On the Tautomerism of N-Substituted Pyrazolones: 1,2-Dihydro-3H-pyrazol-3-ones versus 1H-Pyrazol-3-ols

 , , ,
, , ,
Abstract
:
1. Introduction
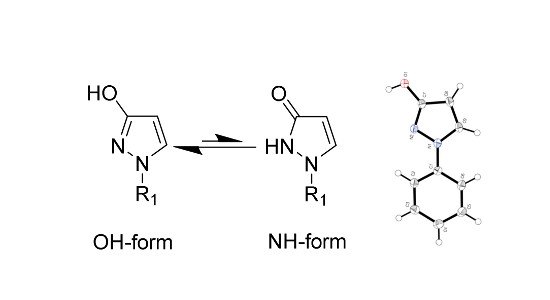
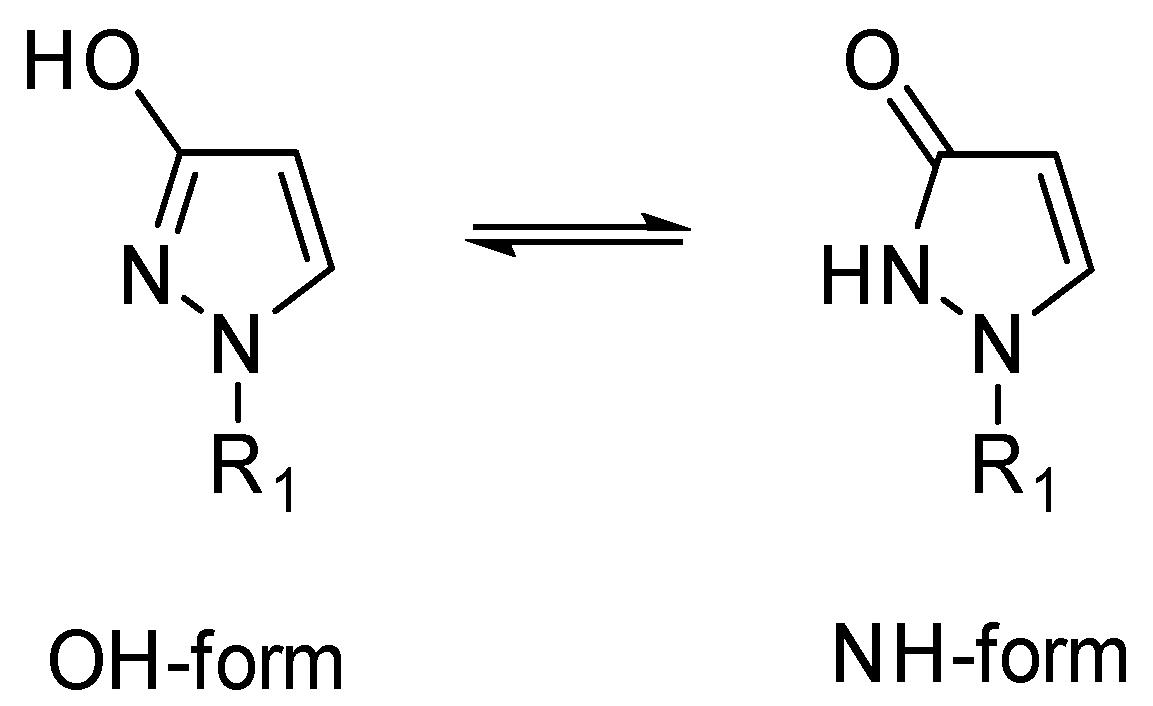
2. Results and Discussion
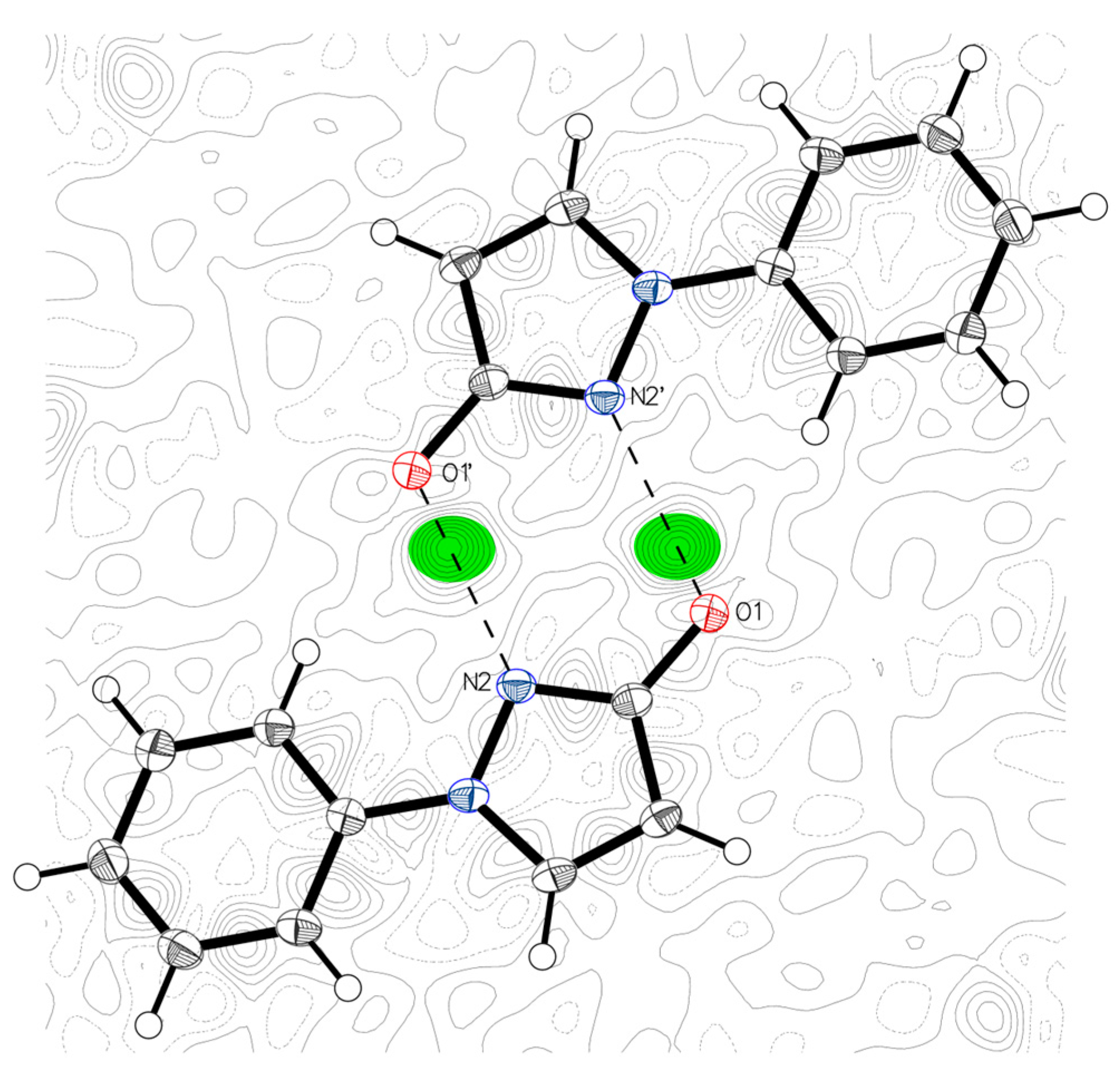
2.1. X-ray Analysis of 1-Phenyl-1,2-dihydro-3H-pyrazol-3-one (1-Phenyl-1H-pyrazol-3-ol) (1)
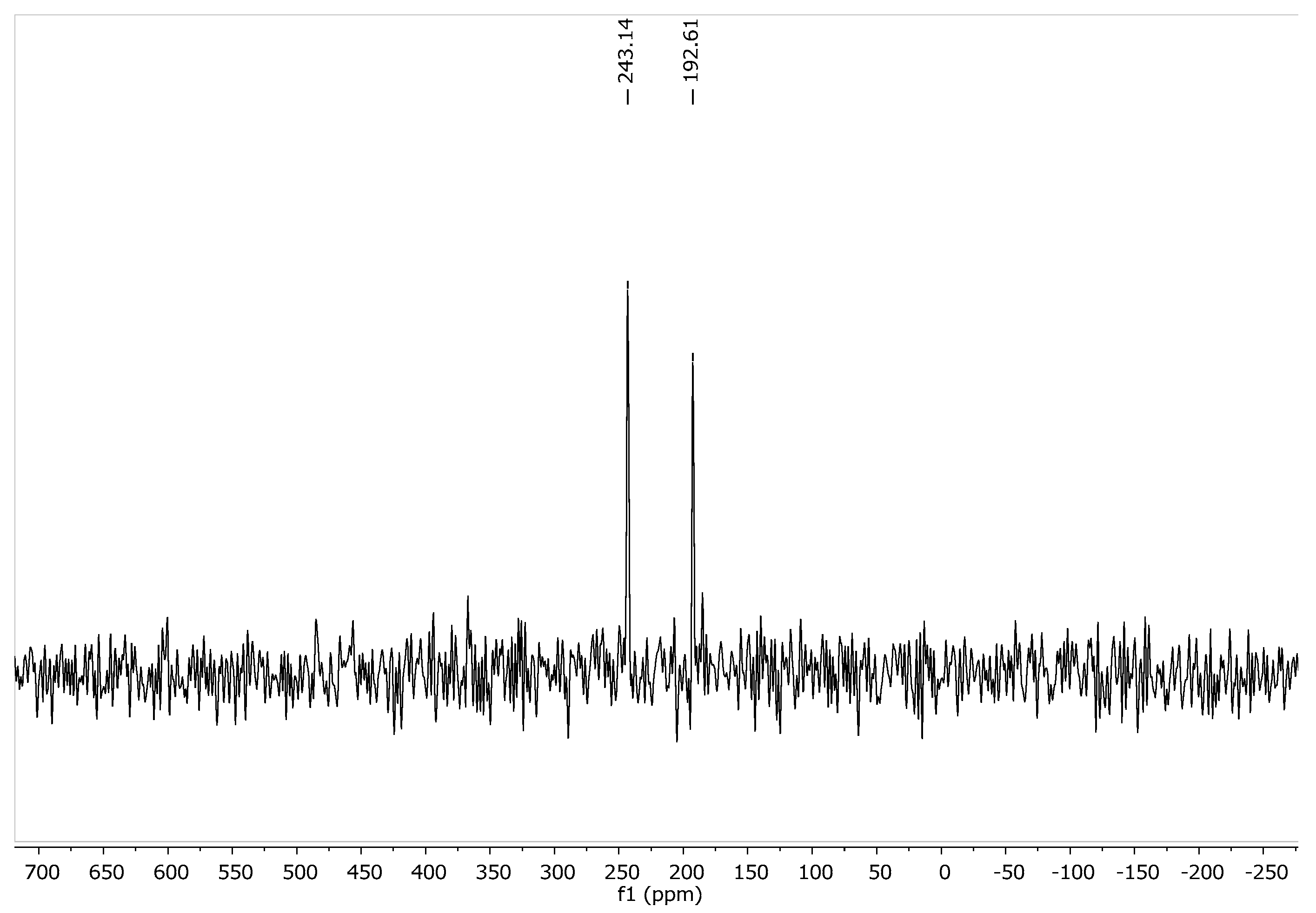
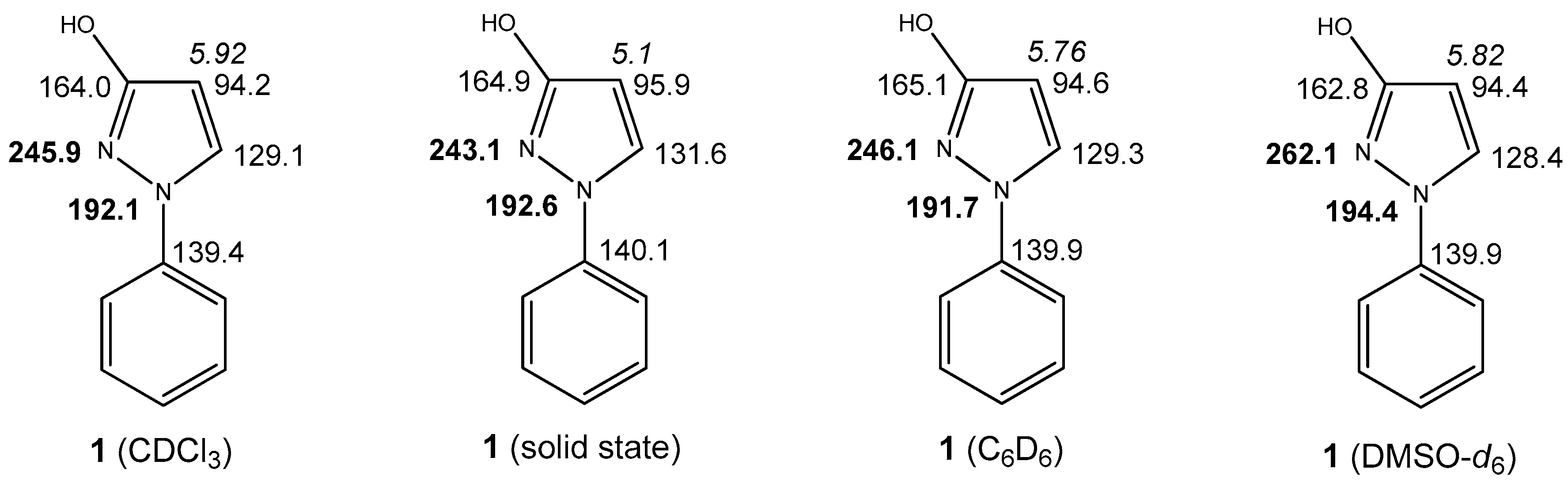
2.2. Solid State NMR (SSNMR) of 1
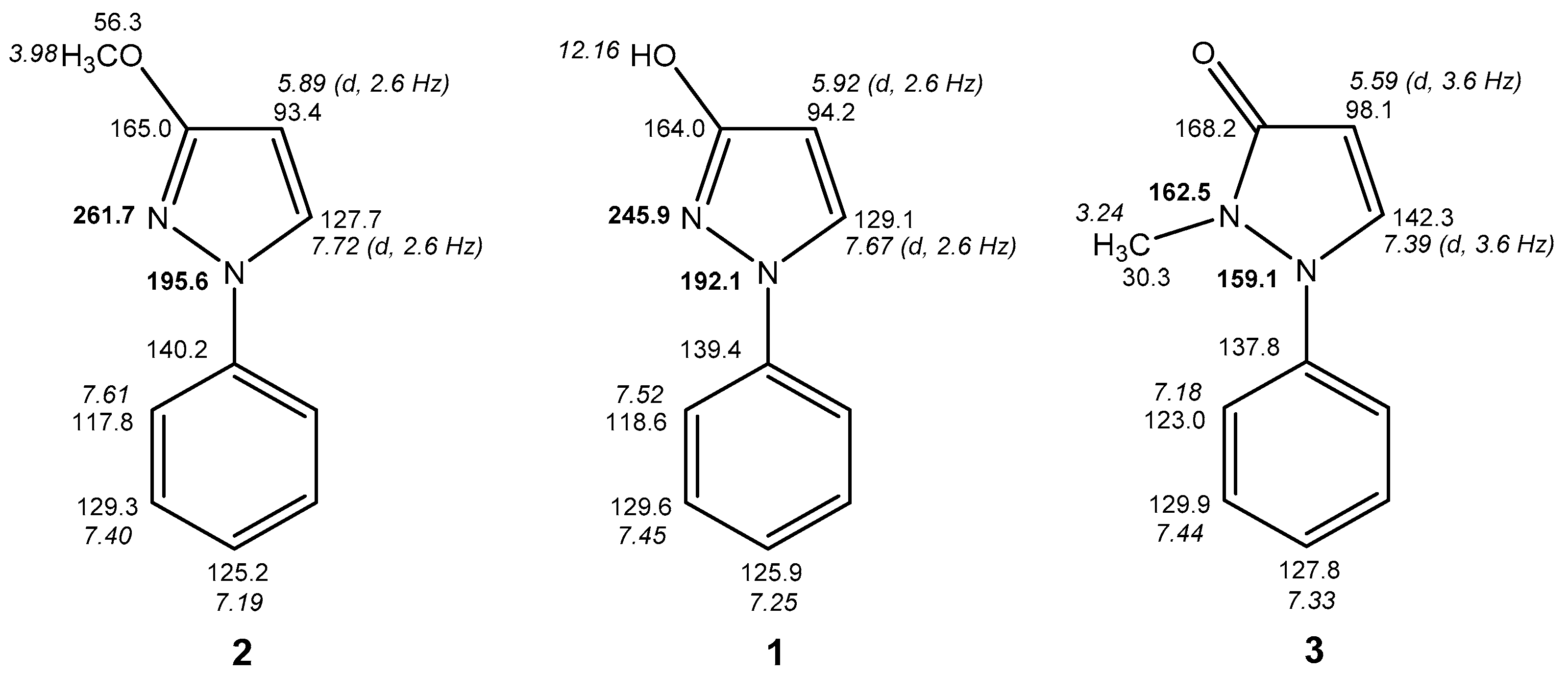
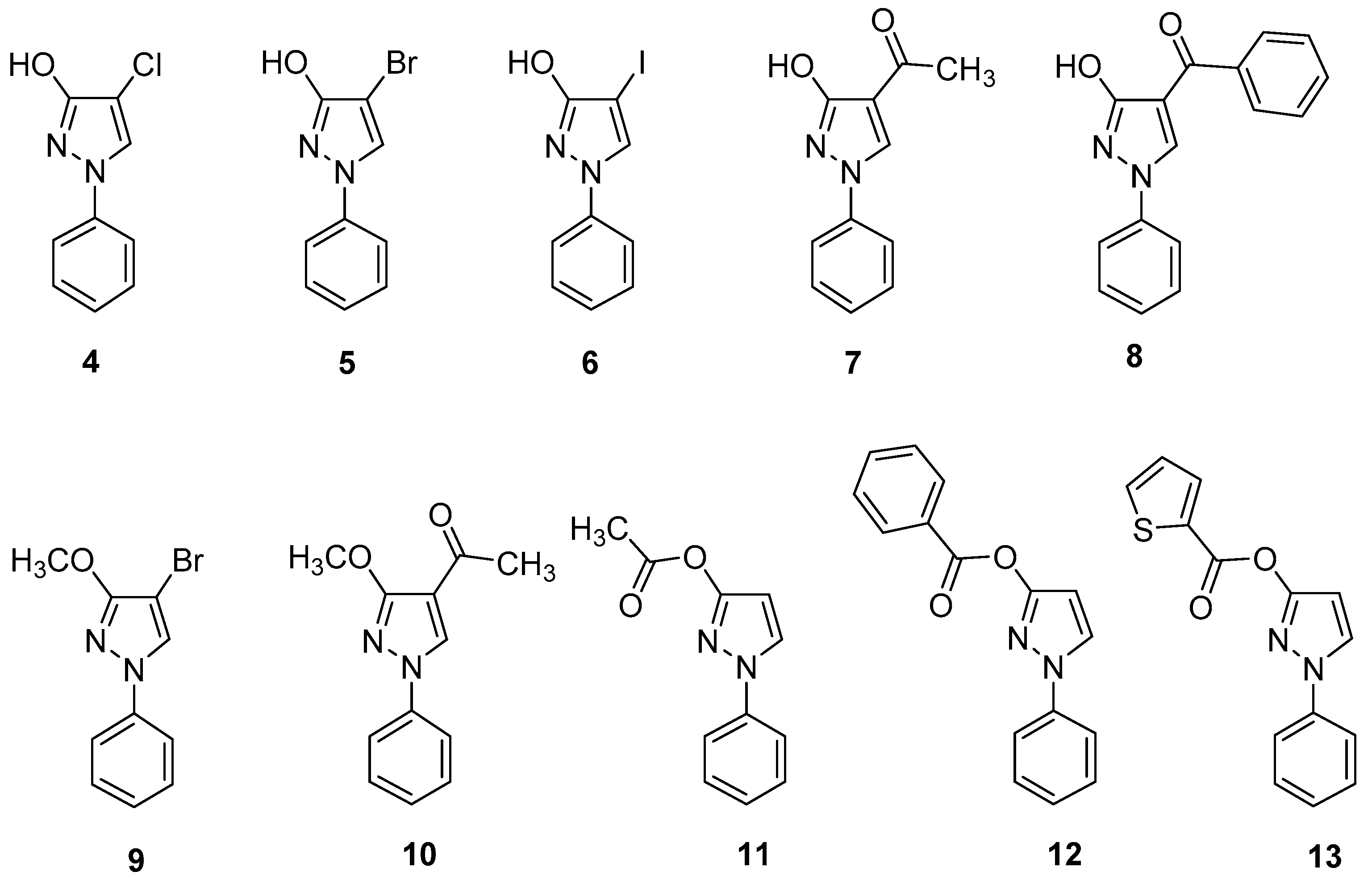
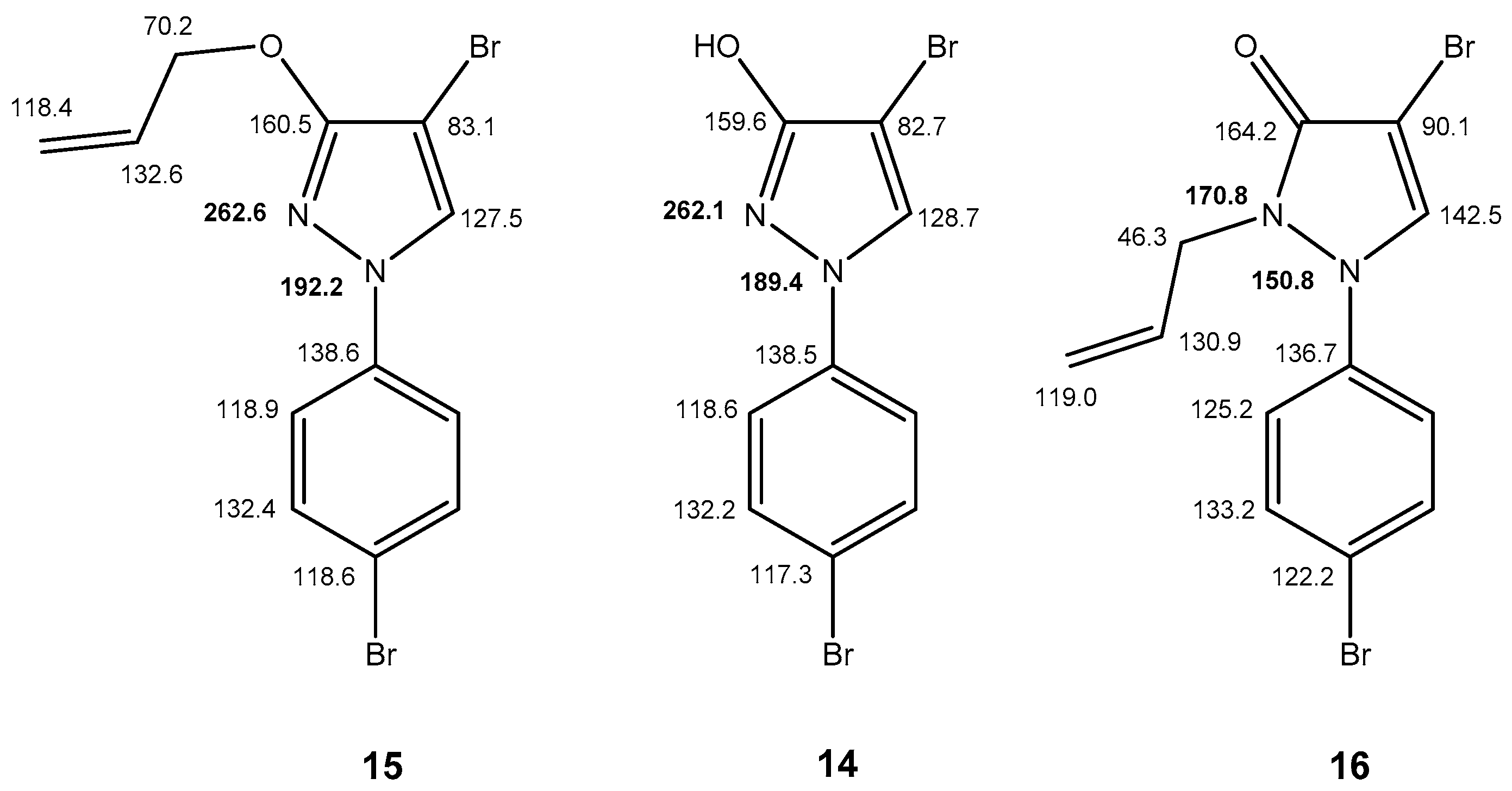
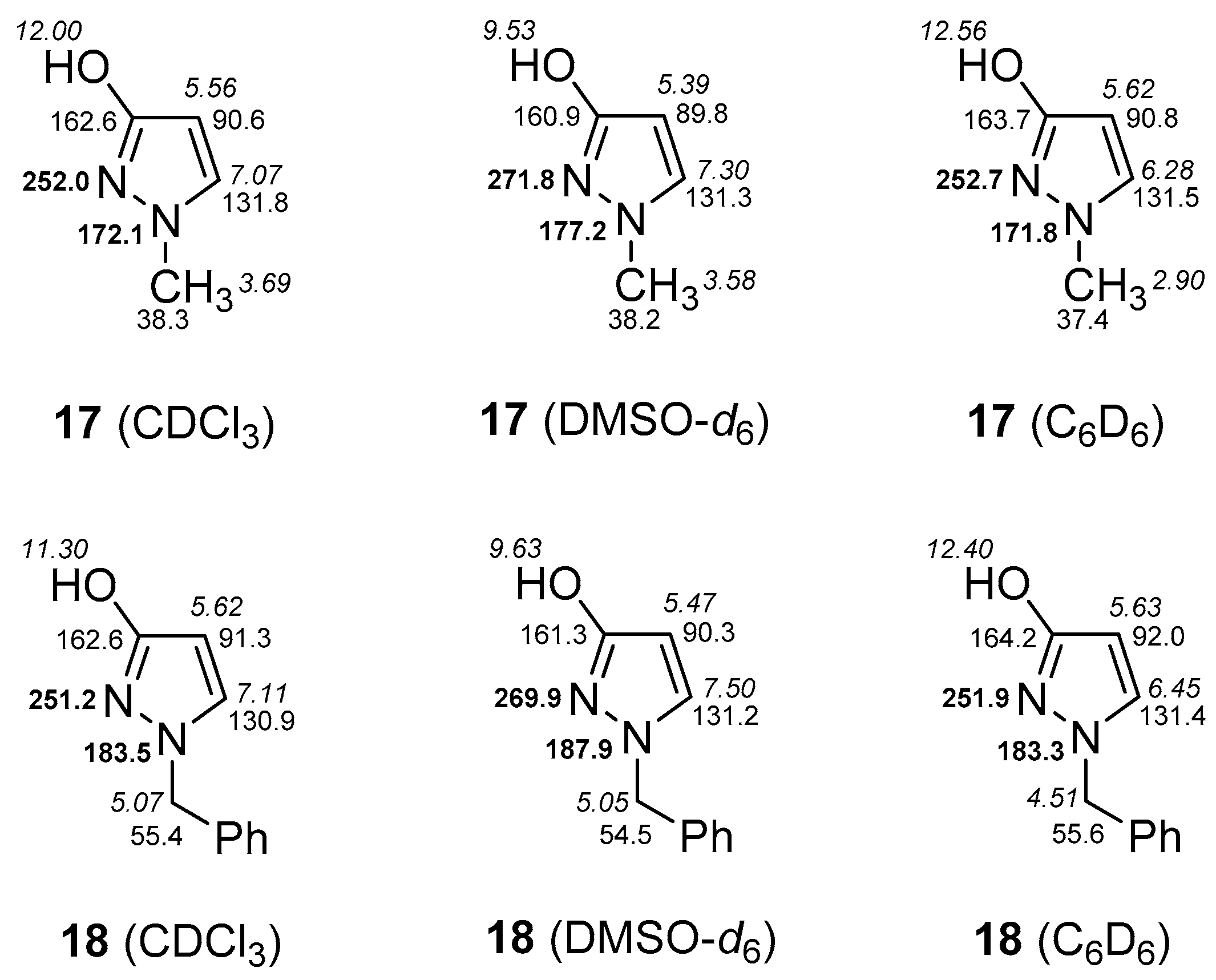
2.3. NMR Spectra in Solution
3. Experimental Section
3.1. General Information
3.2. Data of Investigated Compounds
3.3. X-ray Crystal Structure Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Elguero, J.; Goya, P.; Jagerovic, N.; Silva, A.M.S. Pyrazoles as drugs: Facts and fantasies. In Targets in Heterocyclic Systems; Attanasi, O.A., Spinelli, D., Eds.; Royal Society of Chemistry: Cambridge, UK, 2002; Volume 6, pp. 52–98. [Google Scholar]
