A Facile Oxidative Opening of the C-Ring in Luotonin A and Derivatives

1
Department of Pharmaceutical Chemistry, University of Vienna, Althanstraße 14, A-1090 Vienna, Austria
2
Institute of Cancer Research, Medical University of Vienna, Borschkegasse 8a, A-1090 Vienna, Austria
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(9), 1540; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22091540
Submission received: 7 August 2017
/
Revised: 11 September 2017
/
Accepted: 11 September 2017
/
Published: 12 September 2017
(This article belongs to the Collection Heterocyclic Compounds)
Abstract
:An oxidative ring opening reaction of the central ring C in the alkaloid Luotonin A and two of its derivatives was found to occur upon heating with an excess amine and potassium carbonate in dimethylsulfoxide (DMSO) solution in the presence of air oxygen. The structure of the novel amide-type products was elucidated and a possible mechanism for this reaction is proposed. Four of the new compounds show moderate in vitro anticancer activity towards human colon adenocarcinoma cells.
1. Introduction
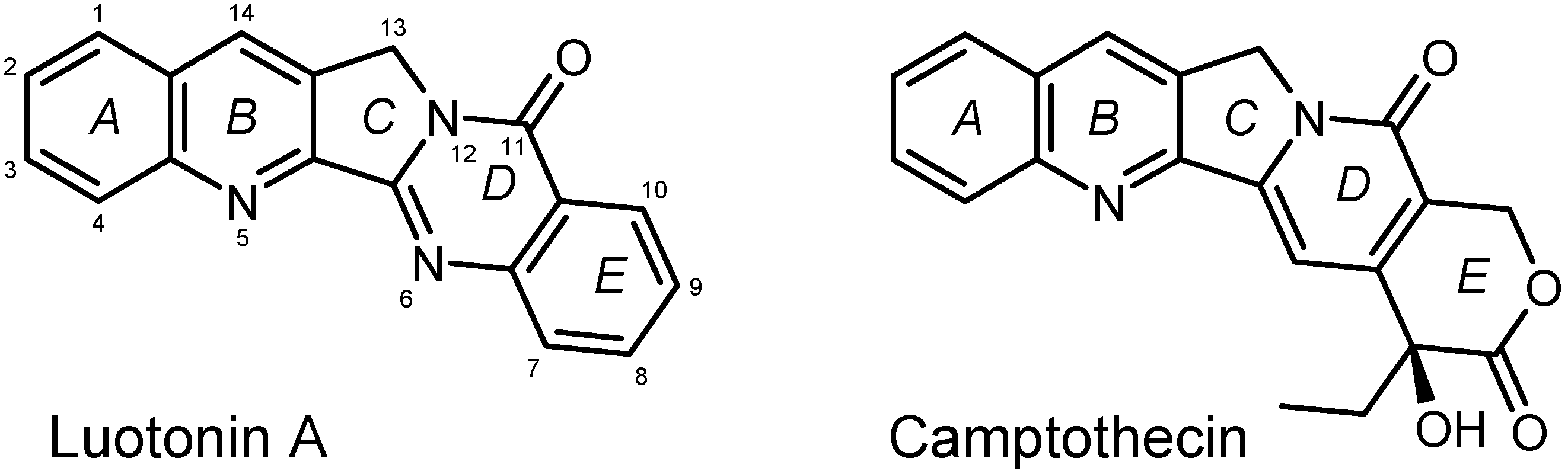
In search of new anticancer agents, structural modifications of the alkaloid Luotonin A (first isolated in 1997 [1]) have found considerable interest during the past two decades [2,3,4,5,6,7,8,9,10,11,12]. Like its more prominent (and more potent) naturally occurring relative, Camptothecin (CPT) [13,14,15], Luotonin A can act as a topoisomerase 1 poison by stabilizing the DNA/Topo1 complex [2,16]. Due to its aromatic E-ring (see Figure 1), Luotonin A is principally not associated with bladder toxicity which is a specific adverse effect of CPT-derived drugs like Topotecan or Irinotecan with their labile lactone structure as ring E [14]. On the other hand, the antineoplastic activity of the Luotonin A core structure is markedly lower than that of CPT [16] and thus has stimulated efforts to improve topoisomerase 1 toxicity by various modifications of the lead structure.
Several different strategies have been described for the total synthesis of Luotonin A and related compounds, as summarized in a review article [17]. Among the approaches aiming at variation of the substitution pattern of ring A, there have been some recent investigations of our group in which we made use of two orthogonal cycloaddition routes [18,19,20]: the first route is based on a pathway that was previously explored by Zhou et al. [21] for the synthesis of the parent compound and we found it to be well suited for the introduction of substituents into positions 2 or 4, whereas the second route offers a convenient access to 1- or 3-substituted congeners. Although a number of new Luotonin A derivatives could be made available via these two routes directly, additional modifications by replacement of existing substituents were envisaged in order to further extend our compound library. In particular, nucleophilic substitution of a fluoro functionality with amines appeared quite promising in view of the reported facile transformation of 8-fluoroquinolines into the corresponding 8-(di)alkylaminoquinolines via nucleophilic substitution [22,23]. Herein, we report on the synthesis of a hitherto unknown 4-fluoro derivative of Luotonin A (containing the 8-fluoroquinoline motif) and the observation of an unprecedented oxidative opening of ring C on attempted nucleophilic substitution reactions with primary or secondary amines.
2. Results and Discussion
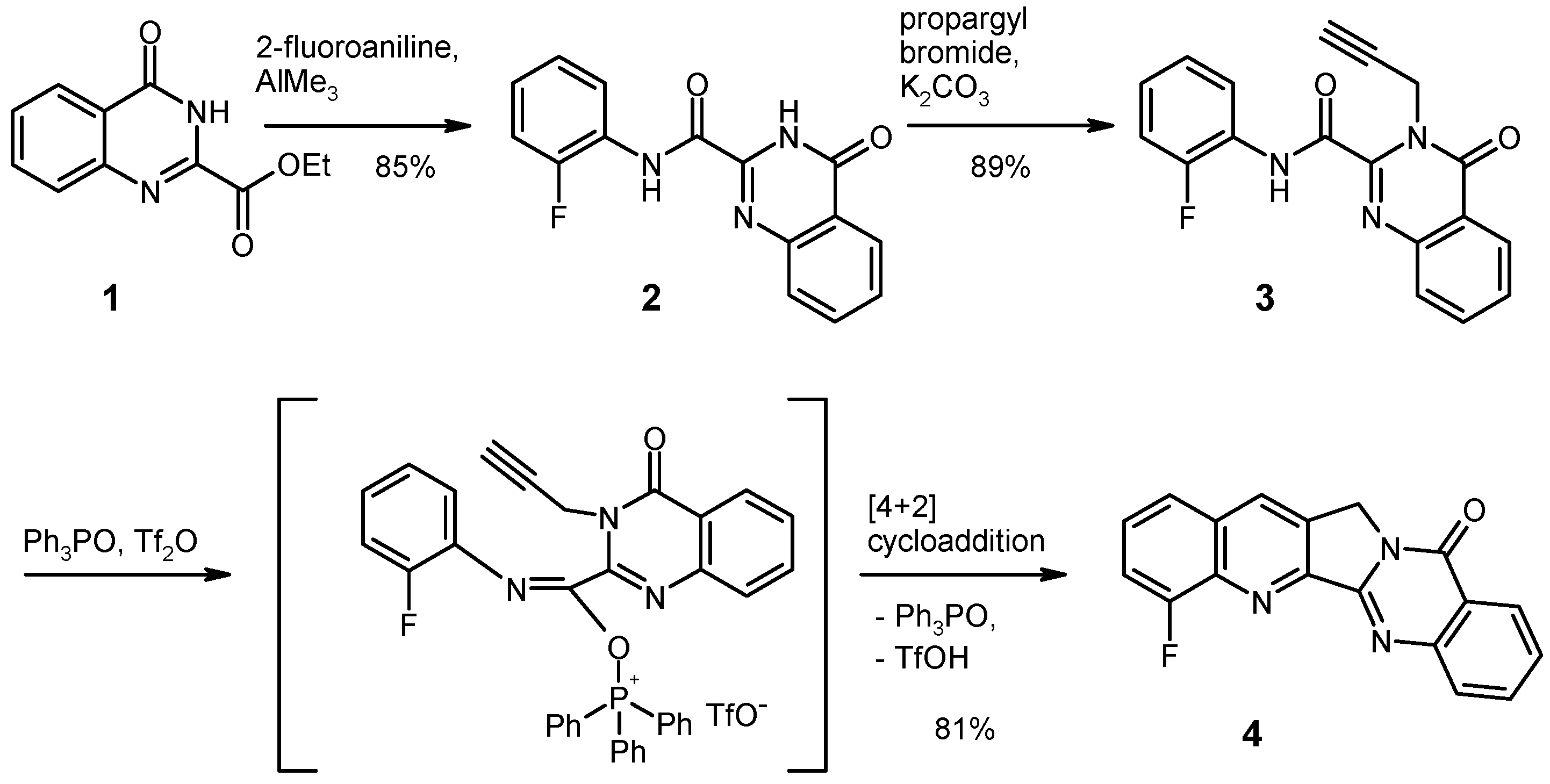
Among the four possible sites at ring A, position 4 (corresponding to position 8 of the quinoline core) appeared most interesting for the envisaged nucleophilic replacement of a fluorine atom with an amino substituent, as there have been several reports describing the preferential attack of nitrogen nucleophiles at this position in various di- and trifluoroquinolines [24,25,26,27]. We therefore commenced our investigations by preparing the hitherto unknown 4-fluoro derivative of Luotonin A via the pathway depicted in Scheme 1, starting from the quinazolinonecarboxylic acid ester 1 [21,28]. Weinreb amidation [29] with 2-fluoroaniline/trimethylaluminium gave the N-arylamide 2 in good yield, selective propargylation at the quinazoline N-3 afforded the key intermediate 3. On treatment of the latter with bis(triphenyl)oxodiphosphonium triflate (Hendrickson’s reagent [30]) in dry dichloromethane at room temperature, an intramolecular [4+2] cycloaddition smoothly afforded the pentacyclic compound 4 in excellent overall yield.
