Virtual Screening against Phosphoglycerate Kinase 1 in Quest of Novel Apoptosis Inhibitors

 ,
,
Abstract
:
1. Introduction
2. Results and Discussions
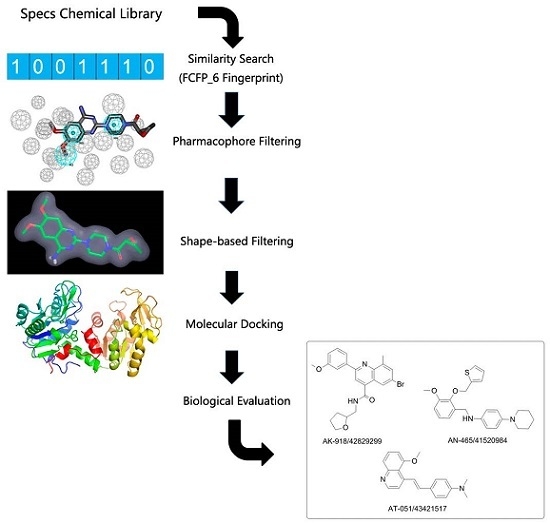
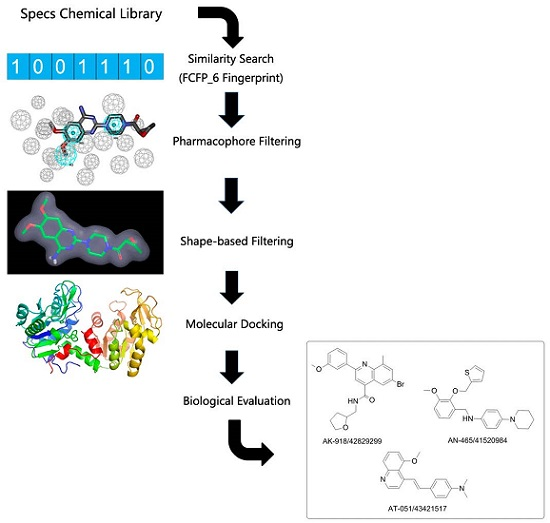
2.1. FCFP_6 Fingerprint-Based Similarity Search
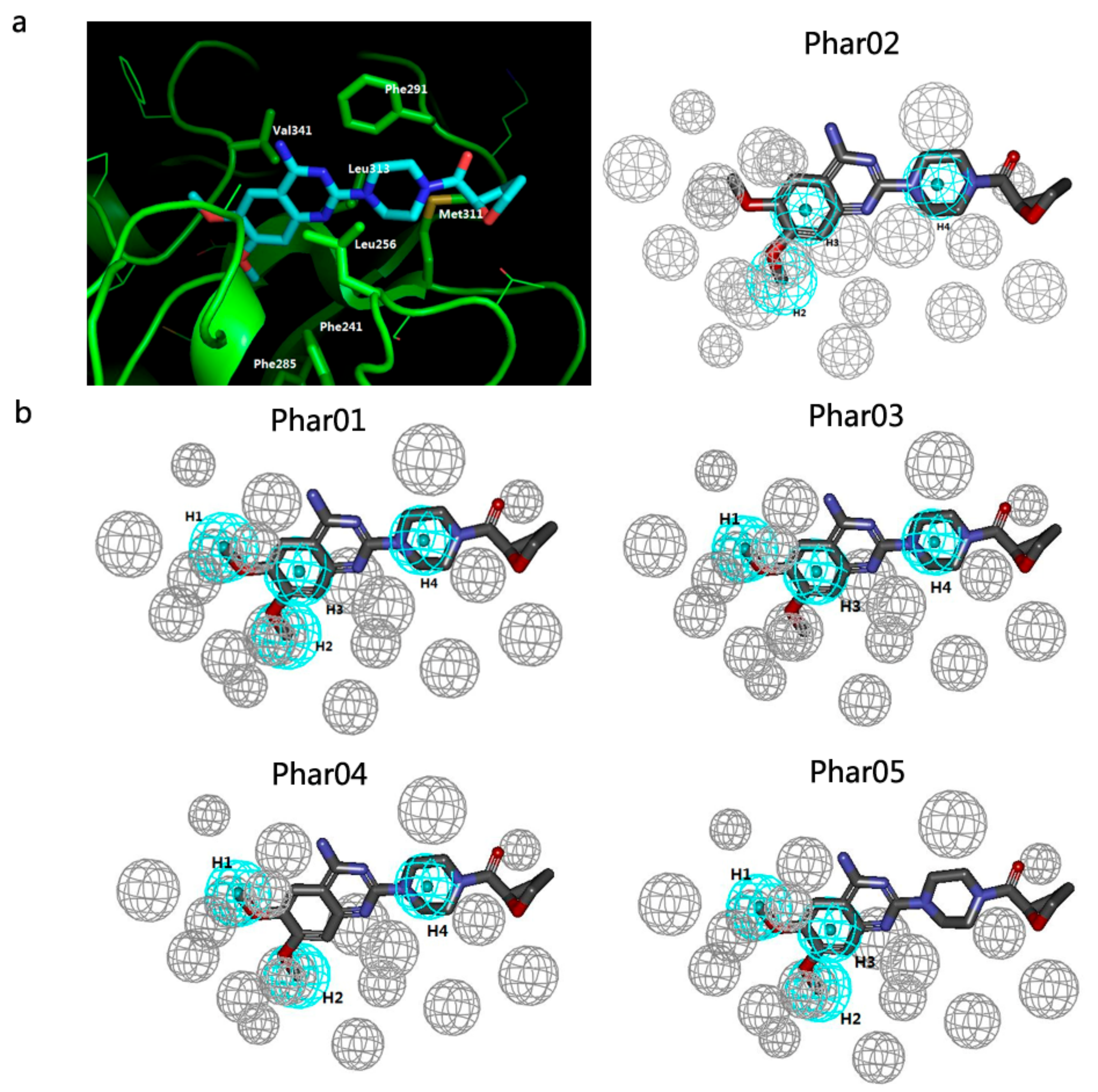
2.2. Pharmacophore Models and Database Filtering
2.3. Shape-Based Model and Database Filtering
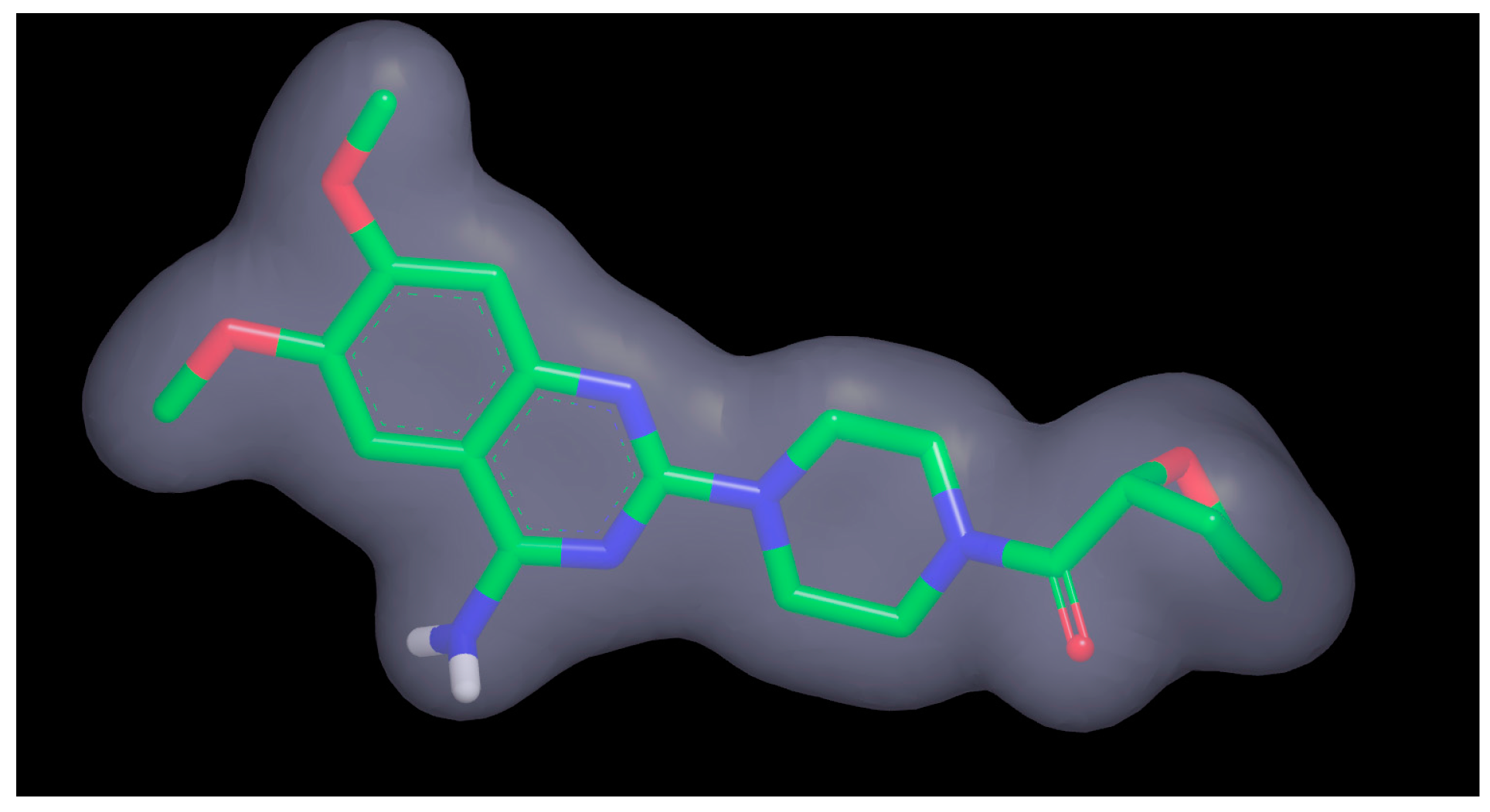
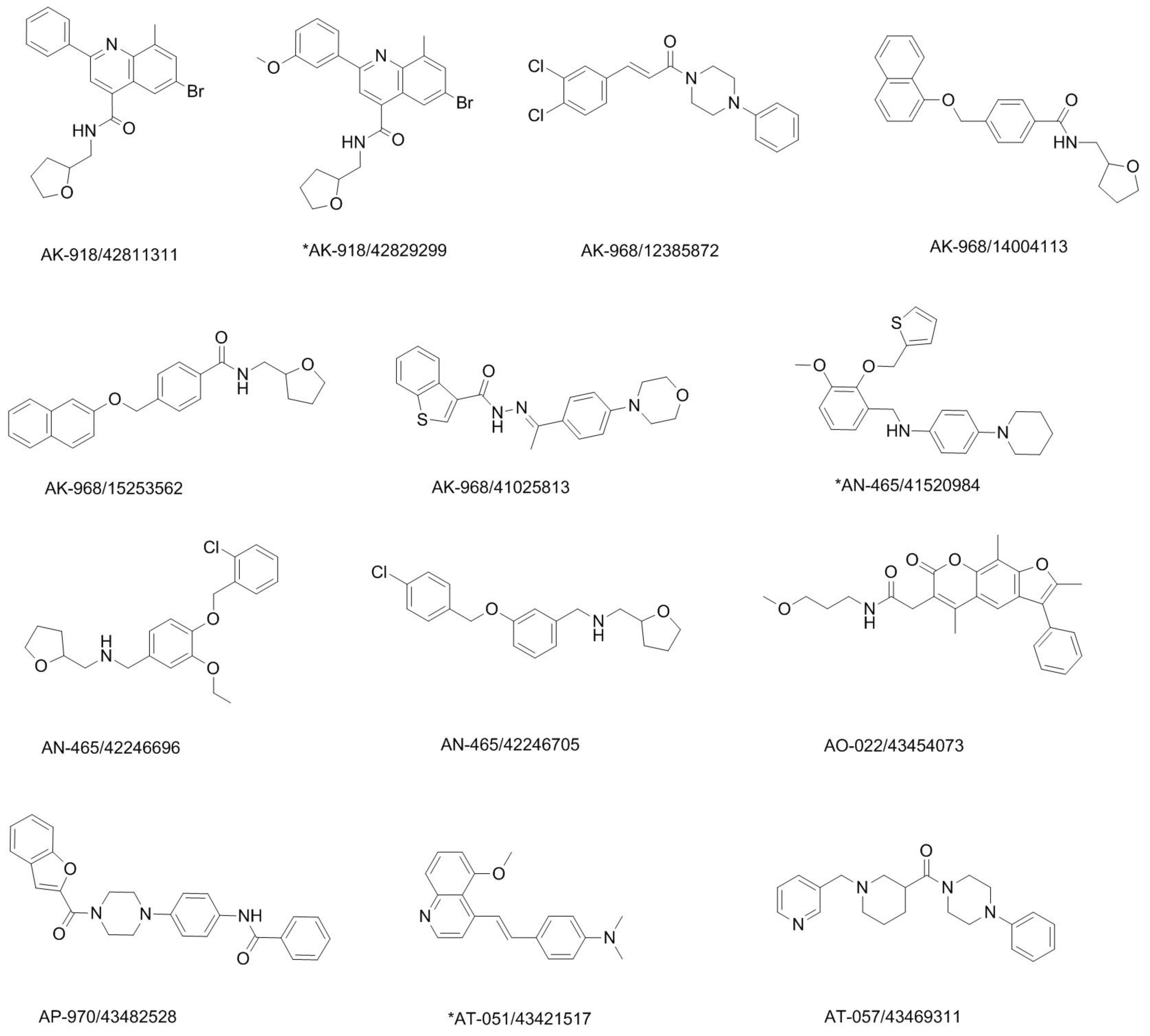
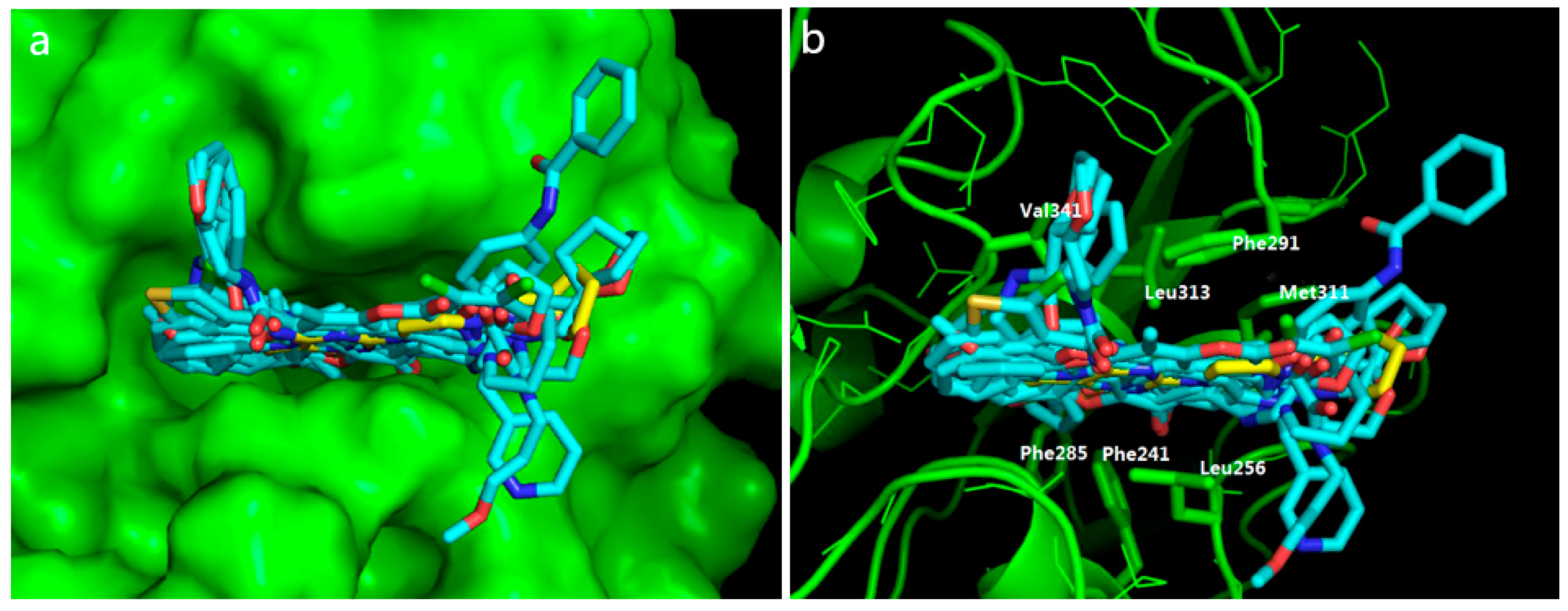
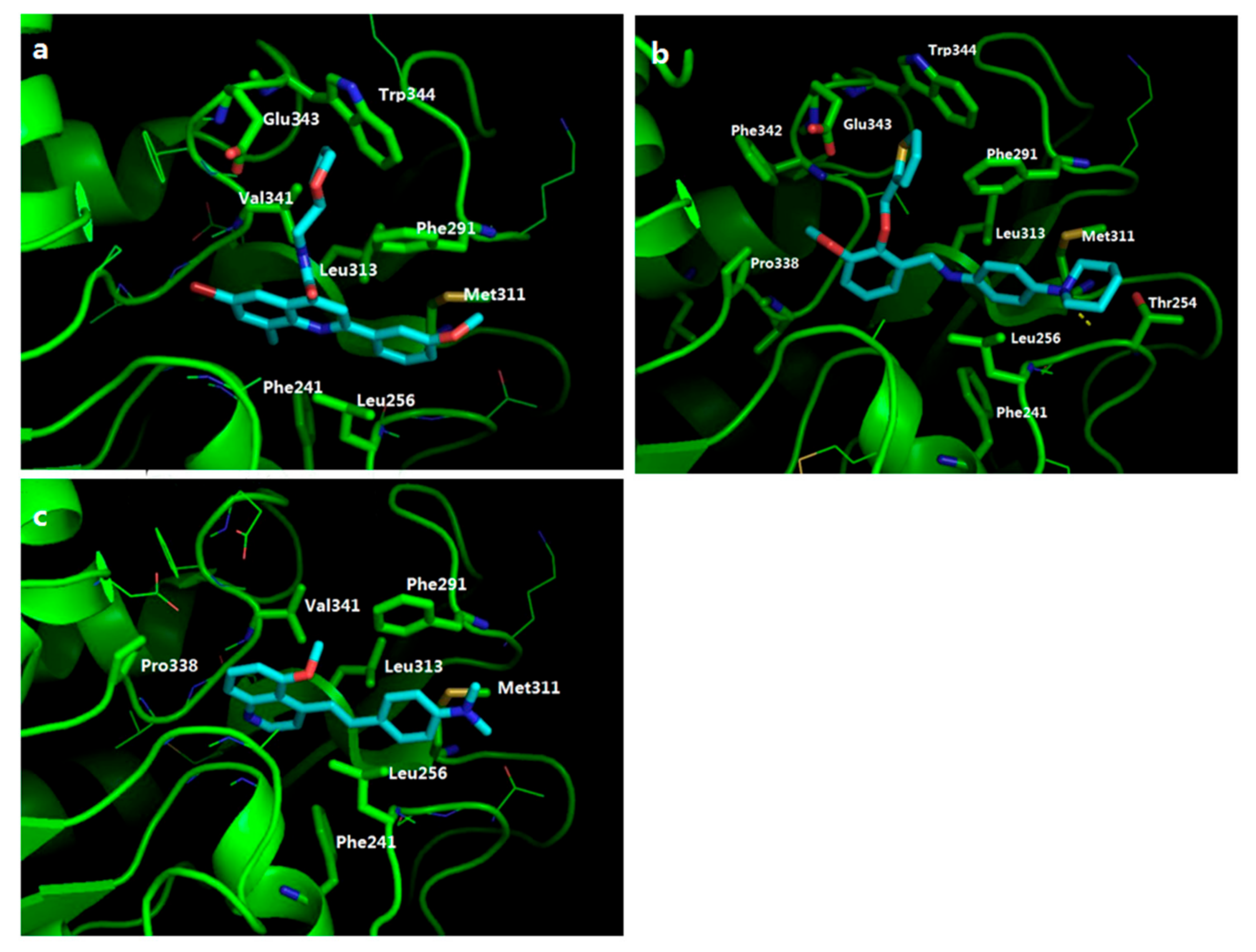
2.4. Potential Hits from Molecular Docking
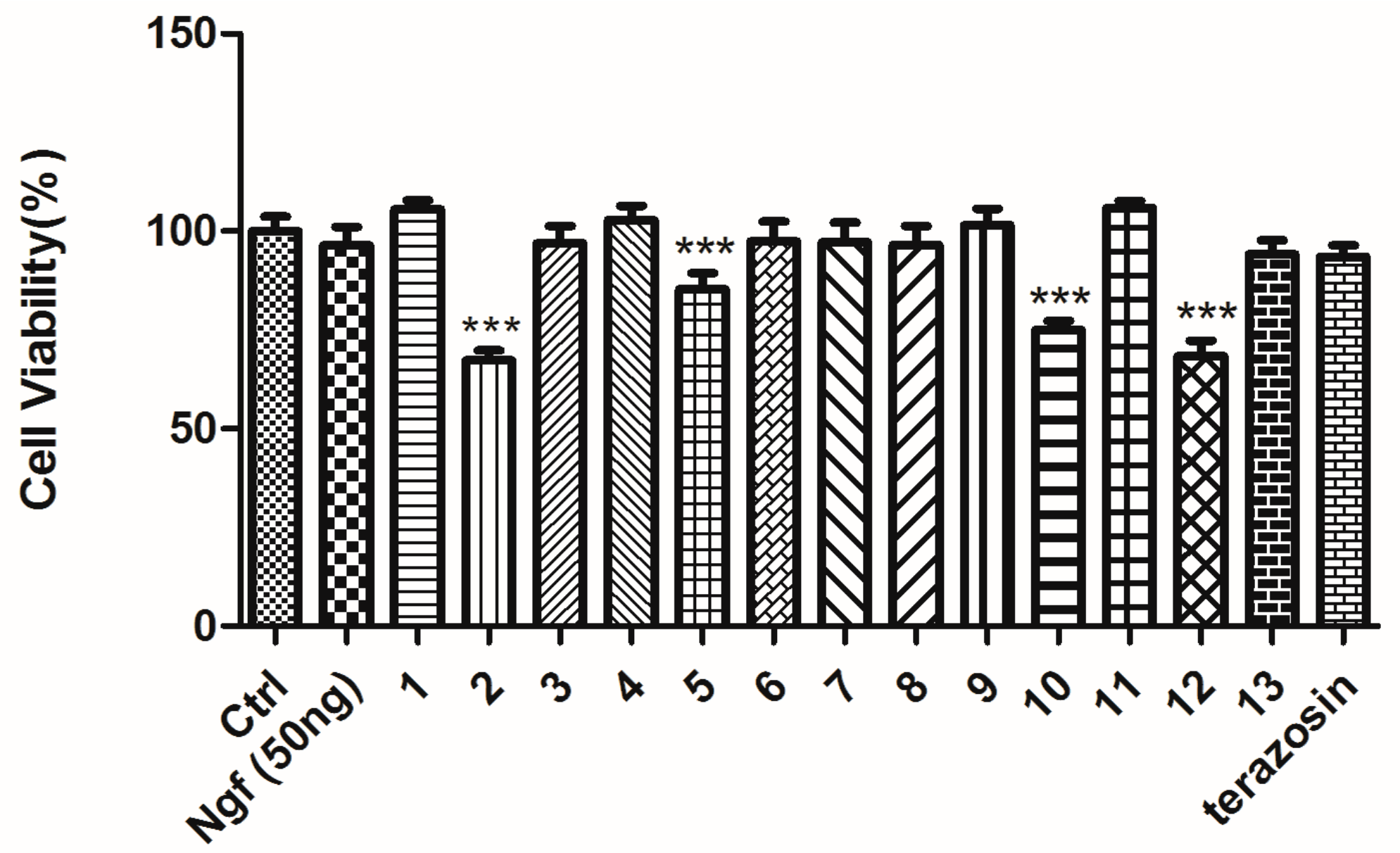
2.5. Protective Effect against Rotenone-Induced PC12 Cell Death
3. Materials and Methods
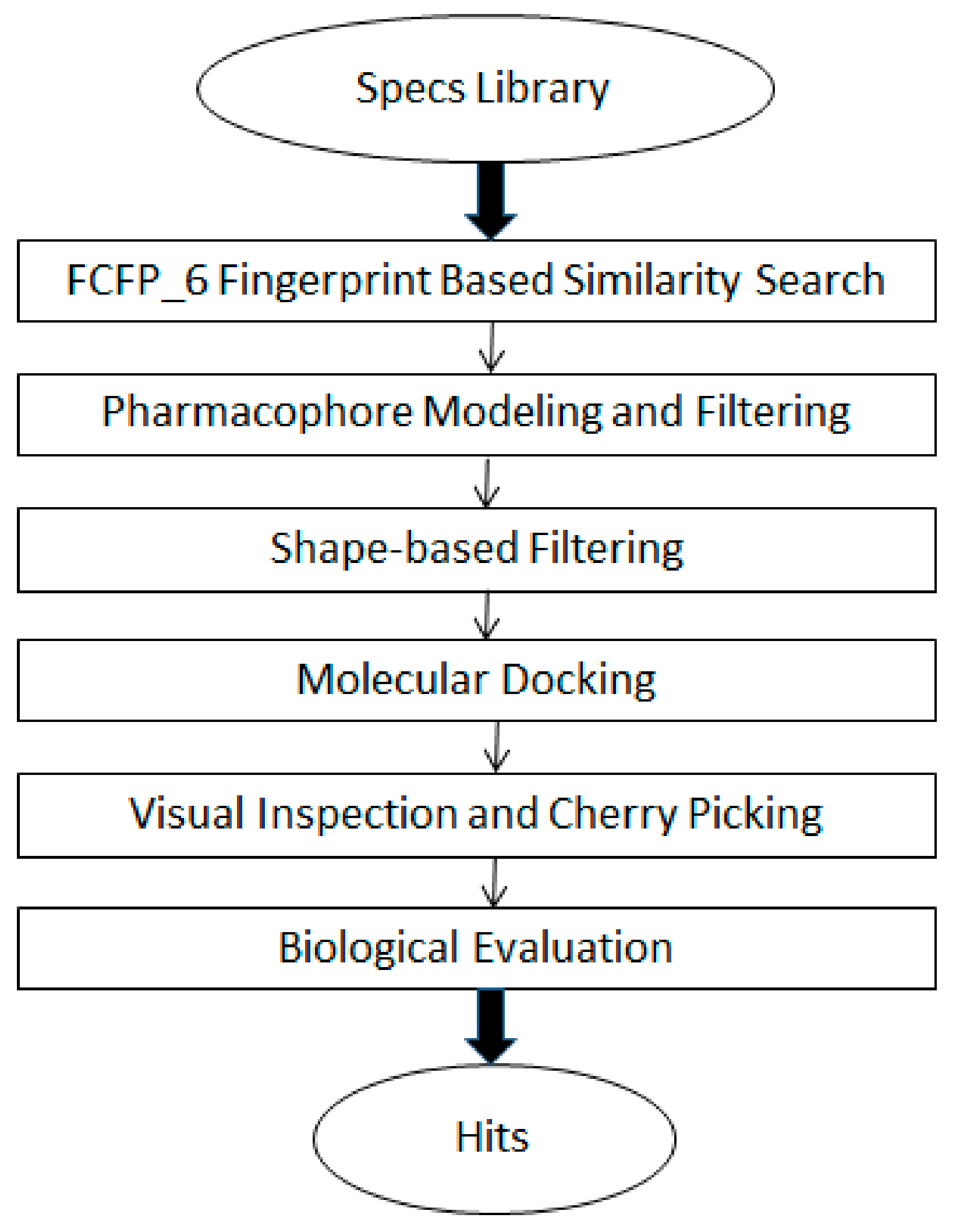
3.1. The General Workflow for Drug Discovery
3.2. FCFP_6 Fingerprint-Based Similarity Search
3.3. Pharmacophore Modeling and Filtering
3.3.1. Receptor-Ligand Pharmacophore Generation
3.3.2. Construction of a Multi-Conformation Ligand Database
3.3.3. Pharmacophore Filtering
3.4. Shape-Based Filtering
3.5. Molecular Docking