- Perez-Fernandez, R.; Goya, P.; Elguero, J. A review of recent progress (2002–2012) on the biological activities of pyrazoles. ARKIVOC 2014, ii, 233–293. [Google Scholar]
- Schmidt, A.; Dreger, A. Recent advances in the chemistry of pyrazoles. Properties, biological activities, and syntheses. Curr. Org. Synth. 2011, 15, 1423–1463. [Google Scholar] [CrossRef]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to Mid-2010: A fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Lamberth, C. Pyrazole Chemistry in Crop Protection. Heterocycles 2007, 71, 1467–1502. [Google Scholar] [CrossRef]
- Holzer, W.; Claramunt, R.M.; Lopez, C.; Alkorta, I.; Elguero, J. A study in desmotropy. Solid State Nucl. Magn. Reson. 2008, 34, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Elguero, J.; Marzin, C.; Katritzky, A.R.; Linda, P. The Tautomerism of Heterocycles; Academic Press: New York, NY, USA, 1976; ISBN 012020651X. [Google Scholar]
- Minkin, V.I.; Garnovskii, A.D.; Elguero, J.; Katritzky, A.R.; Denisko, O.V. The tautomerism of heterocycles: Five-membered rings with two or more heteroatoms. Adv. Heterocycl. Chem. 2000, 76, 157–323. [Google Scholar]
- Holzer, W.; Mereiter, K.; Plagens, B. 4-Acyl-5-methyl-2-phenylpyrazolones: NMR and X-ray structure investigations. Heterocycles 1999, 50, 799–818. [Google Scholar] [CrossRef]
- Holzer, W.; Hallak, L. Synthesis and NMR spectroscopic investigations with 3-amino-, 3-hydroxy-, and 3-methoxy-4-acyl-1-phenyl-2-pyrazolin-5-ones. Heterocycles 2004, 63, 1311–1334. [Google Scholar] [CrossRef]
- Holzer, W.; Kautsch, C.; Laggner, C.; Claramunt, R.M.; Pérez-Torralba, M.; Alkorta, I.; Elguero, J. On the Tautomerism of Pyrazolones: The Geminal 2J[Pyrazole C-4,H-3(5)] Spin Coupling Constant as a Diagnostic Tool. Tetrahedron 2004, 60, 6791–6805. [Google Scholar] [CrossRef]
- Guillou, S.; Janin, Y.L. 5-Iodo-3-Ethoxypyrazoles: An Entry Point to New Chemical Entities. Chem. Eur. J. 2010, 16, 4669–4677. [Google Scholar] [CrossRef] [PubMed]
- Arbačiauskienė, E.; Laukaitytė, V.; Holzer, W.; Šačkus, A. Metal-free intramolecular alkyne-azide cycloaddition leading to the novel pyrazolo[4,3-f][1,2,3]triazolo[5,1-c][1,4]oxazepine ring system. Eur. J. Org. Chem. 2015, 5663–5670. [Google Scholar]
