3.2. Procedure to Synthesize Compounds 2–18, 20–27 and 29, 30
Dimethyl pyrazolo[1,5-a]pyridine-2,3-dicarboxylate (
2) [
34]: In a 100 mL flask, K
2CO
3 (0.87 g, 6.3 mmol, 1.4 equiv.) and DMAD (0.96 g, 6.8 mmol, 1.5 equiv.) were added to a solution of 1-aminopyridinium iodide (
1, 1.0 g, 4.5 mmol) in anhydrous DMF (10 mL). The mixture was stirred at r.t. for 18 h. The solvent was evaporated in vacuo, then, the residue was purified by flash chromatography (EtOAc/petroleum ether, 2/8) to give
2 as yellow solid (744 mg, 76%). M.p. 64–66 °C; R
f = 0.3 (EtOAc/petroleum ether, 3/7); IR (ATR diamond, cm
−1): 3111, 1730, 1685, 1484, 1364, 1213, 1079, 995, 814;
1H-NMR (400 MHz, CDCl
3): δ
= 8.50 (d,
J = 7.0 Hz, 1H), 8.15 (d,
J = 7.5 Hz, 1H), 7.45 (t,
J = 7.0 Hz, 1H), 7.02 (t,
J = 7.5 Hz, 1H), 4.01 (s, 3H), 3.91 (s, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 163.2 (CO), 162.6 (CO), 147.3 (Cq), 141.4 (Cq), 129.2 (CH), 127.9 (CH), 119.9 (CH), 115.2 (CH), 102.7 (Cq), 53.0 (OCH
3), 51.7 (OCH
3) (
1H-NMR and
13C-NMR of compounds
2–1
8,
20–
27,
29,
30 are in
Supplementary Materials); HRMS (ESI):
m/
z [M + H]
+ calcd for C
11H
11N
2O
4: 235.0713; found: 235.0716.
Pyrazolo[1,5-a]pyridine-2-dicarboxylic acid (
3) [
34]: To a mixture of
2 (1.0 g, 4.3 mmol) in methanol (25 mL) was added a solution of NaOH 2 M in water (5.0 equiv.) and the mixture was refluxed for 1 h. The solvent was evaporated and the obtained residue was neutralized with an aqueous solution of 12 N HCl. After filtration, the precipitate obtained was washed with water (50 mL), and dried, to give
3 as a white solid (880 mg, quantitative). M.p. 216–218 °C; R
f =0.1 (EtOAc/petroleum ether, 9/1); IR (ATR diamond, cm
−1): 3475, 3341, 1697, 1506, 782;
1H-NMR (400 MHz, DMSO-
d6): δ = 8.89 (d,
J = 7.0 Hz, 1H), 8.17 (d,
J = 7.0 Hz, 1H), 7.64 (t,
J = 7.0 Hz, 1H). 7.26 (t,
J = 7.0 Hz, 1H).
13C-NMR (101 MHz, DMSO-
d6): δ = 164.7 (CO), 164.6 (CO), 148.2 (Cq), 141.5 (Cq), 130.4 (CH), 129.4 (CH), 119.7 (CH), 116.2 (CH), 102.5 (Cq); HRMS:
m/
z [M + H]
+ calcd for C
9H
7N
2O
4: 207.0400, found: 207.0400.
2-Methoxycarbonylpyrazolo[1,5-a]pyridine-3-carboxylic acid (4): To a solution of 3 (0.5 g, 2.43 mmol) in DMSO/MeOH (1/1, 10 mL) was gradually added a SOCl2 solution (0.193 mL, 2.67 mmol, 1.1 equiv.) in methanol (10 mL) and the mixture was stirred at reflux for 72 h. The volatiles were evaporated and 15 mL of water were added. The resulting precipitate was filtered, then washed with water (3 × 25 mL) and dried to give 4 as a white solid (0.41 g, 77%). M.p. 214–216 °C; Rf =0.3 (EtOAc/petroleum ether, 9/1); IR (ATR diamond, cm−1): 3012, 2744, 1730, 1440, 771; 1H-NMR (400 MHz, DMSO-d6): δ = 8.86 (d, J = 7.0 Hz, 1H), 8.10 (d, J = 7.0 Hz, 1H), 7.64 (t, J = 7.0 Hz, 1H), 7.24 (t, J = 7.0 Hz, 1H), 3.90 (s, 3H); 13C-NMR (101 MHz, DMSO-d6): δ = 164.2 (CO), 163.3 (CO), 147.9 (Cq), 140.8 (Cq), 130.3 (CH), 129.4 (CH), 119.3 (CH), 116.1 (CH), 102.4 (Cq), 53.3 (OCH3); HRMS: m/z [M + H]+ calcd for C10H9N2O4: 221.0553, found: 221.0556.
