, S.; Sánchez-Morales, N.; Alcántara-Flores, J.L.; Gutiérrez-Pérez, R.; Zamorano-Ulloa, R.; Escudero, R.; de Hoz, M.J.R.; Reyes-Ortega, Y. ESR and magnetic studies of octahedral [Fe(III)(Cl)(pcd)(H2O)(DMSO)] (pcd = pyridine-2,6-dicarboxylato) compound showing Fe(III) species with different spin states in solution. J. Mol. Struct. 2013, 1040, 22–26. [Google Scholar] [CrossRef]
- Reyes-Ortega, Y.; Alvarez-Toledano, C.; Ramírez-Rosales, D.; Sánchez-Sandoval, A.; González-Vergara, E.; Zamorano-Ulloa, R. Pinch-porphyrins, new spectroscopic and kinetic models of peroxidases. Dalton Trans. 1998, 667–674. [Google Scholar] [CrossRef]
- Drago, R.S. Physical Methods for Chemists, 2nd ed.; Saunders College Publishing: Philadelphia, PA, USA, 1992. [Google Scholar]
- Dunford, B. Heme Peroxidases; Wiley-VCH: New York, NY, USA, 1999. [Google Scholar]
- Reed, J.C.A.; Mashiko, T.; Bentley, S.P.W.; Kastner, M.E.; Scheidt, W.R.; Spartalian, K.; Lang, G. The missing heme spin state and a model for cytochrome c’. The mixed S = 3/2, 5/2 intermediate spin ferric porphyrin: perchlorate(meso-tetraphenylporphinato)iron(III). J. Am. Chem. Soc. 1979, 101, 2948–2958. [Google Scholar] [CrossRef]
- Jennifer, S.; Stillman, M.J.; Dunford, H.B. Photochemical reactions of horseradish peroxidase compounds I and I1 at room temperature and 10° K. Biochemistry 1975, 14, 3183–3188. [Google Scholar]
- Reed, C.A.; Guiset, F. A “Magnetochemical” Series. Ligand Field Strengths of Weakly Binding Anions Deduced from S = 3/2, 5/2 Spin State Mixing in Iron(III) Porphyrins. J. Am. Chem. Soc. 1996, 118, 3281. [Google Scholar] [CrossRef]
- Jones, P.; Dunford, H.B. On the mechanism of compound I formation from peroxidases and catalases. J. Theory Biol. 1977, 69, 457–470. [Google Scholar] [CrossRef]
- Jones, P.; Dunford, H.B. The mechanism of compound I formation revisited. J. Inorg. Biochem. 2005, 99, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Mason, H.S.; Morrison, M. Oxidases and Related Redox Systems; University Park Press: Baltimore, MD, USA, 1973; Volume 1, pp. 388–401. [Google Scholar]
- Sanchez-Sandoval, A.; Ramirez-Rosales, D.; Zamorano-Ulloa, R.; Alvarez-Toledano, C.; Moya-Cabrera, M.; Reyes-Ortega, Y. New pinch-porphyrin complexes with quantum mixed spin ground state S = 3/2, 5/2, of iron(III) and their catalytic activity as peroxidase. Biophys. Chem. 2003, 106, 253–265. [Google Scholar] [CrossRef]
- Saunders, B.C.; Holmes-Siedle, A.G.; Stark, B.P. Peroxidase; Butterworths: London, UK, 1964; Volume 3, p. 1015. [Google Scholar]
- Traylor, T.G.; Lee, W.A.; Stynes, D.V. Model compound studies related to peroxidases. Mechanisms of reactions of hemins with peracids. J. Am. Chem. Soc. 1984, 106, 755–764. [Google Scholar] [CrossRef]
- Bonagura, C.A.; Bhaskar, B.; Shimizu, H.; Li, H.; Sundaramoorthy, M.; McRee, D.E.; Goodim, B.G.; Poulos, T.L. High-Resolution Crystal Structures and Spectroscopy of Native and Compound I Cytochrome c Peroxidase. Biochem. 2003, 42, 5600–5608. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.C.; Audi, R.; Ogusucu, R.; Monteiro, G.; Soares Netto, L.E.; Augusto, O. Horseradish peroxidase compound I as a tool to investigate reactive protein-cysteine residues: From quantification to kinetics. Free Radic. Biol. Med. 2011, 50, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Hewson, W.D.; Hager, L.P. Oxidation of horseradish peroxidase compound II to compound I. J. Biol. Chem. 1979, 254, 3182–3186. [Google Scholar] [PubMed]
