15N-, 13C- and 1H-NMR Spectroscopy Characterization and Growth Inhibitory Potency of a Combi-Molecule Synthesized by Acetylation of an Unstable Monoalkyltriazene

Abstract
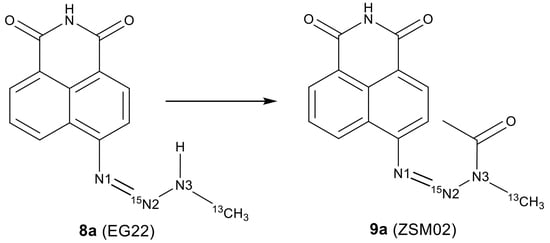
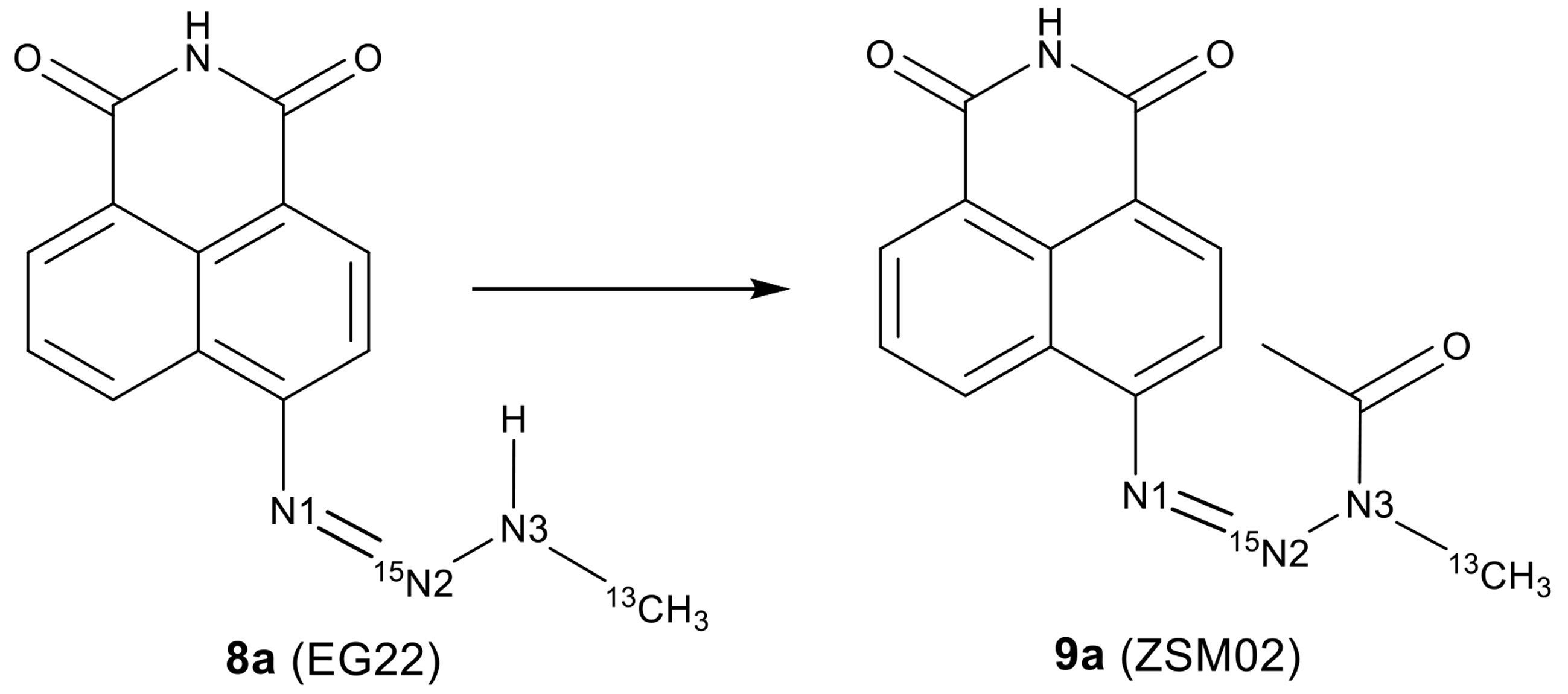
:
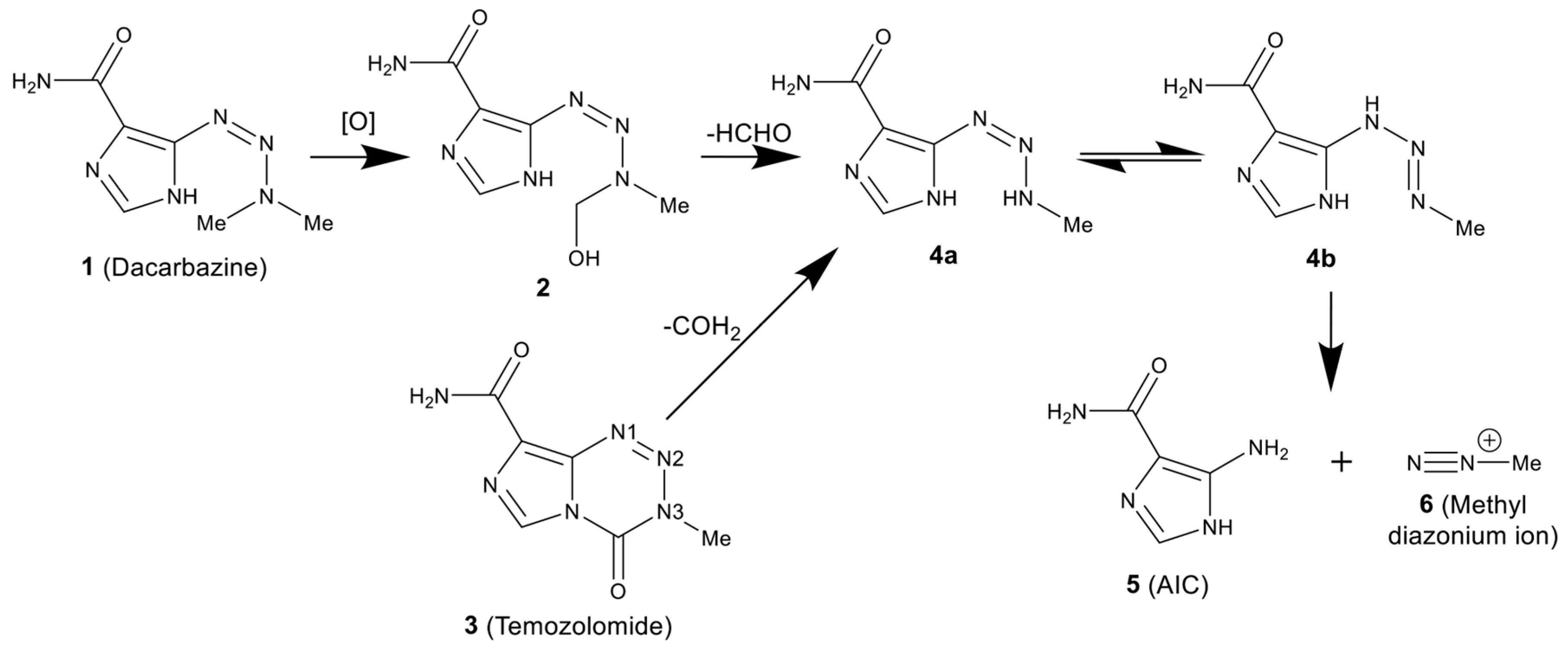
1. Introduction
2. Results and Discussion
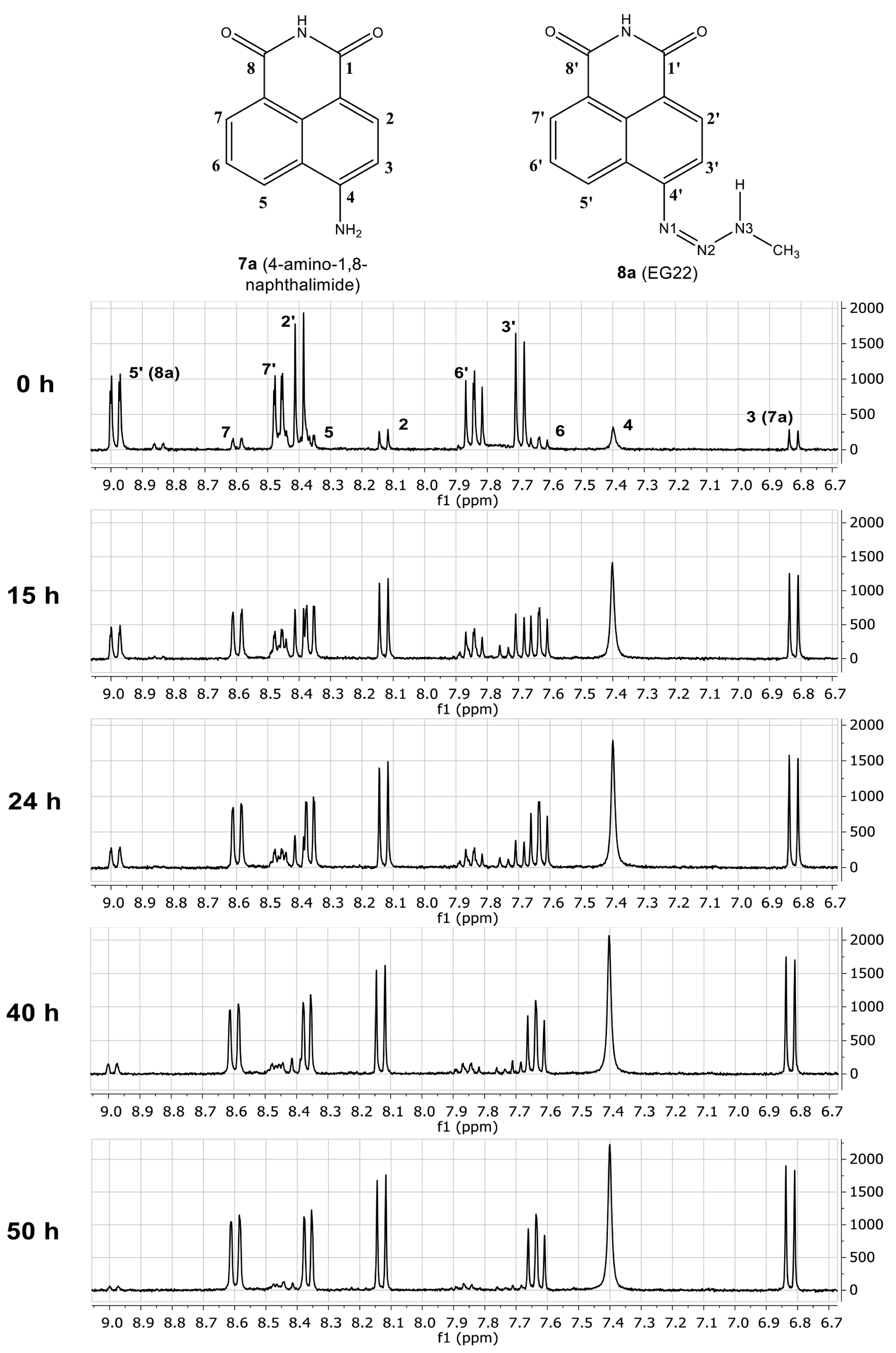
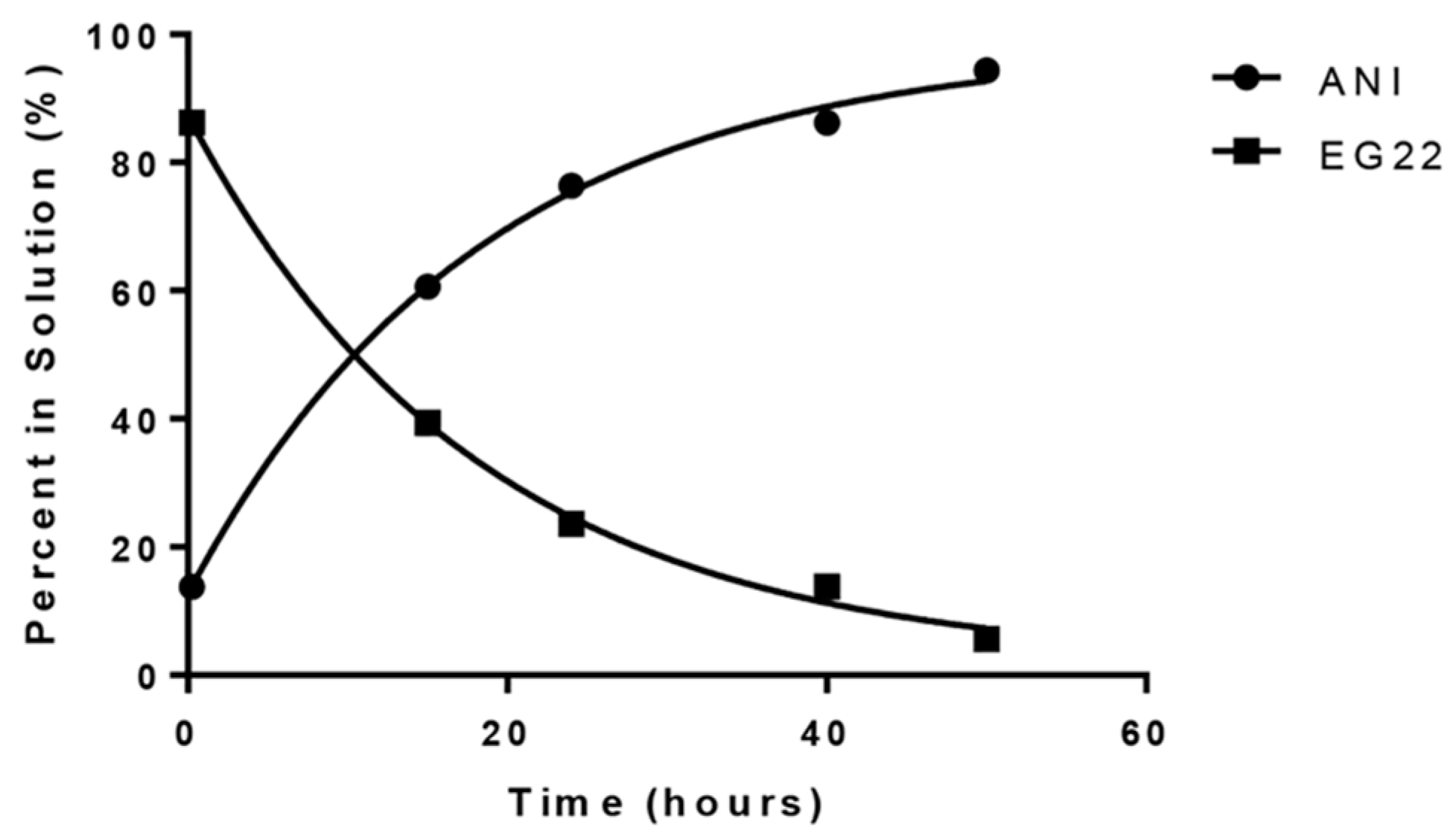
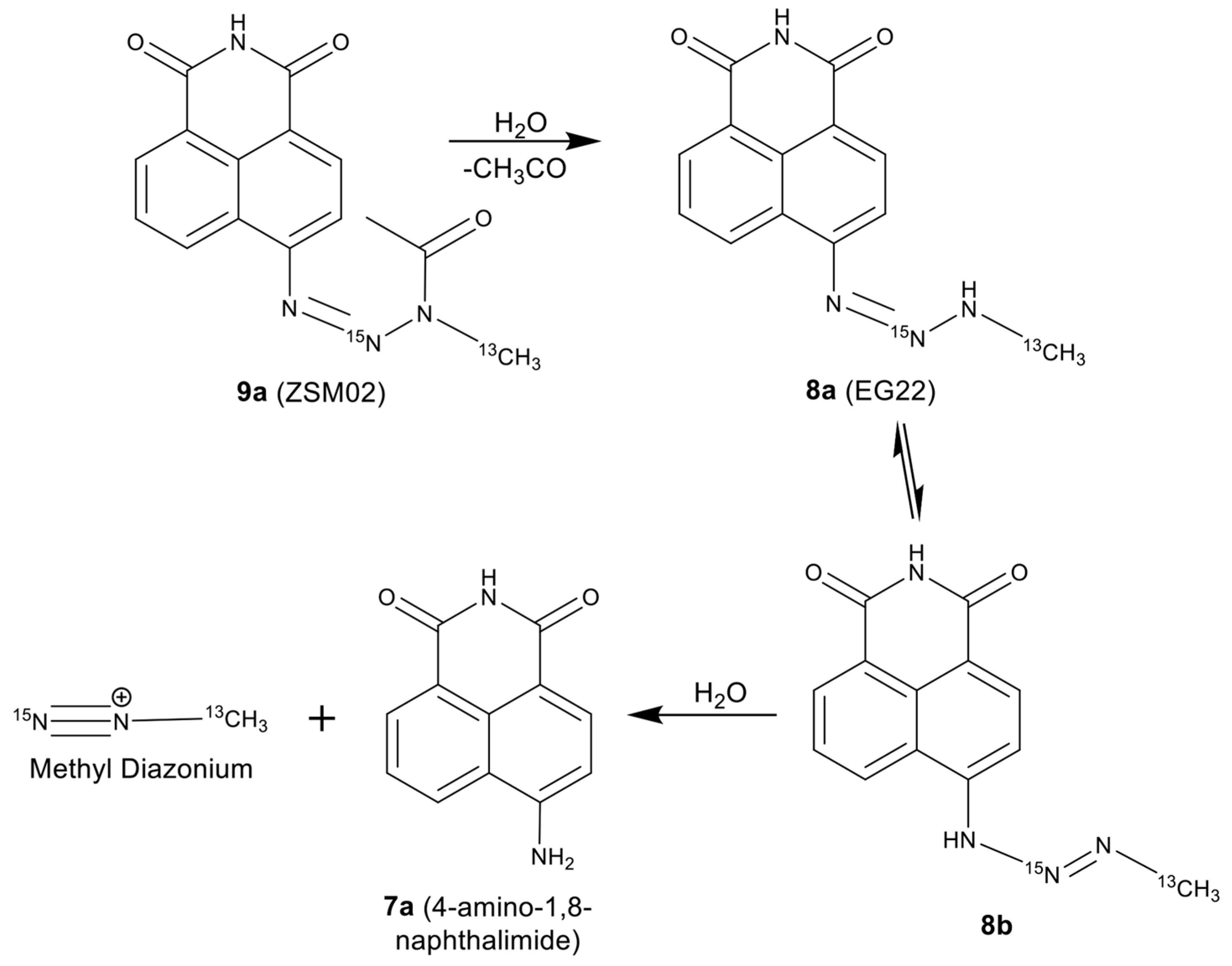
2.1. Stability of EG22 in DMSO
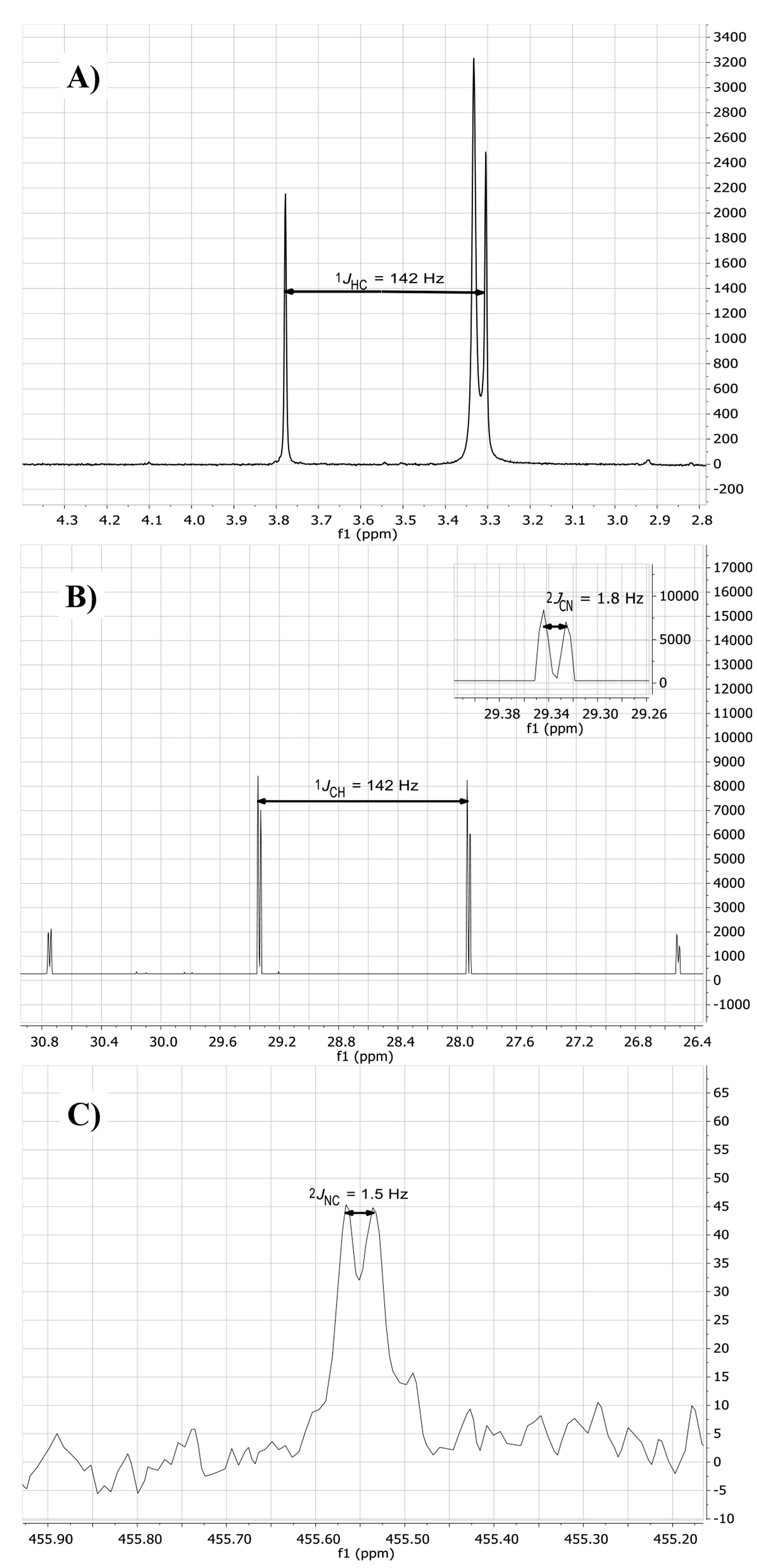
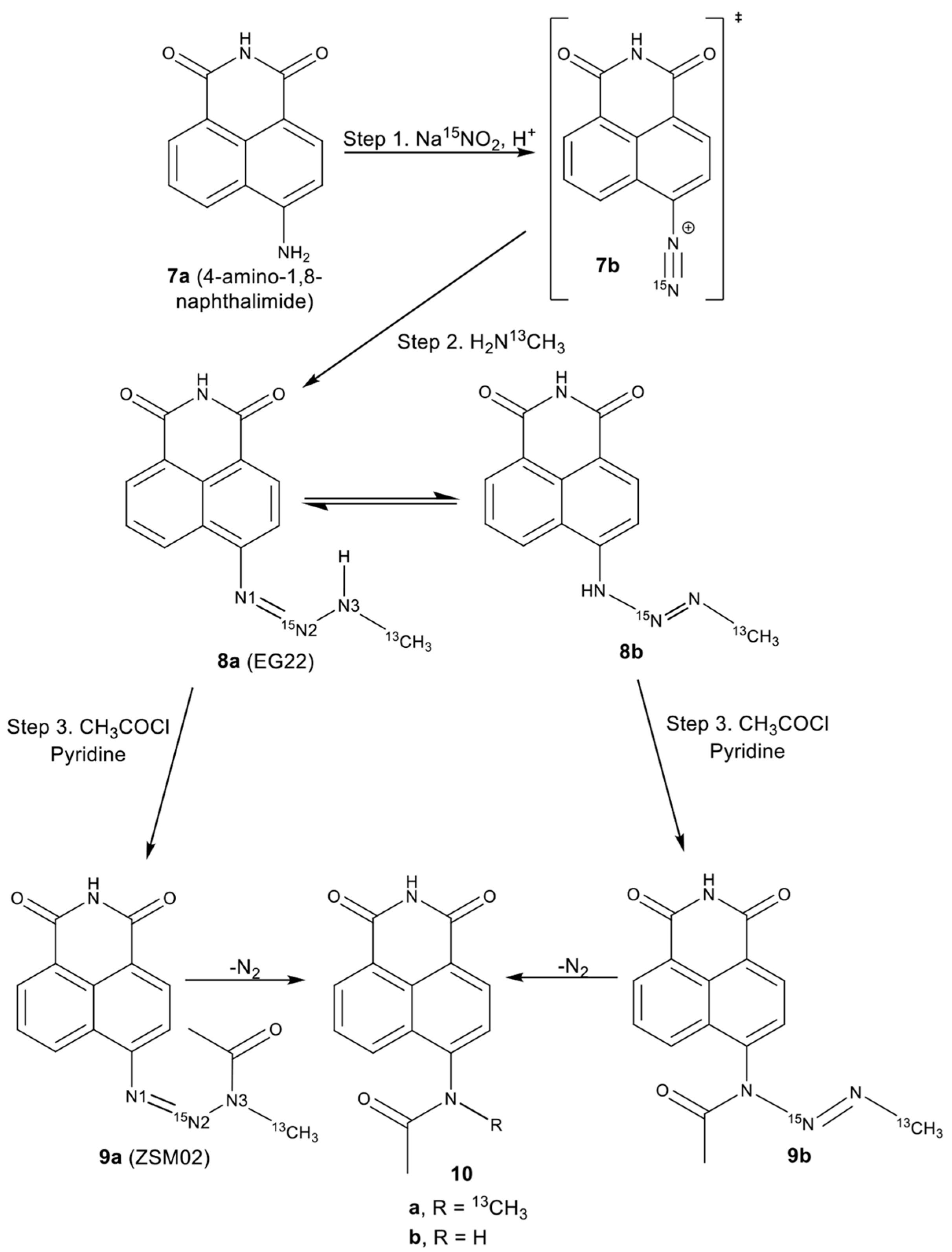
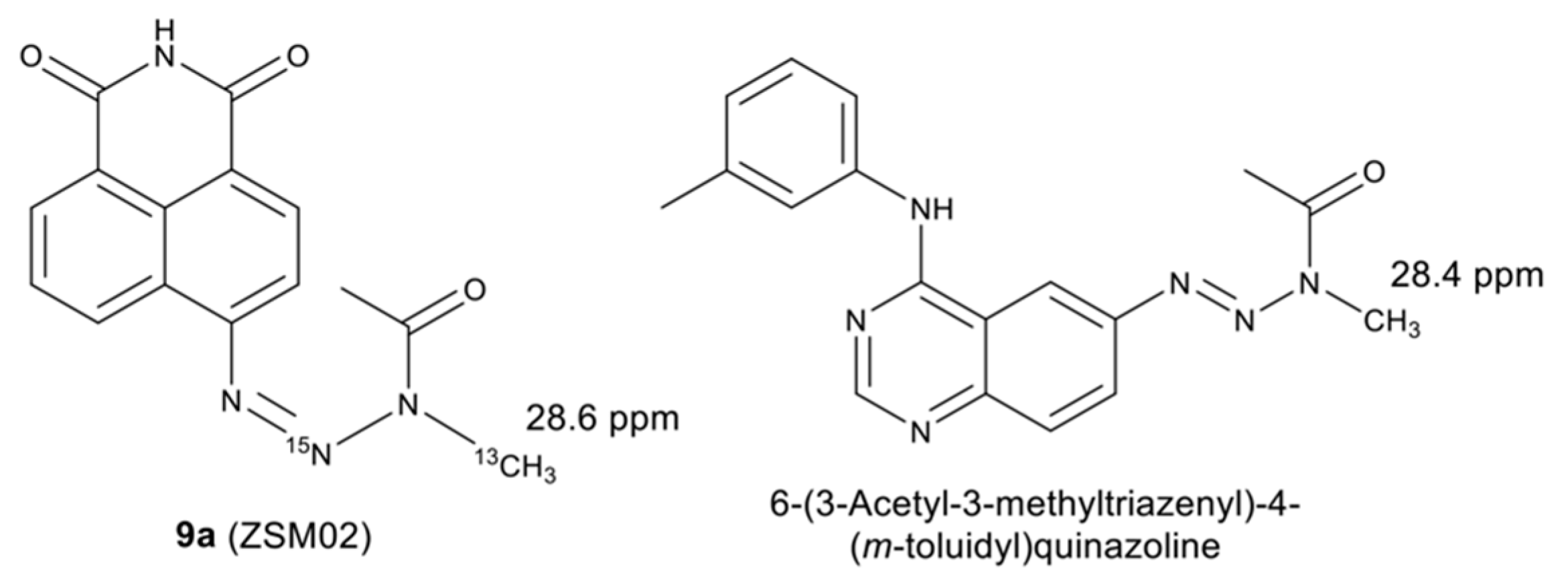
2.2. Isotopic Labelling and NMR Spectroscopy
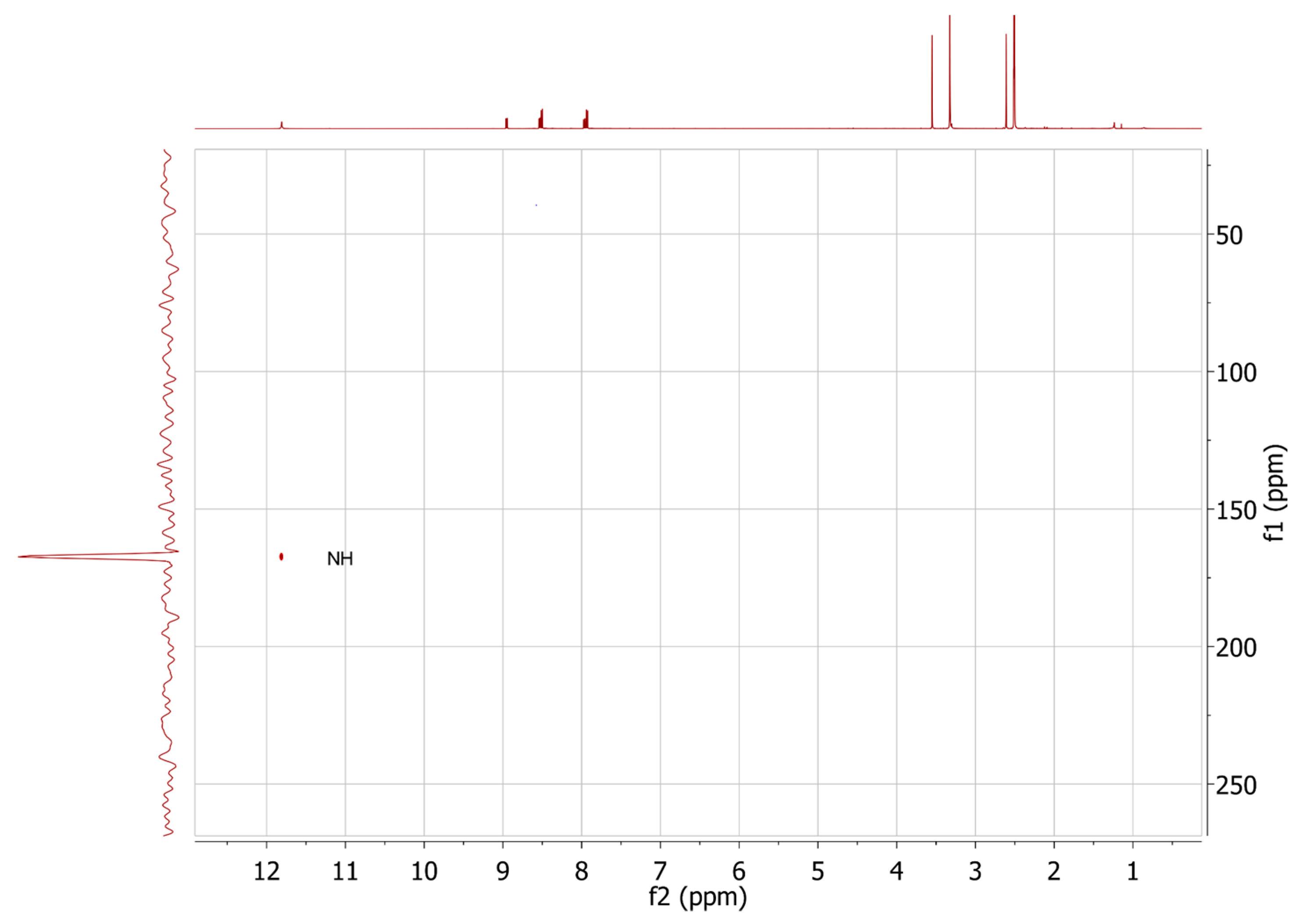
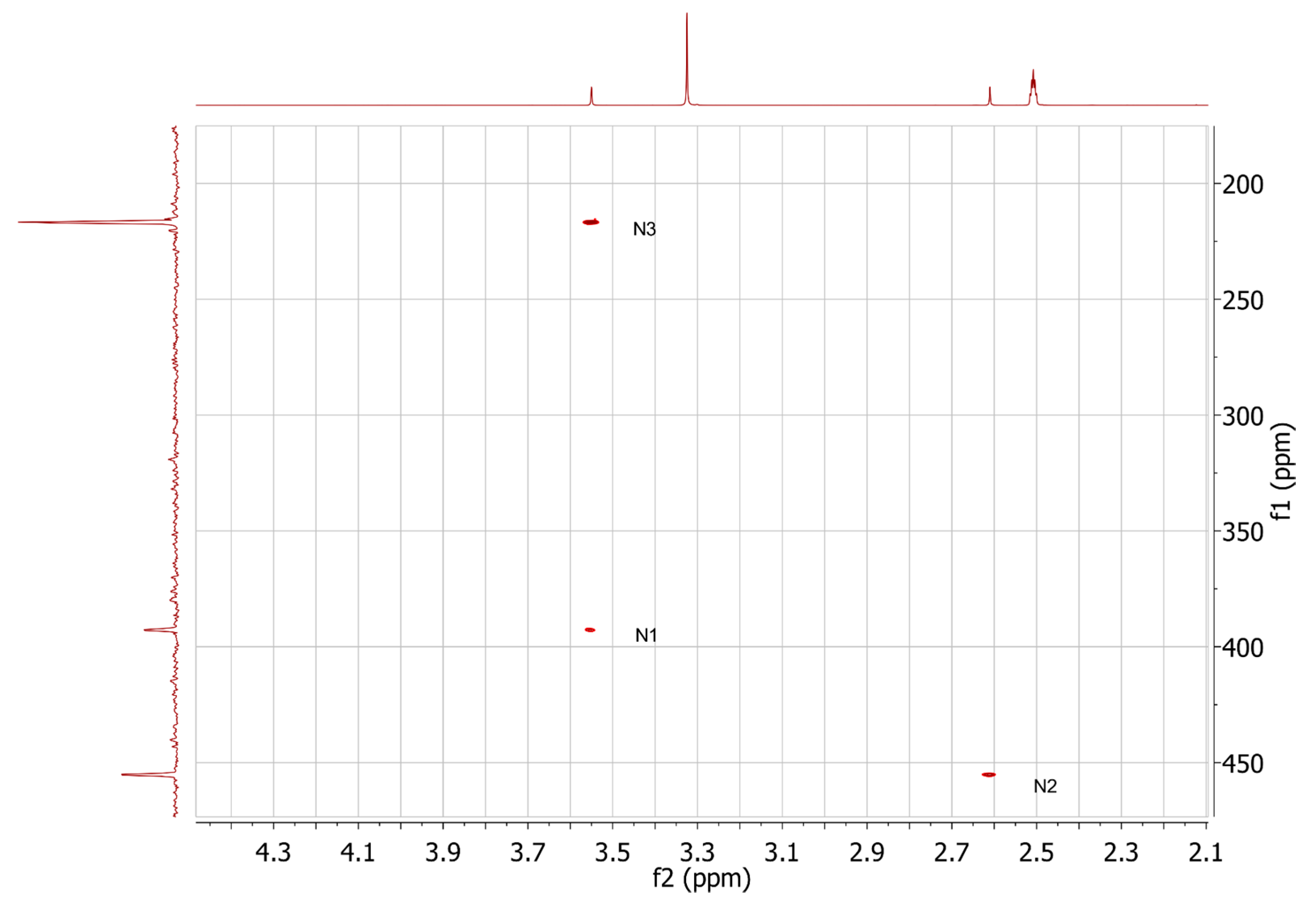
2.3. HMBC and HSQC Analysis
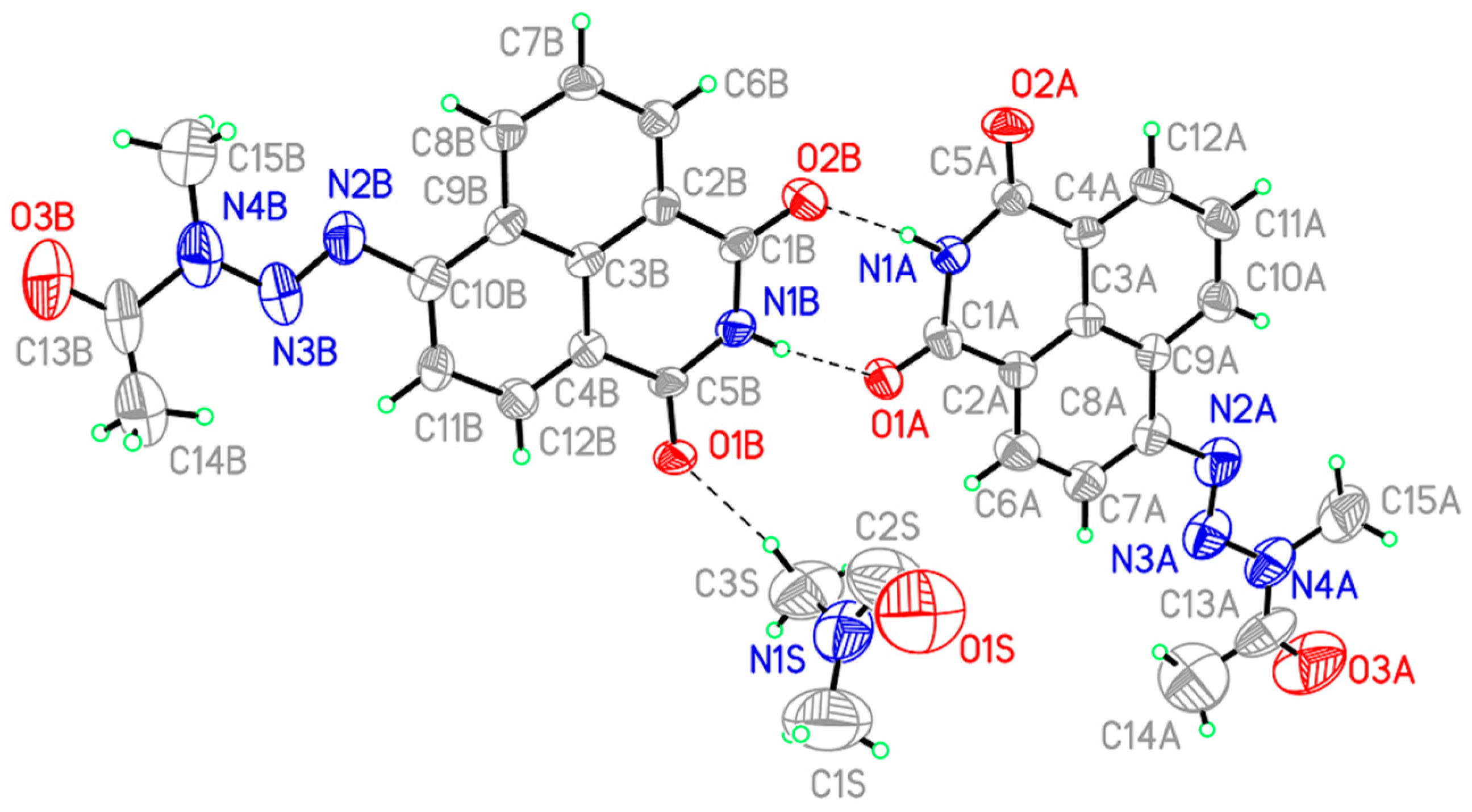
2.4. Determination of the 3D Structure