- Li, Y.; Liu, R.; Yan, Z.; Zhang, X.; Zhu, H. Synthesis, Crystal Structure and Fungicidal Activities of New Type Oxazolidinone-Based Strobilurin Analogues. Bull. Korean Chem. Soc. 2010, 31, 3341–3347. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.-Y.; Chen, N.-Q.; Lue, K.-Z.; Xiong, X.-H.; Li, J. One-Pot Regioselective Synthesis of Novel Oximino Ester-Containing 1-Aryl-4-chloro-3-oxypyrazoles as Potential Fungicides. Helv. Chim. Acta 2014, 97, 1269–1282. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Chen, N.; Lv, K.; Zhou, C.; Xiong, X.; Li, F. Synthesis and Fungicidal Activity of Novel Chloro-Containing 1-Aryl-3-oxypyrazoles with an Oximino Ester or Oximino Amide Moiety. Molecules 2014, 19, 8140–8150. [Google Scholar] [CrossRef] [PubMed]
- Elguero, J.; Katritzky, A.R.; Denisko, O.V. Prototropic Tautomerism of Heterocycles: Heteroaromatic Tautomerism—General Overview and Methodology. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2000; Volume 76, pp. 1–84. [Google Scholar]
- Sanz, D.; Claramunt, R.M.; Alkorta, I.; Elguero, J. The use of chemical shifts vs. coupling constants for studying tautomerism: A combined experimental and theoretical approach. Struct. Chem. 2007, 18, 703–708. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Karelson, M.; Harris, P.A. Prototropic tautomerism of heteroaromatic compounds. Heterocycles 1991, 32, 329–369. [Google Scholar] [CrossRef]
- Von Philipsborn, W.; Müller, R. 15N-NMR Spectroscopy-New Methods and Application. Angew. Chem. Int. Ed. Engl. 1986, 25, 383–413. [Google Scholar] [CrossRef]
- Gil, V.M.S.; von Philipsborn, W. Effect of electron lone-pairs on nuclear spin-spin coupling constants. Magn. Reson. Chem. 1989, 27, 409–430. [Google Scholar] [CrossRef]
- Berger, S.; Braun, S.; Kalinowski, H.-O. NMR-Spektroskopie von Nichtmetallen: 15N-NMR-Spektroskopie; Thieme: Stuttgart, Germany; New York, NY, USA, 1992; Volume 2, pp. 42–43. ISBN 3-13-769101-X. [Google Scholar]