3.6. Visual Inspection and Cherry-Picking of Potential Hits
3.7. Biological Evaluation
3.7.1. Reagents
3.7.2. Cell Culture and Drug Treatment
3.7.3. Cell Viability Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Pgk1 | phosphoglycerate kinase 1 |
| VS | virtual screening |
| caspases | cysteine aspartate-specific proteases |
| HSPs | heat shock proteins |
| hPgk1 | human Pgk1 |
| ROS | reactive oxygen species |
| NGF | nerve growth factor |
| DS2016 | discovery studio 2016 |
| GFA | genetic function approximation |
| GA | genetic algorithm |
| DMSO | dimethyl sulfoxide |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide |
| ATCC | American type culture collection |
| DMEM | Dulbecco’s modified Eagle’s medium |
| OD | optical density |
References
- Engelberg-Kulka, H.; Amitai, S.; Kolodkin-Gal, I.; Hazan, R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006, 2, e135. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Moran, L.B. Mechanisms of cell death in neurodegenerative diseases: Fashion, fiction, and facts. Brain Pathol. 2002, 12, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis 2009, 14, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L.; Brunner, M.; Ramos, L.; Deitch, E.A. Scientific and clinical challenges in sepsis. Curr. Pharm. Des. 2009, 15, 1918–1935. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef]
- Yuan, J.Y.; Horvitz, H.R. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev. Biol. 1990, 138, 33–41. [Google Scholar] [CrossRef]
- Li, Z.; Pan, Y.; Zhong, W.; Zhu, Y.; Zhao, Y.; Li, L.; Liu, W.; Zhou, H.; Yang, C. Synthesis and evaluation of N-acyl-substituted 1,2-benzisothiazol-3-one derivatives as caspase-3 inhibitors. Bioorg. Med. Chem. 2014, 22, 6735–6745. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Lu, M.; Yan, Z.; Tang, X.; Sun, B.; Liu, W.; Zhou, H.; Yang, C. 1,2-benzisothiazol-3-one derivatives as a novel class of small-molecule caspase-3 inhibitors. Bioorg. Med. Chem. 2014, 22, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Tian, Z.; Yan, Z.; Wu, L.; Ma, Y.; Wang, Q.; Liu, W.; Zhou, H.; Yang, C. Design, synthesis and evaluation of 1,2-benzisothiazol-3-one derivatives as potent caspase-3 inhibitors. Bioorg. Med. Chem. 2013, 21, 2960–2967. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, L.; Chen, Z.; Zheng, S.; Shu, H.; Li, J.; Jiang, H.; Liu, S. A novel class of small-molecule caspase-3 inhibitors prepared by multicomponent reactions. Eur. J. Med. Chem. 2012, 54, 232–238. [Google Scholar] [CrossRef] [PubMed]
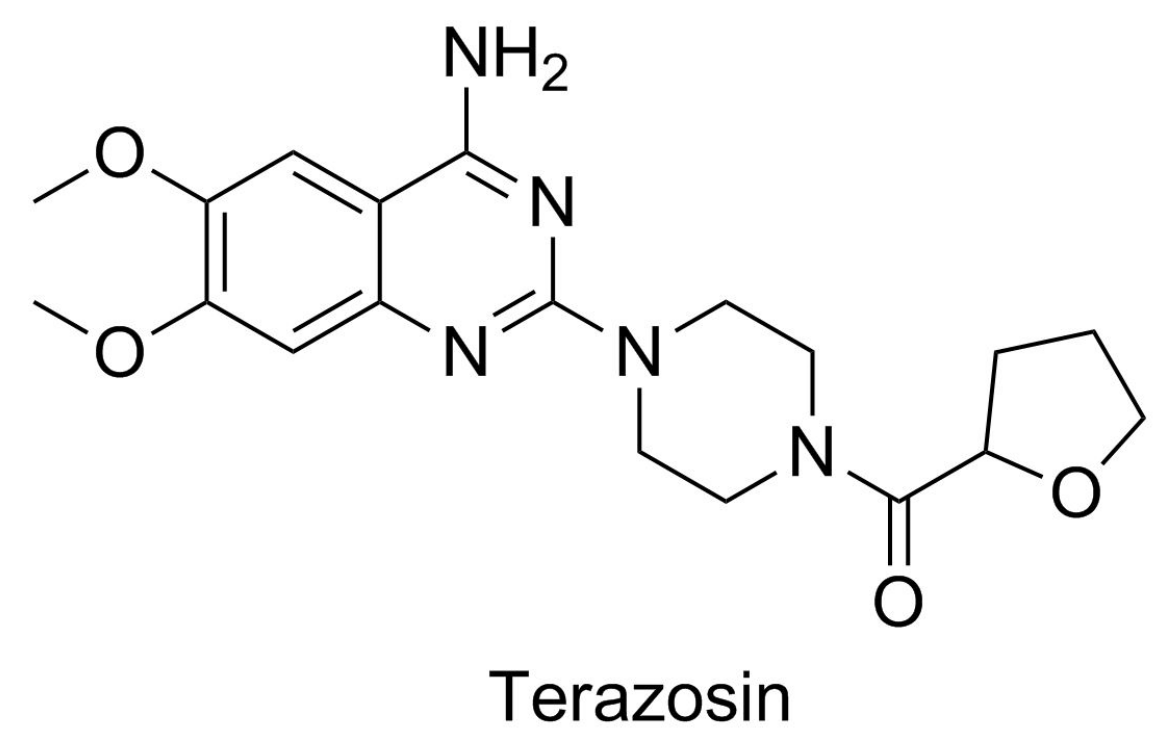
- Chen, X.; Zhao, C.; Li, X.; Wang, T.; Li, Y.; Cao, C.; Ding, Y.; Dong, M.; Finci, L.; Wang, J.H.; et al. Terazosin activates Pgk1 and Hsp90 to promote stress resistance. Nat. Chem. Biol. 2015, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wijeratne, E.M.; Xu, Y.M.; Scherz-Shouval, R.; Marron, M.T.; Rocha, D.D.; Liu, M.X.; Costa-Lotufo, L.V.; Santagata, S.; Lindquist, S.; Whitesell, L.; et al. Structure-activity relationships for withanolides as inducers of the cellular heat-shock response. J. Med. Chem. 2014, 57, 2851–2863. [Google Scholar] [CrossRef] [PubMed]
- Kil, Y.S.; Choi, S.K.; Lee, Y.S.; Jafari, M.; Seo, E.K. Chalcones from Angelica keiskei: Evaluation of Their Heat Shock Protein Inducing Activities. J. Nat. Prod. 2015, 78, 2481–2487. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W.H. Small heat shock proteins and protection against injury. Ann. N. Y. Acad. Sci. 1999, 874, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, R.; Riordan, M.; Thullin, G.; van Why, S.; Siegel, N.J.; Kashgarian, M. The maximal cytoprotective function of the heat shock protein 27 is dependent on heat shock protein 70. Biochim. Biophys. Acta 2011, 1813, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, T.; Zhou, L.; Zhou, L.; Xing, G.; Chen, Y.; Xin, Y. Schisandrin B attenuates acetaminophen-induced hepatic injury through heat-shock protein 27 and 70 in mice. J. Gastroenterol. Hepatol. 2014, 29, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Roesslein, M.; Froehlich, C.; Jans, F.; Piegeler, T.; Goebel, U.; Loop, T. Dobutamine mediates cytoprotection by induction of heat shock protein 70 in vitro. Life Sci. 2014, 98, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Miyamae, Y.; Shigemori, H.; Isoda, H. Neuroprotective effect of 3,5-di-O-caffeoylquinic acid on SH-SY5Y cells and senescence-accelerated-prone mice 8 through the up-regulation of phosphoglycerate kinase-1. Neuroscience 2010, 169, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, C.; Torella, M.; Petrera, A.; Palermo, V.; Falcone, C. PGK1, the