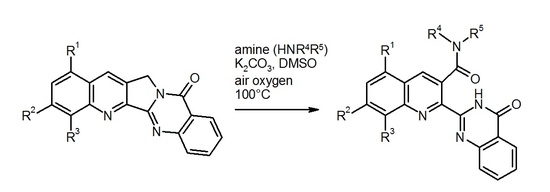
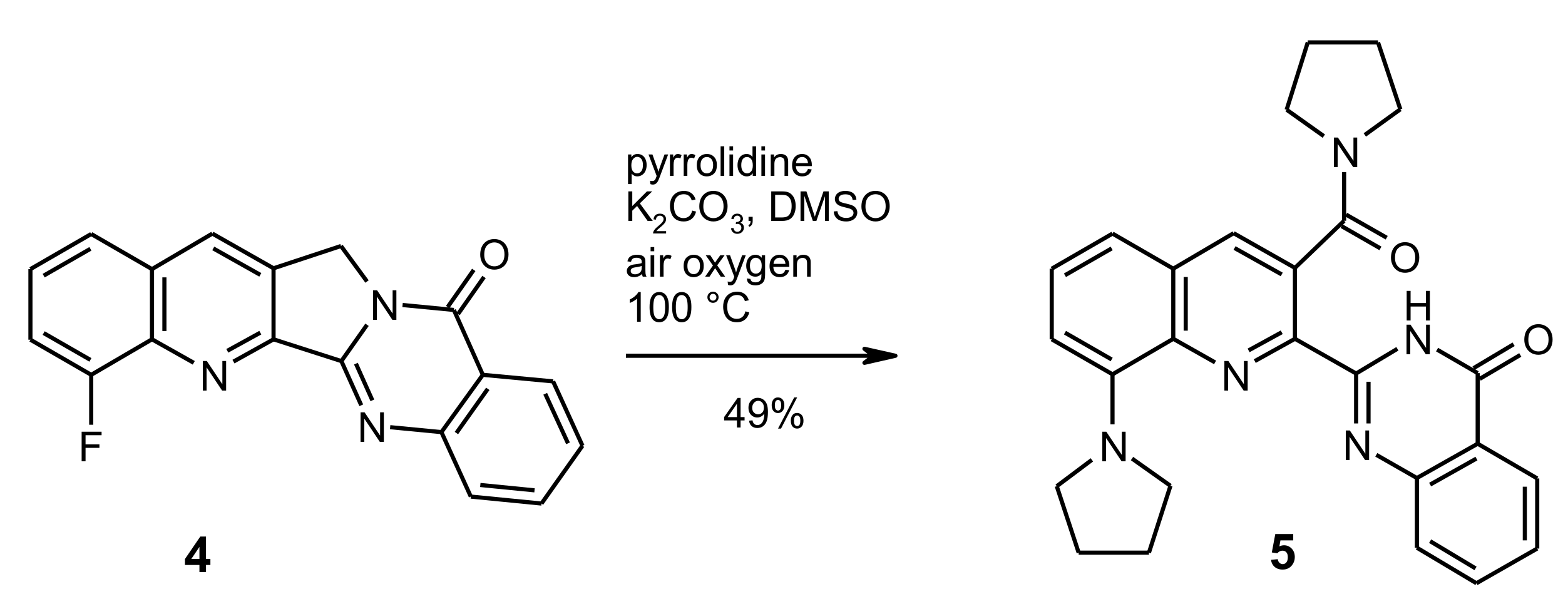
In order to achieve the desired nucleophilic fluorine substitution, we chose the conditions that had been described in the patent literature [22,23] for the reaction of 8-fluoroquinoline derivatives with various amines. Thus, the fluoro compound 4 was heated to 100 °C with an excess of pyrrolidine in dimethylsulfoxide (DMSO) solution in the presence of potassium carbonate. As indicated by TLC, the starting material was completely consumed after 24 h and a single new spot was observed. After evaporation of volatiles and extractive work-up, the crude product was purified by medium-pressure liquid chromatography to afford the new compound in 49% yield as red-coloured crystals. 1H-NMR-Spectroscopic characterisation surprisingly revealed the presence of two pyrrolidinyl residues in the molecule and the absence of the methylene unit that is part of ring C in the Luotonin A skeleton. On the other hand, the 13C-NMR spectrum showed an additional carbonyl resonance (besides the quinazolinone C=O group at 161.4 ppm) at lower field (168.6 ppm). Mass spectrometry (EI-MS (electron-impact ionization) and ESI-TOF (electrospray ionization, time-of-flight) high-resolution MS) indicated a molecular formula of C26H25N5O2. Based on the combined spectroscopic evidence, it can be concluded that (a) the expected nucleophilic substitution of the fluorine atom with pyrrolidine has indeed taken place and (b) the C-ring of the pentacyclic scaffold has undergone an oxidative ring opening, involving another pyrrolidine unit that is now attached to the quinoline core via an amide function (structure 5, Scheme 2).
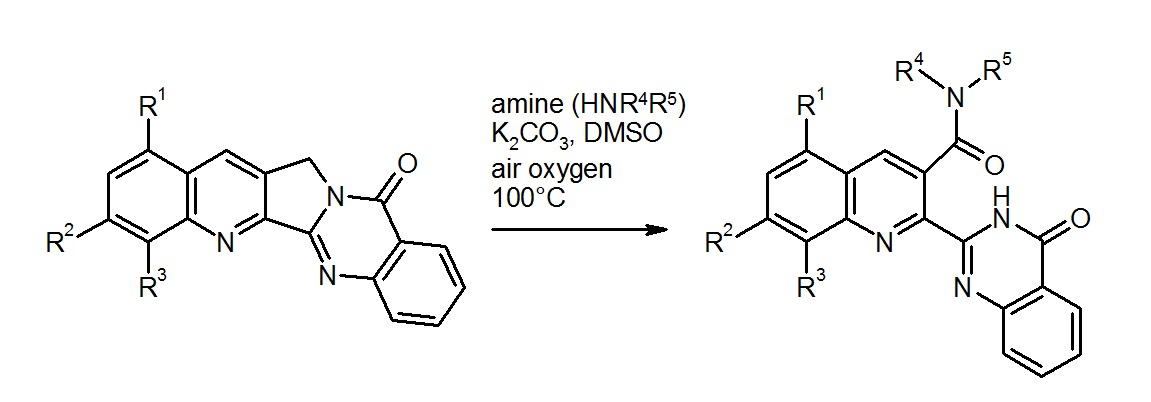
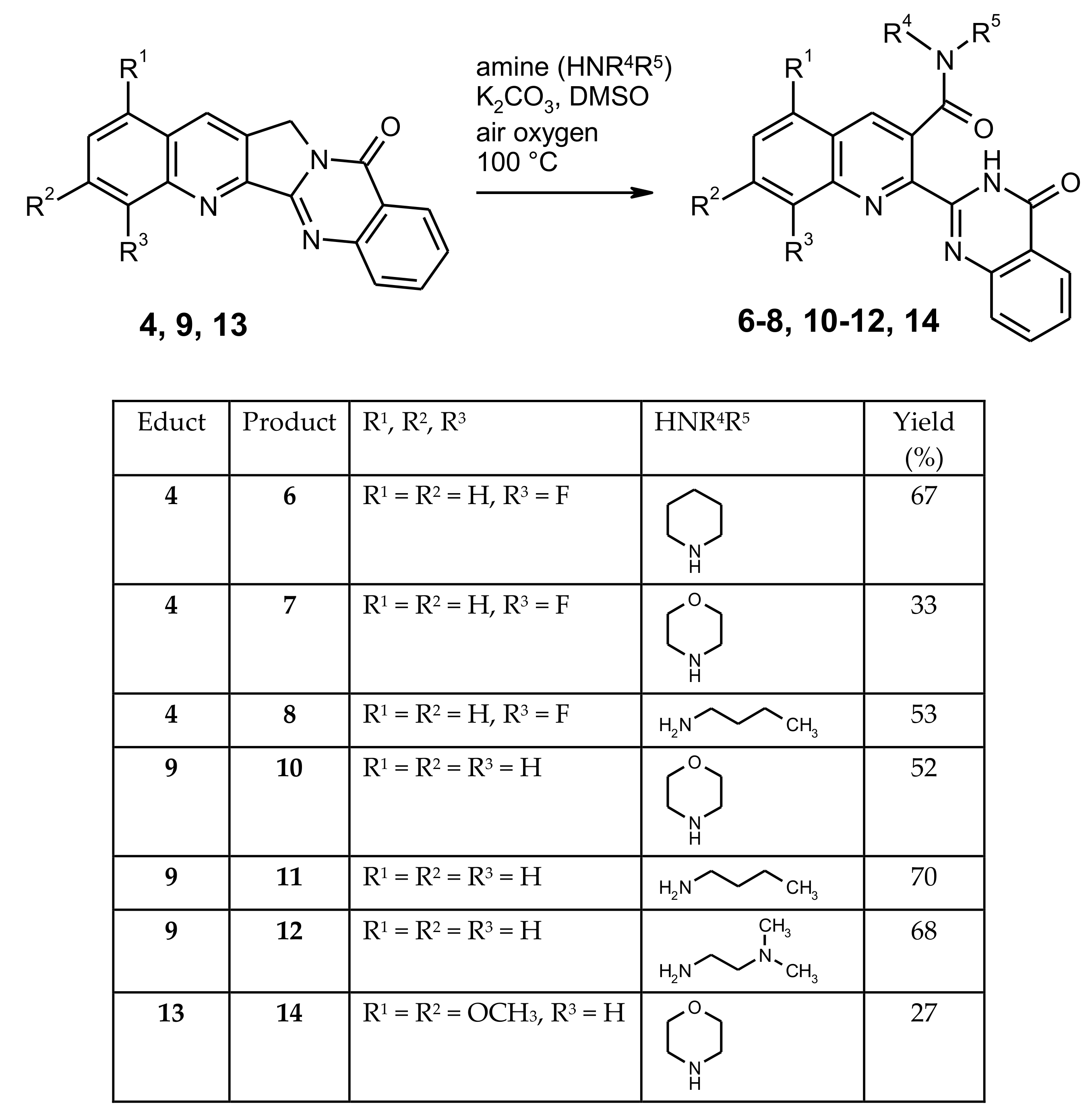
To explore the scope of this unexpected reaction, the fluoro compound 4 was reacted with other amines under the same conditions (100 °C, DMSO as solvent, and K2CO3 as a base). Interestingly, it turned out that only pyrrolidine is reactive enough to effect the fluorine substitution under these conditions, whereas piperidine, morpholine, and n-butylamine left the fluoro function untouched, but all led to the formation of ring-opened products with a (tertiary or secondary) carboxamide group (compounds 6–8, Scheme 3). An identical reaction behavior was found with the alkaloid Luotonin A itself (compound 9), affording the amides 10–12 on treatment with morpholine, n-butylamine, or N,N-dimethylethylenediamine, respectively. Also with the electron-rich 1,3-dimethoxy derivative [18] of Luotonin A (compound 13), an analogous oxidative ring C opening was observed, furnishing the morpholide 14, albeit in moderate yield. All of the isolated carboxamides share some characteristic NMR properties such as the carbonyl resonance in the 13C-NMR spectra at 168–169 ppm (see supplementary material). The position of the secondary or tertiary amide function at C-3 of the quinoline nucleus is evidenced by HMBC (Heteronuclear Multiple Bond Correlation) crosspeaks between this carbonyl signal and the H-4 resonance as well as the NCH2 protons in the amide part. The spatial proximity between the latter two kinds of protons is further proven by a corresponding crosspeak in the NOESY (Nuclear Overhauser Effect Spectroscopy) spectrum. Additionally based on HSQC (Heteronuclear Single-Quantum Correlation) and COSY (Correlation Spectroscopy) experiments, all signals in the 1H- and 13C-NMR spectra could be assigned and are in full agreement with the proposed structures. As with compound 5, the molecular formulae of all new compounds were confirmed by [M + H]+ or [M + Na]+ peaks in the high-resolution ESI-TOF mass spectra.