Methyl 3-(tert-butoxycarbonylamino)pyrazolo[1,5-a]pyridine-2-carboxylate (5): To a mixture of 4 (1.0 g, 4.5 mmol) in THF (15 mL) at −10 °C, was added triethylamine (597 mg, 0.82 mL, 5.9 mmol, 1.3 equiv.) and the mixture was stirred for 5 min. Ethyl chloroformate (739 mg, 6.8 mmol, 1.5 equiv.) was then added and the mixture was stirred for 1.5 h at −10 °C. A solution of NaN3 (0.501 g, 7.7 mmol, 1.7 equiv.) in water (2 mL) was added and the reaction mixture was stirred for 1.5 h at r.t. After filtration and evaporation, (15 mL) of water was added, and then the mixture was extracted with ethyl acetate (3 × 20 mL). After evaporation of the solvent, tert-butanol (20 mL) was added and the mixture was refluxed for 6 h. The reaction mixture was concentrated under reduced pressure. The product was purified by column flash chromatography (EtOAc/petroleum ether, 3/7); to give 5 as a white solid (0.85 g, 65%). M.p. 183–185 °C; Rf = 0.5 (EtOAc/petroleum ether, 4/6); IR (ATR diamond, cm−1): 3412, 3090, 2975, 1542, 782; 1H-NMR (400 MHz, CDCl3): δ = 8.33 (d, J = 7.1 Hz, 1H), 8.13 (d, J = 7.1 Hz, 1H), 8.03 (s, NH), 7.05 (t, J = 7.1, 1H), 6.86 (t, J = 7.1Hz, 1H), 4.02 (s, 3H), 1.54 (s, 9H); 13C-NMR (101 MHz, CDCl3:) δ = 164.1 (CO), 153.0 (CO), 132.5 (Cq), 128.1 (CH), 121.8 (CH), 121.7 (CH), 116.3 (Cq), 114.8 (CH), 99.8 (Cq), 80.8 (CO), 52.2 (OCH3), 28.7 (3XCH3); HRMS: m/z [M + H]+ calcd for C14H18N3O4: 292.1293, found: 292.1291.
Methyl 3-aminopyrazolo[1, 5-a]pyridine-2-carboxylate (6): A solution of 3-N-Boc-aminoester 5 (0.5 g, 1.071 mmol) in a mixture of CH2Cl2/TFA (2/1) (15 mL) was stirred at r.t. for 4 h. After complete disappearance of the starting material, the volatiles were removed in vacuo then an aq. satd. solution of K2CO3 (10 mL) was slowly added. The residue was extracted with EtOAc (3 × 20 mL). The solvent was evaporated under reduced pressure and the crude material was purified by column chromatography (EtOAc/petroleum ether, 3/7) to give 6 as a yellow solid in quantitative manner. M.p. 186–184 °C, Rf = 0.4 (EtOAc/petroleum ether, 5/5); IR (ATR diamond, cm−1): 3404, 3286, 1696, 1127, 910, 716; 1H-NMR (400 MHz, CDCl3): δ = 8.30 (d, J = 7.0 Hz, 1H), 7.44 (d, J = 7.0 Hz, 1H), 7.0 (t, J = 7.0 Hz, 1H), 6.80 (t, J = 7.0 Hz, 1H), 4.06 (s, 2H), 4.01 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 164.6 (CO), 130.0 (Cq), 129.1 (Cq), 128.1 (CH), 126.0 (Cq), 119.8 (CH), 117.5 (CH), 114.8 (CH), 51.9 (OCH3); HRMS: m/z [M + H]+ calcd for C9H10N3O2: 192.0765, found: 192.0767.