gene encoding the glycolitic enzyme phosphoglycerate kinase, acts as a multicopy suppressor of apoptotic phenotypes in S. cerevisiae. Yeast 2009, 26, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Morera, S.M.; Bianchi, P.; Fermo, E.; Zanella, A.; Galizzi, A.; Valentini, G. Molecular insights on pathogenic effects of mutations causing phosphoglycerate kinase deficiency. PLoS ONE 2012, 7, e32065. [Google Scholar] [CrossRef] [PubMed]
- Gondeau, C.; Chaloin, L.; Lallemand, P.; Roy, B.; Perigaud, C.; Barman, T.; Varga, A.; Vas, M.; Lionne, C.; Arold, S.T. Molecular basis for the lack of enantioselectivity of human 3-phosphoglycerate kinase. Nucleic Acids Res. 2008, 36, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Cliff, M.J.; Bowler, M.W.; Varga, A.; Marston, J.P.; Szabo, J.; Hounslow, A.M.; Baxter, N.J.; Blackburn, G.M.; Vas, M.; Waltho, J.P. Transition state analogue structures of human phosphoglycerate kinase establish the importance of charge balance in catalysis. J. Am. Chem. Soc. 2010, 132, 6507–6516. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, P.; Chaloin, L.; Roy, B.; Barman, T.; Bowler, M.W.; Lionne, C. Interaction of human 3-phosphoglycerate kinase with its two substrates: Is substrate antagonism a kinetic advantage? J. Mol. Biol. 2011, 409, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Huang, J.Y.; Yuan, Y.H.; Yan, J.Q.; Wang, Y.N.; Chu, S.F.; Zhu, C.G.; Guo, Q.L.; Shi, J.G.; Chen, N.H. 20C, a bibenzyl compound isolated from Gastrodia elata, protects PC12 cells against rotenone-induced apoptosis via activation of the Nrf2/ARE/HO-1 signaling pathway. Acta Pharmacol. Sin. 2016, 37, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Y.; Yuan, Y.H.; Hu, J.F.; Zhao, Q.; Zhang, D.M.; Chen, N.H. Protective effect of Bu-7, a flavonoid extracted from Clausena lansium, against rotenone injury in PC12 cells. Acta Pharmacol. Sin. 2011, 32, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Salinas, M.; Diaz, R.; Abraham, N.G.; Ruiz de Galarreta, C.M.; Cuadrado, A. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J. Biol. Chem. 2003, 278, 13898–13904. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017, 45, D955–D963. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 1 June 2016).