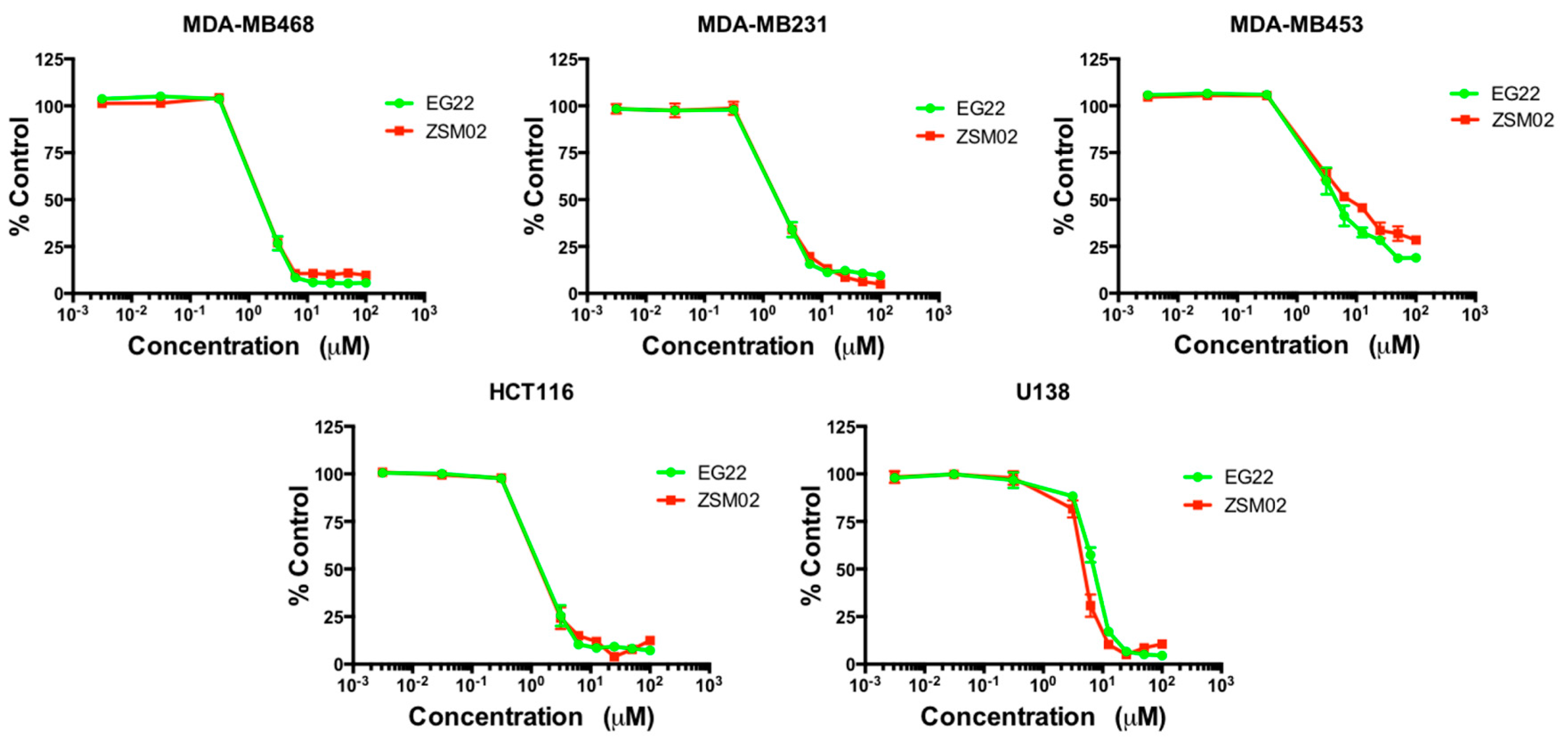
2.5. Growth Inhibitory Potency
3. Material and Methods
3.1. General Information
3.2. Chemical Synthesis
3.3. NMR Acquisition
3.4. X-ray Crystallography
3.5. Cell Culture
3.6. Growth Inhibition Assay
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Nashan, D.; Müller, M.L.; Grabbe, S.; Wustlich, S.; Enk, A. Systemic therapy of disseminated malignant melanoma: An evidence-based overview of the state-of-the-art in daily routine. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Margison, G.P. Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol. 2003, 4, 37–44. [Google Scholar] [CrossRef]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA damage induced by alkylating agents and repair pathways. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Dolan, M.E. Role of Cytochrome P450 Isoenzymes in Metabolism of O6-Benzylguanine. Clin. Cancer Res. 2001, 7, 4239–4244. [Google Scholar] [PubMed]