- Tanaka, M.; Matsuura, K.; Yoshioka, S.; Takahashi, S.; Ishimori, S.; Hori, S.; Morishima, I. Activation of hydrogen peroxide in horseradish peroxidase occurs within ~200 ms observed by a new freeze-quench device. Biophys. J. 2003, 84, 1998–2004. [Google Scholar] [CrossRef]
- Newcomb, M.; Zhang, R.; Chandrasena, R.E.P.; Halgrimson, J.A.; Horner, J.H.; Makris, T.M.; Sligar, S.G. Cytochrome P450 compound I martin newcomb. J. Am. Chem. Soc. 2006, 128, 4580–4581. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Ogawa, T.; Inoue, K.; Masuda, T. Characterization of cytosolic tetrapyrrole-binding proteins in Arabidopsis thaliana. Photochem. Photobiol. Sci. 2008, 7, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peter, S.; Kinne, M.; Hofrichter, M.; Groves, T. Detection and kinetic characterization of a highly reactive heme−thiolate peroxygenase compound I. J. Am. Chem. Soc. 2012, 134, 12897–12900. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Bonilla, W.; Cortes-Figuero, J.E.; Souto-Bachiller, F.A.; Rodriguez, L.; Lopez-Garriga, J. Formation of compound I and compound II ferryl species in the reaction of hemoglobin I from Lucina pectinata with hydrogen peroxide. Arch. Biochem. Biophys. 2001, 390, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, K.; Gold, A.R.; Austin, N.L.; Ball, M. Compound I and compound II analogues from porpholactones. Inorg. Chem. 1997, 36, 4555–4566. [Google Scholar] [CrossRef] [PubMed]
- Oszajca, M.; Franke, A.; Drzewiecka-Matuszek, A.; Brindell, M.; Stochel, G.; van Eldik, R. Temperature and pressure effects on C−H abstraction reactions involving compound I and II mimics in aqueous solution. Inorg. Chem. 2014, 53, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- La Mar, G.N.; de Ropp, J.S.; Smith, K.M.; Langry, K.C. Proton nuclear magnetic resonance investigation of the electronic structure of compound I of horseradish peroxidase. J. Biol. Chem. 1981, 256, 237–243. [Google Scholar] [PubMed]
- Asokan, A.; de Ropp, J.S.; Newmyer, S.; de Montellano, P.O.; La Mar, G.L. Solution 1H NMR of the molecular and electronic structure of the heme cavity and substrate binding pocket of high-spin ferric horseradish peroxidase: Effect of His42Ala mutation. J. Am. Chem. Soc. 2001, 123, 4243–4254. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Pease, E.A.; Tien, M.; Turano, P. 1H NMR Investigation of Manganese Peroxidase from Phanerochaete chrysosporium. Biochemistry 1992, 31, 10009–10017. [Google Scholar] [CrossRef] [PubMed]
- Kurland, R.J.; Little, R.G.; Davis, D.G.; Ho, C. Proton Magnetic Resonance Study of High- and Low-Spin Hemin Derivatives. Biochemistry 1971, 10, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Budd, D.L.; La Mar, G.N.; Langry, K.C.; Smith, K.M.; Nayyir-Mazhir, R. lH-NMR study of high-spin ferric natural porphyrin derivatives as models of methemoproteins. J. Am. Chem. Soc. 1979, 101, 6091–6096. [Google Scholar] [CrossRef]