- Sprague, P.W. Automated chemical hypothesis generation and database searching with Catalyst®. Perspect. Drug Discov. Des. 1995, 3, 1–20. [Google Scholar] [CrossRef]
- Grant, J.A.; Gallardo, M.A.; Pickup, B.T. A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J. Comput. Chem. 1996, 17, 1653–1666. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the hit compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SPECS ID | Chemscore (GOLD) | 2D Similarity (FCFP_6) | FitValue (Catalyst) | ShapeTanimoto (ROCS) | ||||
|---|---|---|---|---|---|---|---|---|
| Score | Rank | Score | Rank | Score | Rank | Score | Rank | |
| AK-918/42811311 | 34.820 | 50 | 0.178 | 1640 | 1.974 | 7269 | 0.704 | 3407 |
| AK-918/42829299 | 35.759 | 25 | 0.205 | 312 | 2.869 | 2683 | 0.687 | 3899 |
| AK-968/12385872 | 34.693 | 53 | 0.149 | 7964 | 2.530 | 5683 | 0.804 | 368 |
| AK-968/14004113 | 35.193 | 31 | 0.180 | 1429 | 2.564 | 5495 | 0.770 | 1232 |
| AK-968/15253562 | 35.815 | 23 | 0.192 | 707 | 2.978 | 470 | 0.743 | 2095 |
| AK-968/41025813 | 34.493 | 57 | 0.150 | 7767 | 2.323 | 6411 | 0.758 | 1615 |
| AN-465/41520984 | 37.517 | 2 | 0.147 | 9044 | 2.727 | 4366 | 0.726 | 2666 |
| AN-465/42246696 | 34.955 | 40 | 0.162 | 3868 | 2.255 | 6597 | 0.770 | 1229 |
| AN-465/42246705 | 33.160 | 143 | 0.158 | 4696 | 2.853 | 2946 | 0.749 | 1919 |
| AO-022/43454073 | 37.586 | 1 | 0.155 | 5656 | 2.841 | 3091 | 0.787 | 719 |
| AP-970/43482528 | 35.452 | 26 | 0.157 | 4923 | 2.754 | 4125 | 0.714 | 3071 |
| AT-051/43421517 | 33.379 | 122 | 0.149 | 8073 | 2.997 | 52 | 0.748 | 1946 |
| AT-057/43469311 | 32.052 | 221 | 0.162 | 3838 | 2.205 | 6741 | 0.755 | 1711 |
| Compound No. | Specs ID | Cell Viability (% of Control) |
|---|---|---|
| control a | 100.0 ± 4.203 | |
| model b | 73.84 ± 4.632 ### | |
| NGF c | 114.5 ± 3.704 *** | |
| 1 | AN-465/41520984 | 89.66 ± 5.859 *** |
| 2 | AO-022/43454073 | 53.01 ± 3.772 *** |
| 3 | AK-968/15253562 | 78.43 ± 5.533 |
| 4 | AK-918/42829299 | 88.04 ± 4.888 *** |
| 5 | AP-970/43482528 | 68.09 ± 4.812 |
| 6 | AK-968/14004113 | 74.21 ± 3.454 |
| 7 | AK-918/42811311 | 72.97 ± 3.662 |
| 8 | AK-968/12385872 | 78.97 ± 7.843 |
| 9 | AN-465/42246696 | 74.33 ± 10.64 |
| 10 | AK-968/41025813 | 62.47 ± 7.143 *** |
| 11 | AT-051/43421517 | 87.97 ± 7.818 *** |
| 12 | AN-465/42246705 | 80.22 ± 6.887 |
| 13 | AT-057/43469311 | 78.37 ± 6.860 |
| terazosin d | 72.25 ± 5.490 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, J.; Feng, B.; Shao, Q.; Yuan, Y.; Wang, X.S.; Chen, N.; Wu, S. Virtual Screening against Phosphoglycerate Kinase 1 in Quest of Novel Apoptosis Inhibitors. Molecules 2017, 22, 1029. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22061029
Xia J, Feng B, Shao Q, Yuan Y, Wang XS, Chen N, Wu S. Virtual Screening against Phosphoglycerate Kinase 1 in Quest of Novel Apoptosis Inhibitors. Molecules. 2017; 22(6):1029. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22061029
Chicago/Turabian StyleXia, Jie, Bo Feng, Qianhang Shao, Yuhe Yuan, Xiang Simon Wang, Naihong Chen, and Song Wu. 2017. "Virtual Screening against Phosphoglycerate Kinase 1 in Quest of Novel Apoptosis Inhibitors" Molecules 22, no. 6: 1029. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22061029