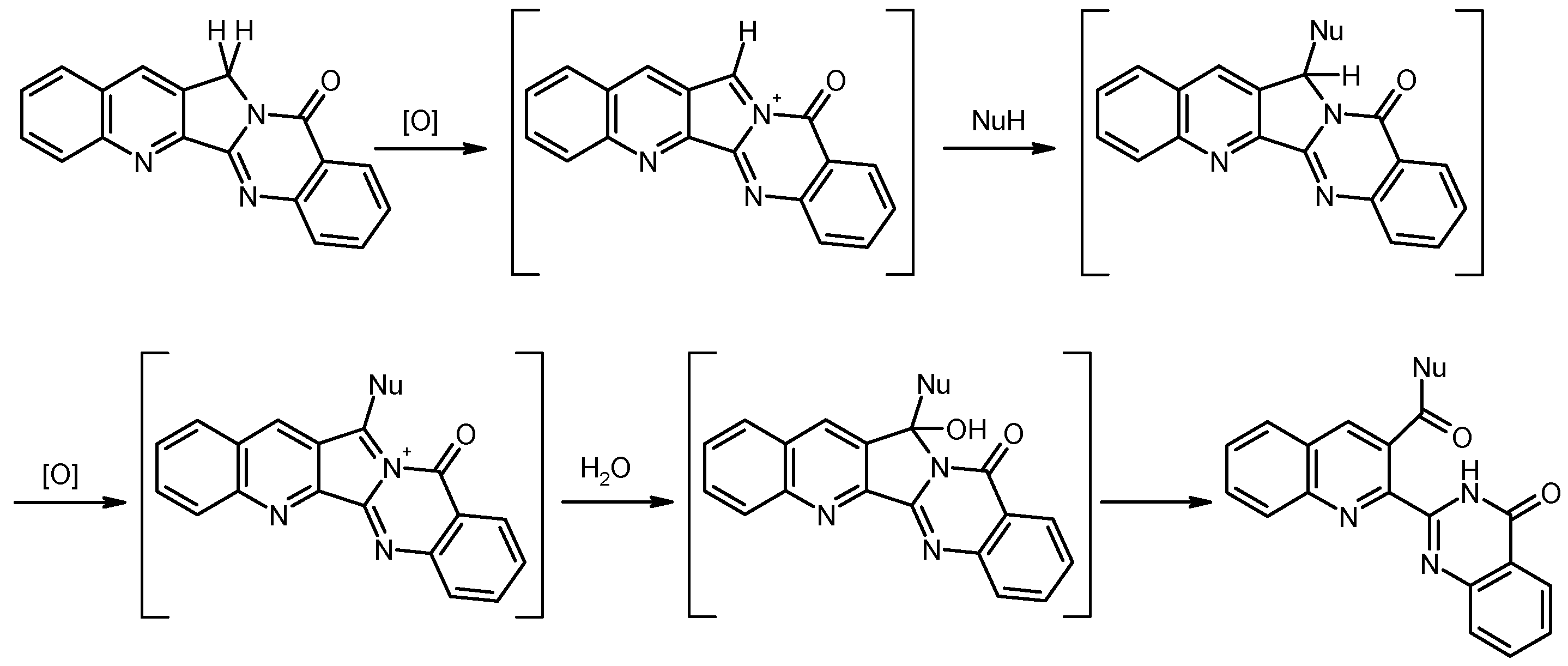
To gain more insight into this remarkable transformation, we varied the solvent and observed essentially the same results when N,N-dimethylformamide (DMF) was used instead of DMSO. However, with 1-propanol as a protic solvent, a complex reaction mixture was obtained from which no defined product could be isolated. Thus, an aprotic dipolar solvent seems to be favorable in this context. As neither the amine component, nor the inorganic base, nor the solvent have oxidative properties to perform the observed oxidation of the benzylic CH2 group of ring C into an amide function, we suspected atmospheric oxygen to play a crucial role in this process. Indeed, when Luotonin A was heated to 100 °C with morpholine/K2CO3 in DMSO or DMF solution under argon atmosphere, the starting material remained essentially unchanged after several hours [31], whereas a control experiment in the presence of air oxygen showed an almost complete transformation into compound 10 after the same time span. A possible mechanism for this oxidative/nucleophilic ring opening is formulated in Scheme 4: Presumably, the benzylic carbon undergoes a two-step oxidation sequence, involving attack of the amine after the first step and hydrolytic ring cleavage after the second step (promoted by moisture in the solvent or during work-up). To the best of our knowledge, a comparable reaction has not yet been reported for Luotonin A nor for the structurally related CPT.
Although our initial aim, i.e., the synthesis of new A-ring-modified Luotonin A derivatives, was not met, the obtained quinazolinyl-substituted quinoline-3-carboxamides represent interesting and hitherto unknown molecular entities. Therefore, we briefly examined the new compounds for their in vitro growth inhibitory activity towards SW480 human colon adenocarcinoma cells. Indeed, three of the tested compounds (5, 6 and 7) showed moderate activity, slightly exceeding that of the natural lead compound, Luotonin A. Compounds 8, 11, 12 and 14 are clearly inferior and compound 10 is approximately equipotent as compared to the reference (Table 1). At present, no valid conclusions can be drawn with respect to structure-activity relationships and the mode of action.
3. Experimental
3.1. General
Melting points (uncorrected) were determined on a Kofler hot-stage microscope (Leica GmbH, Wetzlar, Germany). 1H-NMR and 13C-NMR spectra were recorded on a Bruker Avance III 400 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) at 400 MHz and 100 MHz, respectively; chemical shifts (ppm) were referenced to residual amounts of undeuterated solvents. Mass spectra (EI) were obtained on a Shimadzu QP5050A DI 50 instrument (Shimadzu Corp., Kyoto, Japan); high-resolution mass spectra (ESI-TOF) were recorded on a Bruker maXis HD spectrometer (Bruker Daltonics GmbH, Bremen, Germany). Column chromatography was carried out on Merck Kieselgel 60 (Merck, Darmstadt, Germany), 0.063–0.200 mm. Medium-pressure liquid chromatography (MPLC) was performed on a Biotage Isolera One system (Biotage AB, Uppsala, Sweden), using Biotage SNAP cartridges (KP-Sil 10 g). Thin layer chromatography was done on Merck aluminium sheets pre-coated with Kieselgel 60 F254 (Merck, Darmstadt, Germany). Microanalyses were performed at the Microanalytical Laboratory, Faculty of Chemistry, University of Vienna. Ethyl 4-oxo-3,4-dihydroquinazoline-2-carboxylate [28] (1), Luotonin A (9) and its dimethoxy derivative (13) were prepared according to literature procedures [18,21]. Amines were purified by distillation prior to use, solvents (analytical grade) were used without further purification.