Methyl 3-(benzoylcarbamothioylamino)pyrazolo[1,5-a]pyridine-2-carboxylate (7): To a solution of the amine 6 (85 mg, 0.44 mmol) in chloroform (5 mL) was added benzoyl isothiocyanate (87 mg, 0.53 mmol, 1.2 equiv.) and the mixture was stirred at r.t. for 12 h. After evaporating the solvent, the crude material was purified by column chromatography on silica gel, (EtOAc/petroleum ether, 2/8) to give the desired product 7 as a yellow solid (125 mg, 80%). M.p. 216–218 °C, IR (ATR diamond, cm−1): 3248, 3033, 1722, 1522, 1190, 750, 668; 1H-NMR (250 MHz, DMSO-d6): δ = 12.49 (s, NH), 11.79 (s, NH), 8.71 (d, J = 7.1 Hz, 1H), 8.00 (d, J = 8.0 HZ, 1H), 7.97 (d, J = 8.0 HZ, 1H), 7.68–7.60 (m, 2H), 7.52 (t, J = 7.1 Hz, 2H), 7.36–7.29 (m, 1H), 7.10 (t, J = 7.1 Hz, 1H), 3.83 (s, 3H); 13C-NMR (101 MHz, DMSO-d6): δ = 181.3 (CO), 172.6 (CS), 168.8 (CO), 162.3 (Cq), 137.9 (Cq), 135.8 (Cq), 133.7 (CH), 129.6 (CH), 129.2 (2 × CH), 129.0 (2 × CH), 124.7 (CH), 119.7 (CH), 115.7 (CH), 113.1 (Cq), 52.0 (OCH3); HRMS: m/z [M + H]+ calcd for C17H15N4O3S: 355.0862, found:355.0859.
2-Thioxo-1H-pyrido[3,4]pyrazolo[4,3-d]pyrimidin-4-one (8): To a solution of 7 (137 mg, 0.39 mmol) in ethanol (10 mL) was added a solution of sodium ethoxide (28.9 mg, 0.425 mmol, 1.1 equiv.). The reaction mixture was refluxed for 8 h. After evaporating the solvent, a mixture of water/acetic acid (4/1) was added. The obtained precipitate was filtered, washed with diethyl ether (2 × 20 mL) and the product 8 was obtained as a white solid (65 mg, 76%). M.p. 314–316 °C; IR (ATR diamond, cm−1): 3212, 2920, 1586, 1362, 839, 785; 1H-NMR (400 MHz, DMSO-d6): δ = 13.26 (s, NH), 12.40 (s, NH), 8.80 (d, J = 7.1 Hz, 1H), 8.16 (d, J = 7.1 Hz, 1H), 7.40 (t, J = 7.1 Hz, 1H), 7.23 (t, J = 7.1 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6): δ = 172.9 (CO), 156.1 (CS), 132.9 (Cq), 129.6 (CH), 127.4 (Cq), 124.3 (CH), 120.8 (Cq), 119.1 (CH), 117.5 (CH); HRMS: m/z [M + H]+ calcd for C9H7N4OS: 219.0332, found: 219.0335.
2-Methylsulfanyl-3H-pyrido[3,4]pyrazolo[4,3-d]pyrimidin-4-one (9): To a solution of 8 (0.51 g, 2.33 mmol) in 10 mL of DMSO and 20 mL of ethanol was added NaOH (93 mg, 2.33 mmol, 1.0 equiv.). Iodomethane was slowly added (0.143 mL, 2.33 mmol, 1.0 equiv.) and the reaction was stirred at r.t. during 2 h. Then, the volatiles were evaporated and water (20 mL) was added. The resulting precipitate was filtered, washed with water (2 × 20 mL) and dried to give 9 as a yellow solid (270 mg, 85%). M.p. 302–304 °C; IR (ATR diamond, cm−1): 3232, 2927, 1596, 1365, 859, 775, 1H-NMR (250 MHz, DMSO-d6): δ = 12.52 (s, 1H), 8.88 (d, J = 7.0 Hz, 1H), 8.07 (d, J = 7.0 Hz, 1H), 7.43 (t, J = 7.0 Hz, 1H), 7.30 (t, J = 7.0 Hz, 1H), 2.61 (s, 3H); 13C-NMR (101 MHz, DMSO-d6): δ = 157.1 (CO), 152.7 (Cq), 135.2 (Cq), 133.5 (Cq), 130.6 (Cq), 129.8 (CH), 124.4 (CH), 118.6 (CH), 117.7 (CH), 13.5 (SCH3); HRMS: m/z [M + H]+ calcd for C10H9N4OS: 233.0489, found: 233.0491.