- Begtrup, M. Hydrogen-1 and carbon-13-NMR spectra of phenyl-substituted azole derivatives. II. Conformational study. Acta Chem. Scand. B 1974, 28, 61–77. [Google Scholar] [CrossRef]
- Carrillo, J.R.; Cossio, F.P.; Diaz-Ortiz, A.; Gomez-Escalonilla, M.J.; de la Hoz, A.; Lecea, B.; Moreno, A.; Prieto, P. A complete model for the prediction of 1H- and 13C-NMR chemical shifts and torsional angles in phenyl-substituted pyrazoles. Tetrahedron 2001, 57, 4179–4187. [Google Scholar] [CrossRef]
- Kleizienė, N.; Arbačiauskienė, E.; Holzer, W.; Šačkus, A. 4-Bromo-3-methoxy-1-phenyl-1H-pyrazole. Molbank 2009, M639. [Google Scholar] [CrossRef]
- Kalinowski, H.-O.; Berger, S.; Braun, S. 13C-NMR Spektroskopie; Thieme: Stuttgart, Germany; New York, NY, USA, 1984; p. 194. [Google Scholar]
- Arbačiauskienė, E.; Vilkauskaite, G.; Eller, G.A.; Holzer, W.; Sackus, A. Pd-Catalyzed Cross-Coupling Reactions of Halogenated 1-Phenylpyrazol-3-ols and Related Triflates. Tetrahedron 2009, 65, 7817–7824. [Google Scholar] [CrossRef]
- O’Brien, D.F.; Gates, J.W., Jr. Some Reactions of 3-Hydroxy-1-phenylpyrazole. J. Org. Chem. 1966, 31, 1538–1542. [Google Scholar] [CrossRef]