3.2. Procedures
N-(2-Fluorophenyl)-4-oxo-3,4-dihydroquinazoline-2-carboxamide (2). To a solution of 2-fluoroaniline (0.889 g, 8 mmol) in dry 1,2-dichloroethane (20 mL) under argon was added drop wise a 2 M solution of AlMe3 (4.0 mL, 8 mmol) in heptane. The mixture was stirred for 30 min at room temperature, then the ester 1 (1.091 g, 5 mmol) was added in one portion, and the mixture was refluxed for 2 h. After cooling to 0 °C, it was then quenched by slow addition of 2 N HCl (20 mL), followed by water (80 mL). The mixture was exhaustively extracted with CH2Cl2 and the combined extracts were washed with water and brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was recrystallized from EtOH to give 2 (1.202 g, 85%) as colorless needles, m.p. 216–217 °C. MS (EI, 70 eV) m/z = 283 (M+, 32%), 236 (18), 146 (91), 119 (100), 118 (18), 91 (15), 90 (31), 83 (12); 1H-NMR (DMSO-d6) δ: 12.62 (br s, 1H, NH), 10.50 (br s, 1H, NH), 8.21 (dd, J = 8.0 Hz, 1.2 Hz, 1H, 5-H), 7.97–7.89 (m, 2H, 7-H, phenyl 6′-H), 7.87 (dd, J = 8.2 Hz, 0.9 Hz, 1H, 8-H), 7.65 (ddd, J = 8.2 Hz, 7.0 Hz, 1.3 Hz, 1H, 6-H), 7.41–7.23 (m, 3H, phenyl 3′-H, 4′-H, 5′-H); 13C-NMR (DMSO-d6) δ: 161.3, 158.0, 154.5 (d, JC–F = 246.9 Hz), 146.6, 145.6, 134.8, 128.3, 127.7, 126.9 (d, JC–F = 7.7 Hz), 126.2, 124.8, 124.7, 124.6, 122.8, 115.8 (d, JC–F = 19.4 Hz). Anal. calcd. for C15H10FN3O2·0.2 H2O: C, 62.80; H, 3.65; N, 14.65. Found: C, 62.79; H, 3.46; N, 14.66. HRMS (ESI-TOF) m/z calcd. for C15H11FN3O2 ([M + H]+): 284.0830. Found: 284.0827.
N-(2-Fluorophenyl)-4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxamide (3). To a solution of the anilide 2 (0.566 g, 2 mmol) in dry DMF (15 mL) were added K2CO3 (0.304 g, 2.2 mmol) and propargyl bromide (0.327 g of an 80% solution in toluene, 2.2 mmol). The mixture was stirred at room temperature for 24 h, then it was poured into water (100 mL) and it was extracted with CH2Cl2 (3 × 100 mL). The combined extracts were washed with water and brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was recrystallized from EtOH to give 3 (0.527 g, 89%) as colorless crystals, m.p. 153–154 °C. MS (EI, 70 eV) m/z = 321 (M+, 21%), 320 (72), 301 (27), 292 (52), 184 (25), 155 (54), 148 (40), 145 (36), 129 (100), 119 (96), 90 (64), 83 (47), 75 (42), 63 (41); 1H-NMR (CDCl3) δ: 9.99 (s, 1H, NH), 8.52–8.41 (m, 1H, phenyl 6′-H), 8.37 (ddd, J = 8.0 Hz, 1.3 Hz, 0.6 Hz, 1H, 5-H), 7.89–7.79 (m, 2H, 7-H, 8-H), 7.62 (ddd, J = 8.2 Hz, 6.5 Hz, 1.9 Hz, 1H, 6-H), 7.24–7.14 (m, 3H, phenyl 3′-H, 4′-H, 5′-H), 5.60 (d, J = 2.5 Hz, 2H, CH2), 2.27 (t, J = 2.5 Hz, 1H, acetylene-H); 13C-NMR (CDCl3) δ: 161.4, 158.3, 153.1 (d, JC–F = 244.0 Hz), 145.0, 144.9, 135.1, 129.3, 128.2, 127.6, 125.7, 125.6 (d, JC–F = 7.5 Hz), 124.9 (d, JC–F = 3.8 Hz), 121.9, 121.7, 115.3 (d, JC–F = 19.1 Hz), 78.9, 72.2, 33.8. Anal. calcd. for C18H12FN3O2·0.2 H2O: C, 66.54; H, 3.85; N, 12.93. Found: C, 66.53; H, 3.64; N, 12.97. HRMS (ESI-TOF) m/z calcd. for C18H13FN3O2 ([M + H]+): 322.0986. Found: 322.0988.
4-Fluoroquinolino[2′,3′:3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (4). To a solution of triphenylphosphine oxide (0.835 g, 3 mmol) in dry CH2Cl2 (22 mL) was drop wise added trifluoromethanesulfonic anhydride (0.25 mL, 1.5 mmol) at 0 °C under argon, and the mixture was stirred at the same temperature for 15 min. Then, compound 3 (0.321 g, 1 mmol) was added in one portion at 0 °C, and the mixture was stirred for 1 h while slowly warming to room temperature. The reaction was quenched by addition of 10% aqueous NaHCO3 (15 mL). The phases were separated and the aqueous layer was exhaustively extracted with CH2Cl2. The combined organic layers were washed with water and brine, dried over Na2SO4 and evaporated. To remove excess triphenylphosphine oxide, the residue was subjected to column chromatography, eluting first with CH2Cl2, then with CH2Cl2/ethyl acetate (19:1). The fraction showing an intense blue fluorescence under UV366 was evaporated under reduced pressure and the residue was recrystallized from EtOH to give 4 (0.244 g, 81%) as colorless needles, m.p. > 310 °C (sublimation). MS (EI, 70 eV) m/z = 304 ([M + 1]+, 20%), 303 (M+, 100), 302 (36), 275 (13), 274 (10), 262 (8), 152 (12), 77 (7); 1H-NMR (CDCl3) δ: 8.49 (d, J = 1.0 Hz, 1H, 14-H), 8.42 (dd, J = 8.0 Hz, 1.1 Hz, 1H, 10-H), 8.13–8.08 (m, 1H, 7-H), 7.86 (ddd, J = 8.3 Hz, 7.2 Hz, 1.6 Hz, 1H, 8-H), 7.75 (d, J = 8.3 Hz, 1H, 1-H), 7.68–7.49 (m, 3H, 2-H, 3-H, 9-H), 5.37 (d, J = 0.8 Hz, 2H, CH2); 13C-NMR (CDCl3) δ: 160.7 (11-C), 158.8 (d, JC–F = 261.4 Hz, 4-C), 152.1 (5b-C), 151.7 (5a-C), 149.4 (6a-C), 140.1 (d, JC–F = 12.8 Hz, 4a-C), 134.8 (8-C), 131.6 (d, JC–F = 2.9 Hz, 14-C), 130.7 (13a-C), 130.3 (14a-C), 129.1 (7-C), 128.7 (d, JC–F = 7.8 Hz, 2-C), 127.8 (9-C), 126.6 (10-C), 123.7 (d, JC–F = 5.0 Hz, 1-C), 121.5 (10a-C), 114.9 (d, JC–F = 18.7 Hz, 3-C), 47.4 (13-C). Anal. calcd. for C18H10FN3O·0.15 H2O: C, 70.65; H, 3.39; N, 13.73. Found: C, 70.63; H, 3.30; N, 13.74. HRMS (ESI-TOF) m/z calcd. for C18H11FN3O ([M + H]+): 304.0881. Found: 304.0882.