- Chen, C.; Xu, T.; Lu, Y.; Chen, J.; Wu, S. The efficacy of temozolomide for recurrent glioblastoma multiforme. Eur. J. Neurol. 2013, 20, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar] [PubMed]
- Hvizdos, K.M.; Goa, K.L. Temozolomide. CNS Drugs 1999, 12, 237–243. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Huang, Y.; Qiu, Q.; Mcnamee, J.P.; Belinsky, G.; Jean-Claude, B.J. The combi-targeting concept: Mechanism of action of the pleiotropic combi-molecule RB24 and discovery of a novel cell signaling-based combination principle. Cell. Signal. 2011, 23, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Rachid, Z.; Brahimi, F.; Katsoulas, A.; Teoh, N.; Jean-Claude, B.J. The combi-targeting concept: Chemical dissection of the dual targeting properties of a series of “combi-triazenes”. J. Med. Chem. 2003, 46, 4313–4321. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Domarkas, J.; Banerjee, R.; Merayo, N.; Brahimi, F.; McNamee, J.P.; Gibbs, B.F.; Jean-Claude, B.J. The combi-targeting concept: In vitro and in vivo fragmentation of a stable combi-nitrosourea engineered to interact with the epidermal growth factor receptor while remaining DNA reactive. Clin. Cancer Res. 2007, 13, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Matheson, S.L.; Brahimi, F.; Jean-Claude, B.J. The combi-targeting concept: Intracellular fragmentation of the binary epidermal growth factor (EGFR)/DNA targeting “combi-triazene” SMA41. Biochem. Pharmacol. 2004, 67, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rachid, Z.; Peyrard, L.; Senhaji Mouhri, Z.; Williams, C.; Jean-Claude, B.J. Positional