- Savenkova, M.I.; Satterlee, J.E.; Erman, J.E.; Siems, W.F.; Helms, G. Expression, purification, characterization, and NMR studies of highly deuterated recombinant cytochrome c peroxidase. Biochemistry 2001, 40, 12123–12131. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tachikawa, H.; Yi, X.; Manoj, K.M.; Hager, L.P. The two-dimensional NMR study of the heme active site structure of chloroperoxidase. J. Biol. Chem. 2003, 278, 7765–7774. [Google Scholar] [CrossRef] [PubMed]
- Balch, A.L.; La Mar, G.N.; Latos-Grazynski, L.; Renner, M.W.; Thanabal, V. Nuclear magnetic resonance studies of axial amine coordination in synthetic ferryl, porphyrin complexes and in ferryl myoglobin. J. Am. Chem. Soc. 1985, 107, 3003–3007. [Google Scholar] [CrossRef]
- Gold, A.; Jayaraj, K.; Doppelt, P.; Weis, R.; Chottard, G.; Bill, E.; Ding, X.; Trautwein, A.X. Oxoferryl complexes of the halogenated (porphinato)iron catalyst (tetrakis(2,6-dichlorophenyl)porphinato)iron. J. Am. Chem. Soc. 1988, 110, 5756–5761. [Google Scholar] [CrossRef]
- Khindaria, A.; Aust, S. EPR detection and characterization of lignin peroxidase porphyrin π-cation radical. Biochemistry 1996, 35, 13107–13111. [Google Scholar] [CrossRef] [PubMed]
- Davydov, R.; Strushkevich, N.; Smil, D.; Yantsevich, A.; Gilep, A.; Usanov, S.; Hoffman, B.H. Evidence that compound I is the active species in both the hydroxylase lyase steps by which P450scc converts cholesterol to pregnenolone: EPR/ENDOR/cryoreduction/annealing studies. Biochemistry 2015, 54, 7089–7097. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.K.; Dalton, D.A.; Rosell, F.I.; Lloyd-Raven, E. Class I heme peroxidases: Characterization of soybean ascorbate peroxidase. Arch. Biochem. Biophy. 1998, 360, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Loginova, N.V.; Kovalchuk, T.V.; Zheldakova, R.A.; Osipovich, N.P.; Sorokin, V.L.; Polozov, G.I.; Ksendzova, G.A.; Glushonok, G.K.; Chernyavskaya, A.A.; Shadyro, O.I. Synthesis and biological evaluation of copper (II) complexes of sterically hindered o-aminophenol derivativesas antimicrobial agents. Bioorg. Med. Chem. Lett. 2006, 16, 5403–5407. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Yoshimura, T.; Kamada, H. ESR studies of A1u and A2u oxoiron(IV) porphyrin π-cation radical complexes. Spin coupling between ferryl iron and A1u/A2u orbitals. Inorg. Chem. 1996, 35, 2373–2377. [Google Scholar] [CrossRef] [PubMed]
- Khindaria, A.; Yamazaki, I.; Aust, S.D. Veratryl alcohol oxidation by lignin peroxidase? Biochemistry 1995, 34, 16860–16869. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Ortega, Y.; Arellano-Merino, C.; Mejía-Rodríguez, J.; Zamorano-Ulloa, R. Activation parameters of pinch-porphyrin complexes, models of peroxidase enzymes. Inf. Technol. 2002, 13, 65–71. [Google Scholar]
- Sample Availability: Samples of the obtained compounds are available from the authors.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Sources | λmax (nm) | Reference |
|---|---|---|
| Horseradish Peroxidase-I | 475, 530 | [7] |
| Horseradish Peroxidase-I | 529, 555 | [18] |
| Cytochrome P450-I | 515, 572 | [19] |
| Cytosolic Heme Binding Protein-I | 540, 547 | [20] |