2-[8-(Pyrrolidin-1-yl)-3-(pyrrolidin-1-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (5). To a solution/suspension of the fluoro compound 4 (0.152 g, 0.5 mmol) and K2CO3 (0.069 g, 0.5 mmol) in DMSO (15 mL) was added pyrrolidine (0.356 g, 5 mmol). The mixture was heated to 100 °C and the color of the solution changed into dark red. Stirring was continued for 24 h at 100 °C, then the mixture was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/MeOH (94:6) to afford compound 5 (108 mg, 49%) as red crystals, m.p. 210–211 °C (CHCl3). MS (EI, 70 eV) m/z = 439 (M+, 9%), 368 (100), 340 (42), 339 (27), 311 (28), 299 (23), 273 (23); 1H-NMR (CDCl3) δ 10.68 (s, 1H, NH), 8.35 (dd, J = 7.9 Hz, 1.1 Hz, 1H, quinazoline 5-H), 8.10 (s, 1H, quinoline 4-H), 7.80–7.73 (m, 1H, quinazoline 7-H), 7.67–7.60 (m, 1H, quinazoline 8-H), 7.55–7.45 (m, 2H, quinazoline 6-H, quinoline 6-H), 7.15 (d, J = 7.2 Hz, 1H, quinoline 5-H), 6.84 (d, J = 7.8 Hz, 1H, quinoline 7-H), 4.00–3.80 (m, 6H, N-arylpyrrolidine 2-H, 2′-H, 5-H, 5′-H, N-acylpyrrolidine 2-H, 2′-H), 3.24 (t, J = 6.7 Hz, 2H, N-acylpyrrolidine 5-H, 5′-H), 2.17–2.10 (m, 4H, N-arylpyrrolidine 3-H, 3′-H, 4-H, 4′-H), 2.06 (quint, J = 6.9 Hz, 2H, N-acylpyrrolidine 3-H, 3′-H), 1.88 (quint, J = 6.8 Hz, 2H, N-acylpyrrolidine 4-H, 4′-H); 13C-NMR (CDCl3) δ: 168.6 (amide C=O), 161.4 (quinazoline 4-C), 149.0 (quinazoline 8a-C), 148.7 (quinazoline 2-C), 146.6 (quinoline 8-C), 139.3 quinoline 2-C), 138.4 (quinoline 8a-C), 136.1 (quinoline 4-C), 134.7 (quinazoline 7-C), 130.8 (quinoline 4a-C), 130.5 (quinoline 3-C), 130.3 (quinoline 6-C), 128.3 (quinazoline 8-C), 127.6 (quinazoline 6-C), 127.0 (quinazoline 5-C), 122.6 (quinazoline 4a-C), 114.9 (quinoline 5-C), 111.3 (quinoline 7-C), 52.6 (N-arylpyrrolidine 2-C, 5-C), 48.6 (N-acylpyrrolidine 5-C), 46.2 (N-acylpyrrolidine 2-C), 26.2 (N-arylpyrrolidine 3-C, 4-C), 26.1 (N-acylpyrrolidine 4-C), 25.0 (N-acylpyrrolidine 3-C). HRMS (ESI-TOF) m/z calcd. for C26H26N5O2 ([M + H]+): 440.2081. Found: 440.2080.
2-[8-Fluoro-3-(piperidin-1-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (6). A mixture of the fluoro compound 4 (0.152 g, 0.5 mmol), K2CO3 (0.069 g, 0.5 mmol) and piperidine (0.426 g, 5 mmol) in DMSO (15 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/MeOH (97:3) to afford 6 (0.134 g, 67%) as dark-red crystals, m.p. 215–216 °C (CHCl3). MS (EI, 70 eV) m/z = 402 (M+, 2%), 318 (26), 289 (16), 171 (21), 119 (36), 84 (100), 69 (26), 64 (21), 57 (41), 56 (44), 55 (67); 1H-NMR (CDCl3) δ 11.11 (s, 1H, NH), 8.38 (ddd, J = 7.9 Hz, 1.5 Hz, 0.5 Hz, 1H, quinazoline 5-H), 8.24 (d, J = 1.4 Hz, 1H, quinoline 4-H), 7.81 (ddd, J = 8.4 Hz, 6.9 Hz, 1.5 Hz, 1H, quinazoline 7-H), 7.76 (ddd, J = 8.2 Hz, 1.4 Hz, 0.5 Hz, 1H, quinazoline 8-H), 7.70 (dd, J = 8.2 Hz, 1.2 Hz, 1H, quinoline 5-H), 7.64 (td, J = 7.9 Hz, 4.7 Hz, 1H, quinoline 6-H), 7.59–7.49 (m, 2H, quinazoline 6-H, quinoline 7-H), 4.44–4.27 (m, 1H, piperidine 2-H), 3.50 (ddd, J = 13.3 Hz, 9.8 Hz, 3.6 Hz, 1H, piperidine 2′-H), 3.32–3.22 (m, 1H, piperidine 6-H), 3.12 (ddd, J = 13.1 Hz, 9.6 Hz, 3.4 Hz, 1H, piperidine 6′-H), 2.01–1.87 (m, 1H, piperidine 3-H), 1.84–1.69 (m, 2H, piperidine 3′-H, 4-H), 1.62–1.50 (m, 2H, piperidine 4′-H, 5-H), 1.48–1.41 (m, 1H, piperidine 5′-H); 13C-NMR (CDCl3) δ: 168.2 (amide C=O), 161.4, (quinazoline 4-C), 158.1 (d, JC–F = 261.3 Hz, quinoline 8-C), 148.6 (quinazoline 8a-C), 147.9 (quinazoline 2-C), 144.7 (quinoline 2-C), 136.8 (d, JC–F = 12.4 Hz, quinoline 8a-C), 136.0 (d, JC–F = 2.9 Hz, quinoline 4-C), 134.7 (quinazoline 7-C), 131.4 (quinoline 3-C), 130.0 (quinoline 4a-C), 129.3 (d, JC–F = 7.8 Hz, quinoline 6-C), 128.5 (quinazoline 8-C), 128.2 (quinazoline 6-C), 127.1 (quinazoline 5-C), 123.4 (d, JC–F = 5.0 Hz, quinoline 5-C), 123.0 (quinazoline 4a-C), 115.3 (d, JC–F = 18.3 Hz, quinoline 7-C), 48.2 (piperidine 6-C), 43.0 (piperidine 2-C), 25.8 (piperidine 5-C), 25.4 (piperidine 3-C), 24.7 (piperidine 4-C). HRMS (ESI-TOF) m/z calcd. for C23H20FN4O2 ([M + H]+) 403.1563. Found 403.1563.
2-[8-Fluoro-3-(morpholin-4-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (7). A mixture of the fluoro compound 4 (0.152 g, 0.5 mmol), K2CO3 (0.069 g, 0.5 mmol) and morpholine (0.436 g, 5 mmol) in DMSO (15 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to column chromatography, eluting with CH2Cl2/MeOH (97:3) to afford 7 (0.067 g, 33%) as yellow crystals, m.p. > 265 °C (sublimation; CHCl3). MS (EI, 70 eV) m/z = 405 ([M + 1]+, 2%), 404 (M+, 8), 319 (85), 318 (100), 146 (20), 119 (43), 57 (22), 56 (23); 1H-NMR (CDCl3) δ 11.10 (s, 1H, NH), 8.41–8.36 (m, 1H, quinazoline 5-H), 8.28 (d, J = 1.4 Hz, 1H, quinoline 4-H), 7.82 (ddd, J = 8.4 Hz, 7.0 Hz, 1.5 Hz, 1H, quinazoline 7-H), 7.77 (ddd, J = 8.2 Hz, 1.4 Hz, 0.6 Hz, 1H, quinazoline 8-H), 7.72 (dd, J = 8.3 Hz, 1.3 Hz, 1H, quinoline 5-H), 7.66 (td, J = 7.9 Hz, 4.6 Hz, 1H, quinoline 6-H), 7.61–7.56 (m, 1H, quinazoline 6-H), 7.56–7.51 (m, 1H, quinoline 7-H), 4.29 (dt, J = 13.0 Hz, 3.0 Hz, 1H, morpholine 5-H), 4.11 (dt, J = 11.7 Hz, 3.6 Hz, 1H, morpholine 6-H), 3.81 (ddd, J = 11.7 Hz, 9.3 Hz, 2.9 Hz, 1H, morpholine 6′-H), 3.74–3.64 (m, 2H, morpholine 2-H, 5′-H), 3.55 (ddd, J = 11.6 Hz, 9.0 Hz, 3.1 Hz, 1H, morpholine 2′-H), 3.38–3.27 (m, 1H, morpholine 3-H), 3.26–3.17 (m, 1H, morpholine 3′-H); 13C-NMR (CDCl3) δ: 168.8 (amide C=O), 161.3 (quinazoline 4-C), 158.0 (JC–F = 260.0 Hz, quinoline 8-C), 148.3 (quinazoline 8a-C), 147.8 (quinazoline 2-C), 144.7 (quinoline 2-C), 136.9 (JC–F = 12.5 Hz, quinoline 8a-C), 136.4 (JC–F = 2.8 Hz, quinoline 4-C), 134.9 (quinazoline 7-C), 130.2 (quinoline 3-C), 129.8 (quinoline 4a-C), 129.6 (JC–F = 7.9 Hz, quinoline 6-C), 128.5 (quinazoline 8-C), 128.4 (quinazoline 6-C), 127.1 (quinazoline 5-C), 123.5 (JC–F = 4.9 Hz, quinoline 5-C), 122.9 (quinazoline 4a-C), 115.6 (JC–F = 18.4 Hz, quinoline 7-C), 66.6 (morpholine 2-C), 66.2 (morpholine 6-C), 47.2 (morpholine 3-C), 42.6 (morpholine 5-C). HRMS (ESI-TOF) m/z calcd. for C22H18FN4O3 ([M + H]+) 405.1357. Found 405.1356.