Isomerization of A Non-Cleavable Combi-Molecule Dramatically Altered Tumor Cell Response Profile. Chem. Biol. Drug Des. 2015, 85, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Senhaji Mouhri, Z.; Elliot; Jean-Claude, B.J. A type I combi-targeting approach for the design of molecules with enhanced potency against BRCA1/2 mutant- and O6-methylguanine-dna methyltransferase (mgmt)-expressing tumour cells. BMC Cancer 2017. accepted. [Google Scholar]
- Isaacs, N.S.; Rannala, E. Kinetics and mechanism of the decomposition of 3-alkyl-1-aryltriazenes by carboxylic acids. J. Chem. Soc. Perkin Trans. 2 1974, 899–902. [Google Scholar] [CrossRef]
- Rachid, Z.; Brahimi, F.; Domarkas, J.; Jean-Claude, B.J. Synthesis of half-mustard combi-molecules with fluorescence properties: Correlation with EGFR status. Bioorg. Med. Chem. Lett. 2005, 15, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Duker, J.M.; Serianni, A.S. (13C) Substituted sucrose: 13C 1H and 13C 13C spin. Carbohydr. Res. 1993, 249, 281–303. [Google Scholar] [CrossRef]
- Jean-Claude, B.J.; Williams, C.I. 15N NMR study of bi-and tricyclic 1,2,3,5-tetrazepin-4-ones. Magn. Reson. Chem. 1998, 36, 87–91. [Google Scholar] [CrossRef]
- Vaughan, K.; Wilman, D.E.V.; Wheelhouse, R.T.; Stevens, M.F.G. A 15N NMR investigation of a series of benzotriazinones and related antitumour heterocycles. Magn. Reson. Chem. 2002, 40, 300–302. [Google Scholar] [CrossRef]
- Iwamura, H.; Albert, K.; Rieker, A. On the tautomerism of 3-alkyl-1-aryltriazenes. Tetrahedron Lett. 1976, 17, 2627–2630. [Google Scholar] [CrossRef]