| Heme-thiolate Peroxygenase-I | 538, 571 | [21] |
| Hemoglobin I (from Lucina pectinate)-I | 519, 650 | [22] |
| Chloro(2-oxa-3-oxotetramesitylporphinato)Fe(III) | 556, 601 | [23] |
| Chloro(2-oxa-3-oxotetrakis(2,6-dichlorophenyl)porphinato)Fe(III) | 560, 610 | [24] |
| [meso-Tetrakis(2,4,6-trimethyl-3-sulfonatophenyl)-porphinato]Fe(III) | 526, 580 | [25] |
| [FePPPic]-I | 520, 690 | This work |
| [FeMPPic]-I | 522, - | This work |
| [FeDPPic]-I | 520, 690 | This work |
| Compound Source | Chemical Shift (ppm) | Reference |
|---|---|---|
| Horseradish Peroxidase-I | 80, 7 | [30] |
| H42A-Horseradish Peroxidase-I | 80, 6.5 | [31] |
| Cytochrome c peroxidase-I | 35, 5 | [32] |
| Chloroperoxidase-I | 48, 8 | [33] |
| Chloro(2-oxa-3-oxotetramesitylporphinato)Fe(III) | 20, 7.6 | [34] |
| Chloro(2-oxa-3-oxotetrakis(2,6-dichlorophenyl)-porphinato)Fe(III) | 20, 8.9 | [34] |
| (py)tetra-m-tolyporphyrin)Fe(III)-I | −38, −17.7 | [28] |
| Manganese Peroxidase-I | 40, 7.7 | [35] |
| Lignin peroxidase-I | 42, 7.4 | [35] |
| [FePPPic]-I | 39, 7.12 | This work |
| [FeMPPic]-I | 29, 7.02 | This work |
| [FeDPPic]-I | 19, 7.06 | This work |
| Compound Source | g Values | Reference |
|---|---|---|
| Horseradish Peroxidase-I | 6.38, 5.38, 2.00 | [35] |
| Pea ascorbate peroxidase-I | 6.04, 5.27, 1.99 | [36] |
| Chloro(2-oxa-3-oxotetramesitylporphinato)Fe(III) | 3.98, 2.00 | [33] |
| Chloro(2-oxa-3-oxotetrakis(2,6-dichlorophenyl)-porphinato)Fe(III) | 3.72, 1.99 | [33] |
| 4,6-di(tert-Butyl)-2-aminophenolCu(II)-I | 2.32, 2.06 | [37] |
| 2-Anilino-4,6-di(tert-butyl)-2-aminophenolCu(II)-I | 2.27, 2.07 | [37] |
| Oxyferrous hp450scc-DHC-I | 2.44, 2.25, 1.91 | [38] |
| Lignin peroxidase-I | 6.6, 1.95 | [39] |
| [FePPPic]-I | 5.88, 4.03, 1.93 | This work |
| [FeMPPic]-I | 5.74, 3.98, 1.93 | This work |
| [FeDPPic]-I | 5.88, 4.10, 1.95 | This work |
| [Pinch-Porphyrin] | ca [Pinch-Porpyrin]f, Methanol, mM | ca [H2O2]f, Aqua, mM |
|---|---|---|
| [FePPPic] | 0.0098 | 0.123 |
| [FeMPPic] | 0.0487 | 1.14 |
| [FeDPPic] | 0.057 | 1.34 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández Anzaldo, S.; Arroyo Abad, U.; León García, A.; Ramírez Rosales, D.; Zamorano Ulloa, R.; Reyes Ortega, Y. Spectroscopic and Kinetic Characterization of Peroxidase-Like π-Cation Radical Pinch-Porphyrin-Iron(III) Reaction Intermediate Models of Peroxidase Enzymes. Molecules 2016, 21, 804. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21070804
Hernández Anzaldo S, Arroyo Abad U, León García A, Ramírez Rosales D, Zamorano Ulloa R, Reyes Ortega Y. Spectroscopic and Kinetic Characterization of Peroxidase-Like π-Cation Radical Pinch-Porphyrin-Iron(III) Reaction Intermediate Models of Peroxidase Enzymes. Molecules. 2016; 21(7):804. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21070804
Chicago/Turabian StyleHernández Anzaldo, Samuel, Uriel Arroyo Abad, Armando León García, Daniel Ramírez Rosales, Rafael Zamorano Ulloa, and Yasmi Reyes Ortega. 2016. "Spectroscopic and Kinetic Characterization of Peroxidase-Like π-Cation Radical Pinch-Porphyrin-Iron(III) Reaction Intermediate Models of Peroxidase Enzymes" Molecules 21, no. 7: 804. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21070804