- Begtrup, M. Reactions between azolium salts and nucleophilic reagents. IX. Cine-substitution of pyrazolium salts. Acta Chem. Scand. 1973, 27, 2051–2074. [Google Scholar] [CrossRef]
- Arbačiauskienė, E.; Martynaitis, V.; Krikštolaitytė, S.; Holzer, W.; Šačkus, A. Synthesis of 3-substituted 1-phenyl-1H-pyrazole-4-carbaldehydes and the corresponding ethanones by Pd-catalysed cross-coupling reactions. ARKIVOC 2011, xi, 1–21. [Google Scholar]
- Mikhaleva, M.A.; Mamaev, V.P. Interaction of 3-hydroxy- and 4-hydroxypyrazoles with benzylidenebisurea. Izv. Sib. Otd. Akad. Nauk SSSR Seriya Khimicheskikh Nauk 1969, 6, 93–98. [Google Scholar]
- Nedzelskyte, E.; Martynaitis, V.; Šačkus, A.; Eller, G.A.; Holzer, W. Synthesis of Mono- and Dibromo-Derivatives of 1-Phenylpyrazol-3-ol. Molbank 2007, M551. [Google Scholar] [CrossRef]
- Sucrow, W.; Mentzel, C.; Slopianka, M. Enehydrazines. 9. 1-Alkyl-3-hydroxypyrazoles from hydrazones or hydrazines. Chem. Ber. 1974, 107, 1318–1328. [Google Scholar] [CrossRef]
- Bruker SAINT, V7.68A; Copyright © 2005–2018 Bruker AXS: Madison, WI, USA, 2012.