N-Butyl-8-fluoro-2-(4-oxo-3,4-dihydroquinazolin-2-yl)quinoline-3-carboxamide (8). A mixture of the fluoro compound 4 (0.100 g, 0.33 mmol), K2CO3 (0.046 g, 0.33 mmol) and n-butylamine (0.241 g, 3.3 mmol) in DMSO (10 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/MeOH (96:4) to afford 8 (0.078 g, 53%) as colorless crystals, m.p. 218–220 °C (methyl tert-butyl ether). MS (EI, 70 eV) m/z = 391 ([M + 1]+, 2%), 390 (M+, 6), 319 (63), 318 (100), 291 (28), 171 (20), 146 (21), 119 (49), 92 (22), 90 (35); 1H-NMR (CDCl3) δ 10.94 (s, 1H, quinazolinone NH), 8.39 (d, J = 1.3 Hz, 1H, quinoline 4-H), 8.22 (d, J = 7.9 Hz, 1H, quinazoline 5-H), 7.76–7.74 (m, 2H, quinazoline 7-H, 8-H), 7.68 (d, J = 7.6 Hz, 1H, quinoline 5-H), 7.62 (td, J = 7.9 Hz, 4.8 Hz, 1H, quinoline 6-H), 7.53–7.46 (m, 1H, quinoline 7-H), 7.46–7.38 (m, 1H, quinazoline 6-H), 6.54 (t, J = 5.6 Hz, 1H, amide NH), 3.71–3.58 (m, 2H, butyl 1-CH2), 1.76 (p, J = 7.5 Hz, 2H, butyl 2-CH2), 1.51 (dq, J = 14.6 Hz, 7.3 Hz, 2H, butyl 3-CH2), 1.01 (t, J = 7.4 Hz, 3H, butyl CH3); 13C-NMR (CDCl3) δ: 168.1 (amide C=O), 161.2 (quianzoline 4-C), 157.7 (d, JC–F = 261.3 Hz, quinoline 8-C), 148.2 (quinazoline 8a-C), 147.6 (quinazoline 2-C), 144.6 (quinoline 2-C), 137.6 (d, JC–F = 2.5 Hz, quinoline 4-C), 136.6 (d, JC–F = 12.4 Hz, quinoline 8a-C), 134.6 (quinazoline 7-C), 131.5 (quinoline 3-C), 129.4 (quinoline 4a-C), 129.2 (d, JC–F = 7.9 Hz, quinoline 6-C), 128.3 (quinazoline 8-C), 127.9 (quinazoline 6-C), 126.6 (quinazoline 5-C), 123.4 (d, JC–F = 4.8 Hz, quinoline 5-C), 122.4 (quinazoline 4a-C), 115.4 (d, JC–F = 18.3 Hz, quinoline 7-C), 40.5 (butyl 1-C), 31.5 (butyl 2-C), 20.4 (butyl 3-C), 13.9 (butyl 4-C). HRMS (ESI-TOF) m/z calcd. for C22H20FN4O2 ([M + H]+) 391.1565. Found 391.1561.
2-[3-(Morpholin-4-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (10). A mixture of Luotonin A (9) (0.029 g, 0.1 mmol), K2CO3 (0.014 g, 0.1 mmol) and morpholine (0.087 g, 1 mmol) in DMSO (5 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (20 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure to afford 10 (0.020 g, 52%) as almost colorless crystals, m.p. > 272 °C (sublimation; CH2Cl2). MS (EI, 70 eV) m/z = 296 (21%), 211 (27), 111 (32), 109 (24), 97 (47), 95 (39), 85 (38), 83 (46), 81 (51), 71 (56), 69 (81), 67 (28), 57 (100); 1H-NMR (CDCl3) δ 11.15 (s, 1H, NH), 8.38 (dd, J = 7.9 Hz, 1.0 Hz, 1H, quinazoline 5-H), 8.25 (s, 1H, quinoline 4-H), 8.20 (d, J = 7.5 Hz, 1H, quinoline 8-H), 7.92 (d, J = 8.1 Hz, 1H, quinoline 5-H), 7.88 (ddd, J = 8.4 Hz, 6.9 Hz, 1.4 Hz, 1H, quinoline 7-H), 7.85–7.75 (m, 2H, quinazoline 7-H, 8-H), 7.72 (ddd, J = 8.2 Hz, 7.0 Hz, 1.2 Hz, 1H, quinoline 6-H), 7.57 (ddd, J = 8.2 Hz, 6.9 Hz, 1.5 Hz, 1H, quinazoline 6-H), 4.29 (dt, J = 13.4 Hz, 3.5 Hz, 1H, morpholine 3-H), 4.10 (dt, J = 11.6 Hz, 3.7 Hz, 1H, morpholine 2-H), 3.86–3.76 (m, 1H, morpholine 2′-H), 3.74–3.63 (m, 2H, morpholine 3′-H, 6-H), 3.55 (ddd, J = 11.7 Hz, 8.8 Hz, 3.2 Hz, 1H, morpholine 6′-H), 3.32–3.24 (m, 2H, morpholine 5-H, 5′-H); 13C-NMR (CDCl3) δ: 169.2 (amide C=O), 161.4 (quinazoline 4-C), 148.6 (quinazoline 8a-C), 148.1 (quinazoline 2-C), 146.5 (quinoline 8a-C), 144.3 (quinoline 2-C), 136.6 (quinoline 4-C), 134.9 (quinazoline 7-C), 131.7 (quinoline 7-C), 129.8 (quinoline 8-C), 129.5 (quinoline 6-C), 129.2 (quinoline 3-C), 128.5 (quinazoline 8-C), 128.4 (quinoline 4a-C), 128.2 (quinazoline 6-C), 127.9 (quinoline 5-C), 127.0 (quinazoline 5-C), 122.8 (quinazoline 4a-C), 66.6 (morpholine 2-C), 66.3 (morpholine 6-C), 47.3 (morpholine 5-C), 42.6 (morpholine 3-C), HRMS (ESI-TOF) m/z calcd. for C22H19N4O3 ([M + H]+) 387.1452. Found 387.1451.
N-Butyl-2-(4-oxo-3,4-dihydroquinazolin-2-yl)quinoline-3-carboxamide (11). A mixture of Luotonin A (9) (0.057 g, 0.2 mmol), K2CO3 (0.028 g, 0.2 mmol) and n-butylamine (0.146 g, 2 mmol) in DMSO (7 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (30 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure to afford 11 (0.052 g, 70%) as orange crystals, m.p. > 227 °C (sublimation; CH2Cl2). MS (EI, 70 eV) m/z = 373 ([M + 1]+, 2%), 372 (M+, 6), 329 (9), 301 (63), 300 (100), 273 (29), 153 (22), 119 (33), 90 (23); 1H-NMR (CDCl3) δ 11.00 (s, 1H, quinazolinone NH), 8.37 (s, 1H, quinoline 4-H), 8.27 (d, J = 8.2 Hz, 1H, quinazoline 5-H), 8.10 (d, J = 8.4 Hz, 1H, quinoline 8-H), 7.88 (d, J = 8.1 Hz, 1H, quinoline 5-H), 7.86–7.80 (m, 1H, quinoline 7-H), 7.79–7.71 (m, 2H, quinazoline 7-H, 8-H), 7.67 (t, J = 7.5 Hz, 1H, quinoline 6-H), 7.48 (ddd, J = 8.1 Hz, 6.2 Hz, 2.1 Hz, 1H, quinazoline 6-H), 6.50 (t, J = 5.2 Hz, 1H, amide NH), 3.69–3.57 (m, 2H, butyl 1-CH2), 1.80–1.68 (m, 2H, butyl 2-CH2), 1.49 (dq, J = 14.8 Hz, 7.4 Hz, 2H, butyl 3-CH2), 1.00 (t, J = 7.4 Hz, 3H, CH3); 13C-NMR (CDCl3) δ: 168.6 (amide C=O), 161.5 (quinazoline 4-C), 148.5 (quinazoline 8a-C), 148.1 (quinazoline 2-C), 146.4 (quinoline 8a-C), 144.5 (quinoline 2-C), 137.9 (quinoline 4-C), 134.7 (quinazoline 7-C), 131.6 (quinoline 7-C), 130.6 (quinoline 3-C), 129.6 (quinoline 8-C), 129.3 (quinoline 6-C), 128.4 (quinazoline 8-C), 128.1 (quinoline 4a-C), 128.0 (quinazoline 6-C), 127.9 (quinoline 5-C), 126.8 (quinazoline 5-C), 122.6 (quinazoline 4a-C), 40.6 (butyl 1-C), 31.6 (butyl 2-C), 20.5 (butyl 3-C), 14.0 (butyl 4-C). HRMS (ESI-TOF) m/z calcd. for C22H20N4NaO2 ([M + Na]+) 395.1478. Found 395.1476.
N-[2-(Dimethylamino)ethyl]-2-(4-oxo-3,4-dihydroquinazolin-2-yl)quinoline-3-carboxamide (12). A mixture of Luotonin A (9) (0.100 g, 0.35 mmol), K2CO3 (0.048 g, 0.35 mmol) and N,N-dimethylethylenediamine (0.309 g, 3.5 mmol) in DMSO (10 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, dried over Na2SO4 and evaporated under reduced pressure to afford 12 as crude product (0.070 g, 52%). The residue was subjected to MPLC, eluting with CH2Cl2/methanol (80 + 20) to afford 12 as colorless crystals, m.p. 185–188 °C (CH2Cl2). MS (EI, 70 eV) m/z = 197 (5%), 155 (18), 137 (46), 110 (34), 108 (12), 83 (14), 82 (13), 57 (13), 43 (100); 1H-NMR (CDCl3) δ 11.15 (br s, 1H, quinazolinone NH), 8.37 (s, 1H, quinoline 4-H), 8.35 (ddd, J = 8.0 Hz, 1.2 Hz, 0.6 Hz, 1H, quinazoline 5-H), 8.15 (d, J = 8.4 Hz, 1H, quinoline 8-H), 7.88 (d, J = 8.1 Hz, 1H, quinoline 5-H), 7.84 (ddd, J = 8.4 Hz, 6.9 Hz, 1.4 Hz, 1H, quinoline 7-H), 7.80–7.73 (m, 2H, quinazoline 7-H, 8-H), 7.67 (ddd, J = 8.1 Hz, 7.0 Hz, 1.1 Hz, 1H, quinoline 6-H), 7.52 (ddd, J = 8.2 Hz, 6.3 Hz, 2.0 Hz, 1H, quinazoline 6-H), 6.81 (br s, 1H, amide NH), 3.72 (q, J = 5.7 Hz, 2H, 1-CH2), 2.62 (t, J = 5.7 Hz, 2H, 2-CH2), 2.19 (s, 6H, CH3); 13C-NMR (CDCl3) δ: 168.8 (amide C=O), 161.5 (quinazoline 4-C), 148.6 (quinazoline 8a-C), 148.2 (quinazoline 2-C), 146.5 (quinoline 8a-C), 144.7 (quinoline 2-C), 137.7 (quinoline 4-C), 134.7 (quinazoline 7-C), 131.6 (quinoline 7-C), 130.5 (quinoline 3-C), 129.6 (quinoline 8-C), 129.2 (quinoline 6-C), 128.4 (quinazoline 8-C), 128.1 (quinaoline 4a-C), 128.0 (quinoline 5-C or quinazoline 6-C), 127.9 (quinazoline 6-C or quinoline 5-C), 126.9 (quinazoline 5-C), 122.7 (quinazoline 4a-C), 57.5 (2-CH2), 45.1 (CH3), 37.9 (1-C7H2). HRMS (ESI-TOF) m/z calcd. for C22H22N5O2 ([M + H]+): 388.1768. Found: 388.1772.