- Banerjee, R.; Rachid, Z.; McNamee, J.; Jean-Claude, B.J. Synthesis of a prodrug designed to release multiple inhibitors of the epidermal growth factor receptor tyrosine kinase and an alkylating agent: A novel tumor targeting concept. J. Med. Chem. 2003, 46, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Wilman, D. X-ray studies on anti-tumour triazenes. Structures of 1-(4-carbamoylphenyl)-3,3-dimethyltriazene 1-oxide and 3, 3-dimethyl-1-(4-nitrophenyl) triazene. Acta Crystallogr. Sect. B Struct. Sci. 1992, 48, 213–217. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXTL: Structure Determination Software Suite, version 6.10, Bruker AXS: Madison, WI, USA, 2001.
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senhaji Mouhri, Z.; Goodfellow, E.; Kelley, S.P.; Stein, R.S.; Rogers, R.D.; J. Jean-Claude, B. 15N-, 13C- and 1H-NMR Spectroscopy Characterization and Growth Inhibitory Potency of a Combi-Molecule Synthesized by Acetylation of an Unstable Monoalkyltriazene. Molecules 2017, 22, 1183. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22071183
Senhaji Mouhri Z, Goodfellow E, Kelley SP, Stein RS, Rogers RD, J. Jean-Claude B. 15N-, 13C- and 1H-NMR Spectroscopy Characterization and Growth Inhibitory Potency of a Combi-Molecule Synthesized by Acetylation of an Unstable Monoalkyltriazene. Molecules. 2017; 22(7):1183. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22071183
Chicago/Turabian StyleSenhaji Mouhri, Zhor, Elliot Goodfellow, Steven P. Kelley, Robin S. Stein, Robin D. Rogers, and Bertrand J. Jean-Claude. 2017. "15N-, 13C- and 1H-NMR Spectroscopy Characterization and Growth Inhibitory Potency of a Combi-Molecule Synthesized by Acetylation of an Unstable Monoalkyltriazene" Molecules 22, no. 7: 1183. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22071183