- Sheldrick, G.M. SHELXS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Huebschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- CCDC 1586020 Contains the Supplementary Crystallographic Data for This Paper. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 16 November 2017).
- Bechtel, F.; Gaultier, J.; Hauw, C. 1-Phenyl-5-methyl-3-pyrazolone, C10H10N2O. Cryst. Struct. Commun. 1973, 2, 473–476. [Google Scholar]
- Ren, X.-Y.; Wang, J.-G.; Li, Y.-Y. 1-(4-Chlorophenyl)-1H-pyrazol-3-ol. Acta Cryst. 2010, E66, o186. [Google Scholar] [CrossRef] [PubMed]
- Bond Lengths in Crystalline Organic Compounds. In Handbook of Chemistry and Physics, 96th Edition 2015/16; CRC Press: Boca Raton, FL, USA, 2015; ISBN 1482260972.
Sample Availability: Samples of the compounds 1, 4 and 5 are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Machine | Source | Temp. | Detector Distance | Time/Frame | #Frames | Frame Width | CCDC |
|---|---|---|---|---|---|---|---|---|
| Bruker | [K] | [mm] | [s] | [°] | ||||
| 1 | X8 | Mo | 100 | 35 | 25 | 3739 | 0.5 | 1586020 |
| Chemical Formula | C9H8N2O | Crystal System | Orthorhombic | |
|---|---|---|---|---|
| Formula weight (g/mol) | 160.17 | Space group | Pbca | |
| Temperature (K) | 100 | Z | 8 | |
| Measurement method | Φ and ω scans | Volume (Å3) | 1509.4(3) | |
| Radiation (Wavelength (Å)) | MoKα (λ = 0.71073) | Unit cell dimensions (Å) and (°) | 13.6517(13) | 90 |
| Crystal size (mm3) | 0.22 × 0.12 × 0.04 | 6.3663(6) | 90 | |
| Crystal habit | clear colourless plate | 17.3673(17) | 90 | |
| Density (calculated) (g/cm3) | 1.41 | Absorption coefficient/(mm−1) | 0.096 | |
| Abs. correction Tmin | 0.703 | Abs. correction Tmax | 0.746 | |
| Abs. correction type | multi-scan | F(000) (e−) | 672 | |
| Index ranges | −19 ≤ h ≤ 19, −9 ≤ k ≤ 8, −24 ≤ l ≤ 24 | Theta range for data collection (°) | 4.69 to 60.5 | |
| Reflections number | 62937 | Data/restraints/parameters | 2243/0/113 | |
| Refinement method | Least squares | Final R indices | all data | R1 = 0.0473, wR2 = 0.1264 |
| Function minimized | Σ w(Fo2 − Fc2)2 | I > 2σ(I) | R1 = 0.0407, wR2 = 0.1202 | |
| Goodness-of-fit on F2 | 1.085 | Weighting scheme | w = 1/(σ2(Fo2) + (0.0657P)2 + 0.6517P) | |
| Largest diff. peak and hole (e Å−3) | 0.39/−0.21 | where P = (Fo2 + 2Fc2)/3 | ||
| Bond Lengths in Crystalline Organic Compounds | Compound 1 | ||||||
|---|---|---|---|---|---|---|---|
| d | m | σ | ql | qu | |||
| in pyrazole: (N1–N2) | 1.366 | 1.366 | 0.019 | 1.350 | 1.375 | N1 N2 single bond | 1.376 |
| in pyrazole: (N2=C3) | 1.329 | 1.331 | 0.014 | 1.315 | 1.339 | N1 C7 double bond | 1.329 |
| in pyrazole: (N1–C5) | 1.357 | 1.359 | 0.012 | 1.347 | 1.365 | N1 C9 single bond | 1.354 |
| in enols: C=C–OH | 1.333 | 1.331 | 0.017 | 1.324 | 1.342 | C7 O1 single bond | 1.339 |
| in phenols: Caromatic-OH | 1.362 | 1.364 | 0.015 | 1.353 | 1.373 | ||
| in lactams: (C=O) | 1.240 | 1.241 | 0.003 | 1.237 | 1.243 | ||
| in benzoquinones: (C=O) | 1.222 | 1.220 | 0.013 | 1.211 | 1.231 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arbačiauskienė, E.; Krikštolaitytė, S.; Mitrulevičienė, A.; Bieliauskas, A.; Martynaitis, V.; Bechmann, M.; Roller, A.; Šačkus, A.; Holzer, W. On the Tautomerism of N-Substituted Pyrazolones: 1,2-Dihydro-3H-pyrazol-3-ones versus 1H-Pyrazol-3-ols. Molecules 2018, 23, 129. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23010129
Arbačiauskienė E, Krikštolaitytė S, Mitrulevičienė A, Bieliauskas A, Martynaitis V, Bechmann M, Roller A, Šačkus A, Holzer W. On the Tautomerism of N-Substituted Pyrazolones: 1,2-Dihydro-3H-pyrazol-3-ones versus 1H-Pyrazol-3-ols. Molecules. 2018; 23(1):129. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23010129
Chicago/Turabian StyleArbačiauskienė, Eglė, Sonata Krikštolaitytė, Aiva Mitrulevičienė, Aurimas Bieliauskas, Vytas Martynaitis, Matthias Bechmann, Alexander Roller, Algirdas Šačkus, and Wolfgang Holzer. 2018. "On the Tautomerism of N-Substituted Pyrazolones: 1,2-Dihydro-3H-pyrazol-3-ones versus 1H-Pyrazol-3-ols" Molecules 23, no. 1: 129. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23010129