2-[5,7-Dimethoxy-3-(morpholin-4-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (14). A mixture of 1,3-dimethoxyquinolino[2′,3′:3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (13) (0.172 g, 0.5 mmol), K2CO3 (0.069 g, 0.5 mmol) and morpholine (0.436 g, 5 mmol) in DMSO (15 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/methanol (95 + 5) to afford 14 (0.060 g, 27%) as yellowish crystals, m.p. > 264 °C (sublimation; CH2Cl2). MS (EI, 70 eV) m/z = 447 ([M + 1]+, 2%), 446 (M+, 8) 361 (78), 360 (100), 333 (42), 302 (22), 119 (26), 90 (32), 86 (25), 57 (30), 56 (57), 55 (27); 1H-NMR (CDCl3) δ 11.14 (s, 1H, NH), 8.50 (d, J = 0.5 Hz, 1H, quinoline 4-H), 8.37 (dd, J = 8.0 Hz, 1.1 Hz, 1H, quinazoline 5-H), 7.80 (ddd, J = 8.3 Hz, 6.8 Hz, 1.5 Hz, 1H, quinazoline 7-H), 7.76 (dd, J = 8.1 Hz, 1.0 Hz, 1H, quinazoline 8-H), 7.55 (ddd, J = 8.2 Hz, 6.9 Hz, 1.5 Hz, 1H, quinazoline 6-H), 7.06 (d, J = 1.6 Hz, 1H, quinoline 8-H), 6.63 (d, J = 2.1 Hz, 1H, quinoline 6-H), 4.29–4.24 (m, 1H, morpholine 3-H), 4.11–4.06 (m, 1H, morpholine 2-H), 4.02 (s, 3H, 7-OCH3), 4.01 (s, 3H, 5-OCH3), 3.84–3.77 (m, 1H, morpholine 2′-H), 3.73–3.66 (m, 2H, morpholine 3′-H, 6-H), 3.59–3.52 (m, 1H, morpholine 6′-H), 3.31–3.23 (m, 2H, morpholine 5-H, 5′-H); 13C-NMR (CDCl3) δ: 169.7 (amide C=O), 163.1 (quinoline 7-C), 161.4 (quinazoline 4-C), 156.1 (quinoline 5-C), 148.9 (quinoline 8a-C), 148.7 (quinazoline 8a-C), 148.3 (quinazoline 2-C), 144.6 (quinoline 2-C), 134.8 (quinazoline 7-C), 131.7 (quinoline 4-C), 128.5 (quinazoline 8-C), 128.0 (quinazoline 6-C), 127.0 (quinazoline 5-C), 126.0 (quinoline 3-C), 122.8 (quinazoline 4a-C), 117.4 (quinoline 4a-C), 100.7 (quinoline 6-C), 99.5 (quinoline 8-C), 66.7 (morpholine 2-C), 66.4 (morpholine 6-C), 56.2 (7-OCH3), 56.0 (5-OCH3), 47.3 (morpholine 5-C), 42.6 (morpholine 3-C). HRMS (ESI-TOF) m/z calcd. for C24H23N4O5 ([M + H]+) 447.1663. Found 447.1662.
3.3. Biological Evaluation
Human colon adenocarcinoma cells (SW480) were seeded in a 96-well plate at a density of 5 × 104 cells per mL. Minimum Essential Medium Eagle (MEM) with 10% FCS (Fetal Calf Serum) was used to conduct the culture in the first 24 h at 37 °C in the presence of 5% CO2. Compounds were then added in defined concentrations to the cultivated cells using DMSO as solvent and diluting this solution with serum-free MEM-BSA (Minimum Essential Medium Eagle, Bovine Serum Albumin). Treated cells were incubated for further 72 h at 37 °C and an atmosphere containing 5% CO2. After this incubation period, the MTT viability assay [32] was performed. This test measures cell viability using the redox potential of living cells. Functional cells reduce a yellow-colored tetrazolium salt to a red formazan derivative, which can be then determined by optical density. After 3 h of incubation, the absorbance of formazan was measured at 450 nm with 620 nm as a reference wavelength.
4. Conclusions
In summary, we have discovered an unprecedented oxidative ring opening reaction of the central ring C in Luotonin A and two of its derivatives simply on heating with an excess amine and potassium carbonate in DMSO solution in the presence of air oxygen. The amide-type reaction products were fully characterized and all 1H- and 13C-NMR signals have been assigned by a combination of HSQC, HMBC, COSY, and NOESY experiments. Four of the new compounds show moderate in vitro anticancer activity towards human colon adenocarcinoma cells.
Supplementary Materials
Spectra (MS, 1H-NMR, 13C-NMR) of all new compounds are available online.
Acknowledgments
We are grateful to Martin Zehl, Judith Wackerlig, and Daniel Dobusch for recording the high-resolution mass spectra.
Author Contributions
A.I. planned and performed synthetic work and biological testing; K.D. performed synthetic work; B.M. supervised the biological testing and helped to design and interpret in vitro experiments; N.H. designed the synthetic part, recorded and interpreted the NMR spectra and wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Ma, Z.-Z.; Hano, Y.; Nomura, T.; Chen, Y.-J. Two new pyrroloquinazolinoquinoline alkaloids from Peganum nigellastrum. Heterocycles 1997, 46, 541–546. [Google Scholar] [CrossRef]
- Cagir, A.; Eisenhauer, B.M.; Gao, R.; Thomas, S.J.; Hecht, S.M. Synthesis and topoisomerase I properties of Luotonin A analogues. Bioorganic Med. Chem. 2004, 12, 6287–6299. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Merlini, L.; Beretta, G.L.; Tinelli, S.; Zunino, F. Synthesis and cytotoxic activity of substituted Luotonin A derivatives. Bioorganic Med. Chem. Lett. 2004, 14, 5757–5761. [Google Scholar] [CrossRef] [PubMed]
- Tangirala, R.; Antony, S.; Agama, K.; Pommier, Y.; Curran, D.P. Total synthesis of Luotonin and a small library of AB-ring substituted analogues by cascade radical annulation of isonitriles. Synlett 2005, 2843–2846. [Google Scholar] [CrossRef]
- Mason, J.J.; Bergman, J. Total synthesis of Luotonin A and 14-substituted analogues. Org. Biomol. Chem. 2007, 5, 2486–2490. [Google Scholar] [CrossRef] [PubMed]
- Nacro, K.; Zha, C.; Guzzo, P.R.; Herr, R.J.; Peace, D.; Friedrich, T.D. Synthesis and topoisomerase poisoning activity of A-ring and E-ring substituted Luotonin A derivatives. Bioorg. Med. Chem. 2007, 15, 4237–4246. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.F.M.M.; Kim, D.-H.; Liang, J.-L.; Lee, E.-S.; Na, Y.-H.; Jun, K.-Y.; Kwon, Y.-J.; Jahng, Y.-D. Synthesis and biological properties of Luotonin A derivatives. Bull. Korean Chem. Soc. 2008, 29, 1988–1992. [Google Scholar] [CrossRef]
- Ju, Y.; Liu, F.; Li, C. Palladium-catalyzed sequential cyanation/N-addition/N-arylation in one-pot: Efficient synthesis of Luotonin A and its derivatives. Org. Lett. 2009, 11, 3582–3585. [Google Scholar] [CrossRef] [PubMed]
- Golubev, A.S.; Bogomolov, V.O.; Shidlovskii, A.F.; Dezhenkova, L.G.; Peregudov, A.S.; Shtil, A.A.; Chkanikov, N.D. Synthesis of fluoromethyl-containing analogs of antitumor alkaloid Luotonin A. Russ. Chem. Bull. 2010, 59, 209–218. [Google Scholar] [CrossRef]
- Boisse, T.; Gavara, L.; Gautret, P.; Baldeyrou, B.; Lansiaux, A.; Goossens, J.-F.; Hénichart, J.-P.; Rigo, B. Toward new Camptothecins. Part 7: Synthesis of Thioluotonin and its 5-methoxycarbonyl derivative. Tetrahedron Lett. 2011, 52, 1592–1596. [Google Scholar] [CrossRef]
- Tseng, M.-C.; Chu, Y.-W.; Tsai, H.-P.; Lin, C.-M.; Hwang, J.; Chu, Y.-H. One-pot synthesis of Luotonin A and its analogues. Org. Lett. 2011, 13, 920–923. [Google Scholar] [CrossRef] [PubMed]
- González-Ruiz, V.; Pascua, I.; Fernández-Marcelo, T.; Ribelles, P.; Bianchini, G.; Sridharan, V.; Iniesta, P.; Ramos, M.T.; Olives, A.I.; Martín, M.A.; et al. B-ring-aryl substituted Luotonin A analogues with a new binding mode to the topoisomerase 1-DNA complex show enhanced cytotoxic activity. PLoS ONE 2014, 9, e95998. [Google Scholar] [CrossRef] [PubMed]
- Du, W. Towards new anticancer drugs: A decade of advances in synthesis of Camptothecins and related alkaloids. Tetrahedron 2003, 59, 8649–8687. [Google Scholar] [CrossRef]
- Pizzolato, J.F.; Saltz, L.B. The Camptothecins. Lancet 2003, 361, 2235–2242. [Google Scholar] [CrossRef]
- Thomas, C.J.; Rahier, N.J.; Hecht, S.M. Camptothecin: Current perspectives. Bioorg. Med. Chem. 2004, 12, 1585–1604. [Google Scholar] [CrossRef] [PubMed]
- Cagir, A.; Jones, S.H.; Gao, R.; Eisenhauer, B.M.; Hecht, S.M. Luotonin A. A naturally occurring human DNA topoisomerase I poison. J. Am. Chem. Soc. 2003, 125, 13628–13629. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.L.; Cha, H.C.; Jahng, Y. Recent advances in the studies on Luotonins. Molecules 2011, 16, 4861–4883. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.; Nuß, S. Weinreb amidation as the cornerstone of an improved synthetic route to A-ring-modified derivatives of Luotonin A. Molecules 2012, 17, 11363–11378. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.; Meng, G.; Roger, S.; Wank, S. An efficient and selective access to 1-substituted and 3-substituted derivatives of Luotonin A. Tetrahedron 2013, 69, 7066–7072. [Google Scholar] [CrossRef]
- Atia, M.; Bogdán, D.; Brügger, M.; Haider, N.; Mátyus, P. Remarkable regioselectivities in the course of the synthesis of two new Luotonin A derivatives. Tetrahedron 2017, 73, 3231–3239. [Google Scholar] [CrossRef]
- Zhou, H.-B.; Liu, G.-S.; Yao, Z.-J. Short and efficient total synthesis of Luotonin A and 22-hydroxyacuminatine using a common cascade strategy. J. Org. Chem. 2007, 72, 6270–6272. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.N.; Stemp, G.; Thompson, M.; Witty, D.R. 3-(Arylsulfonyl)quinolines as 5-HT6 Receptor Antagonists for the Treatment of CNS Disorders. Patent WO 2005113539 A1, 1 December 2005. [Google Scholar]
- Ancliff, R.A.; Hancock, A.P.; Kranz, M.J.; Parr, N.J. Histamine receptor antagonis comprising an azepin corets. Patent WO 2007135081 A1, 29 November 2007. [Google Scholar]
- Safina, L.Y.; Selivanova, G.A.; Bagryanskaya, I.Y.; Shteingarts, V.D. Reaction of quinolines fluorinated at the benzene ring with nitrogen-centered nucleophiles. Russ. Chem. Bull. 2009, 58, 1049–1061. [Google Scholar] [CrossRef]
- Childers, W.E.; Havran, L.M.; Asselin, M.; Bicksler, J.J.; Chong, D.C.; Grosu, G.T.; Shen, Z.; Abou-Gharbia, M.A.; Bach, A.C.; Harrison, B.L.; et al. The synthesis and biological evaluation of quinolyl-piperazinyl piperidines as potent serotonin 5-HT1A antagonists. J. Med. Chem. 2010, 53, 4066–4084. [Google Scholar] [CrossRef] [PubMed]
- Gurskaya, L.Y.; Selivanova, G.A.; Shteingarts, V.D. Interaction of quinolines polyfluorinated on the benzene moiety with sodium and potassium amides in liquid ammonia. J. Fluor. Chem. 2012, 136, 32–37. [Google Scholar] [CrossRef]
- Skolyapova, A.D.; Selivanova, G.A.; Tretyakov, E.V.; Bogdanova, T.F.; Shchegoleva, L.N.; Bagryanskaya, I.Y.; Gurskaya, L.Y.; Shteingarts, V.D. Interaction of polyfluorinated 2-chloroquinolines with ammonia. Tetrahedron 2017, 73, 1219–1229. [Google Scholar] [CrossRef]
- Baker, B.R.; Almaula, P.I. Nonclassical antimetabolites. X.1,2 A facile synthesis of 4-quinazolone-2-carboxylic acid and the structure of Bogert’s ammonium salt. J. Org. Chem. 1962, 27, 4672–4674. [Google Scholar] [CrossRef]
- Lipton, M.F.; Basha, A.; Weinreb, S.M. Conversion of esters to amides with dimethylaluminum amides: N,N-dimethylcyclohexanecarboxamide. Org. Synth. 1979, 59, 49–53. [Google Scholar] [CrossRef]
- Hendrickson, J.B.; Singer, M.; Hussoin, M. Direct borohydride reduction of alcohols to alkanes with phosphonium anhydride activation. J. Org. Chem. 1993, 58, 6913–6914. [Google Scholar] [CrossRef]
- Another control experiment was performed, starting from the fluoro compound 4 and pyrrolidine under the conditions described for 5, but under argon atmosphere. In this case, slow consumption of 4 was observed by TLC, together with some decomposition and the formation of a red-fluorescent product that is less polar than the ring-opened compound 5. A sample of the reaction product was isolated and identified as 4-(pyrrolidin-1-yl)quinolino[2′,3′:3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one: orange-red solid, m.p. 264–265 °C (CH2Cl2/ethyl acetate). 1H-NMR (CDCl3) δ: 8.41 (dd, J = 8.0 Hz, 1.3 Hz, 1H, 10-H), 8.20 (s, 1H, 14-H), 7.99 (d, J = 8.0 Hz, 1H, 7-H), 7.81 (ddd, J = 8.4 Hz, 7.2 Hz, 1.5 Hz, 1H, 8-H), 7.53 (ddd, J = 8.1 Hz, 6.9 Hz, 1.0 Hz, 1H, 9-H), 7.45 (t, J = 7.8 Hz, 1H, 2-H), 7.13 (d, J = 7.8 Hz, 1H, 1-H), 6.78 (d, J = 7.8 Hz, 1H, 3-H), 5.22 (s, 2H, 13-CH2), 4.02–3.86 (m, 4H, pyrrolidine 2′-CH2, 5′-CH2), 2.19–2.01 (m, 4H, pyrrolidine 3′-CH2, 4′-CH2); 13C-NMR (CDCl3) δ: 161.0 (11-C), 153.3 (5b-C), 149.8 (6a-C), 147.9 (4-C), 146.3 (5a-C), 142.1 (4a-C), 134.4 (8-C), 130.8 (14a-C), 130.6 (14-C), 129.7 (2-C), 129.1 (13a-C), 128.8 (7-C), 127.0 (9-C), 126.5 (10-C), 121.3 (10a-C), 114.7 (1-C), 110.3 (3-C), 52.4 (pyrrolidine 2′-C, 5′-C), 47.3 (13-C), 26.0 (pyrrolidine 3′-C, 4′-C); HRMS (ESI-TOF) m/z calcd. for C22H19N4O ([M + H]+): 355.1553. Found: 355.1556.
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
Figure 1.
Structures of Luotonin A and Camptothecin (CPT).

Scheme 1.
Synthesis of the 4-fluoro derivative (4) of Luotonin A.

Scheme 2.
Reaction of the fluoro compound 4 with pyrrolidine: substitution and ring C opening.

Scheme 3.
Ring opening reactions of compounds 4, 9 and 13 with various amines.

Scheme 4.
Proposed reaction mechanism for the oxidative/nucleophilic opening of ring C (NuH = primary or secondary amine).
Scheme 4.
Proposed reaction mechanism for the oxidative/nucleophilic opening of ring C (NuH = primary or secondary amine).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
In vitro cell growth inhibition (%) of compounds 5–8, 10–12 and 14 towards SW480 cancer cells at a fixed concentration of 40 μM (MTT viability assay [32]).
Table 1.
In vitro cell growth inhibition (%) of compounds 5–8, 10–12 and 14 towards SW480 cancer cells at a fixed concentration of 40 μM (MTT viability assay [32]).
| Compound | Luotonin A | 5 | 6 | 7 | 8 | 10 | 11 | 12 | 14 |
|---|---|---|---|---|---|---|---|---|---|
| % Inhibition | 19 ± 3 | 47 ± 3 | 54 ± 10 | 52 ± 7 | no inhib. | 22 ± 1 | 5 ± 4 | 3 ± 0 | 7 ± 9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ibric, A.; Dutter, K.; Marian, B.; Haider, N. A Facile Oxidative Opening of the C-Ring in Luotonin A and Derivatives. Molecules 2017, 22, 1540. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22091540
AMA Style
Ibric A, Dutter K, Marian B, Haider N. A Facile Oxidative Opening of the C-Ring in Luotonin A and Derivatives. Molecules. 2017; 22(9):1540. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22091540
Chicago/Turabian StyleIbric, Amra, Kathrin Dutter, Brigitte Marian, and Norbert Haider. 2017. "A Facile Oxidative Opening of the C-Ring in Luotonin A and Derivatives" Molecules 22, no. 9: 1540. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22091540