General procedure A for the direct arylation via C-OH bond activation: In a sealed tube of 10 mL, a solution of 9 (100 mg, 0.43 mmol) in dioxane (4 mL) was degassed by argon bubbling for 10 min, then PyBroP (240 mg, 0.51 mmol, 1.2 equiv.) and Et3N (1.29 mmol, 0.17 mL, 3.0 equiv.) were added successively. After 18 h at 80 °C, the reaction mixture was cooled and a solution containing the required (Het)arylboronic acid (2.0 equiv.), Na2CO3 (5.0 equiv.) and PdCl2(dppf).CH2Cl2 (10 mol%) in H2O (1 mL) was added. The sealed tube was stirred and heated at 110 °C for 24 h. After cooling at r.t. the volatiles were evaporated under reduced pressure and the crude material diluted in CH2Cl2 (20 mL). The organic layers were washed with water (10 mL), dried over MgSO4, filtered, and concentrated under vacuum. The crude mixture was purified by column chromatography on silica gel to give the desired mono-arylated compounds.
2-Methylsulfanyl-4-(p-tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (10): Using p-tolylboronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 0.5/9.5) to afford 10 as a yellow solid in a 85% yield. M.p. 206–208 °C; Rf = 0.5 (EtOAc/petroleum ether, 0.5/9.5); IR (ATR diamond, cm−1): 2922, 1635, 1577, 1528, 1435, 1240, 1184, 1002, 799, 752, 634; 1H-NMR (400 MHz, CDCl3): δ = 8.85–8.79 (m, 3H), 8.38 (d, J = 7.0 Hz, 1H), 7.44–7.37 (m, 3H), 7.32 (t, J = 7.0 Hz, 1H), 2.78 (s, 3H), 2.47 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.5 (Cq), 154.7 (Cq), 142.1 (Cq), 138.4 (Cq), 136.9 (Cq), 133.5 (Cq), 133.3 (Cq), 130.3 (2 × CH), 129.5 (2 × CH), 128.9 (CH), 123.0 (CH), 119.7 (CH), 119.2 (CH), 21.8 (CH3), 14.9 (SCH3); HRMS: m/z [M + H]+ calculated for C17H15N4S: 307.1013, found: 307.1011.
4-(4-Methoxyphenyl)-2-methylsulfanyl-pyrido[3,4]pyrazolo[4,3-d]pyrimidine (11): Using 4-methoxy-phenylboronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 0.5/9.5) to afford 11 as a yellow solid in a 87% yield. M.p. 188–190 °C; Rf = 0.5 (EtOAc/petroleum ether, 0.5/9.5); IR (ATR diamond, cm−1): 2924, 2866, 1728, 1433, 1272, 1122, 844, 635; 1H-NMR (250 MHz, CDCl3): δ = 8.93 (d, J = 6.7 Hz, 2H), 8.77 (d, J = 6.7 Hz, 1H), 8.35 (d, J = 6.7 Hz, 1H), 7.3 (t, J = 6.7 Hz, 1H), 7.29 (t, J = 6.7 Hz, 1H), 7.08 (d, J = 6.7 Hz, 2H), 3.90 (s, 3H), 2.76 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.5 (Cq), 162.3 (Cq), 154.1 (Cq), 138.1 (Cq), 136.7 (Cq), 133.3 (Cq), 132.0 (2 × CH), 128.7 (CH), 128.5 (Cq), 122.8 (CH), 119.6 (CH), 119.0 (CH), 114.1 (2 × CH), 55.4 (OCH3), 14.8 (SCH3); HRMS: m/z [M + H]+ calcd for C17H15N4OS: 323.0959, found: 323.0961.
4-(3-Methoxyphenyl)-2-methylsulfanyl-pyrido[3,4]pyrazolo[4,3-d]pyrimidine (12): Using 3-methoxy-phenylboronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 5/5) to afford 12 as a yellow solid in a 67% yield. M.p. 154–156 °C; Rf = 0.4 (EtOAc/petroleum ether, 5/5); IR (ATR diamond, cm−1): 2919, 2851, 1737, 1635, 1495, 1132, 844, 628; 1H-NMR (250 MHz, CDCl3): δ = 8.82 (d, J = 6.9 Hz, 1H), 8.57 (d, J = 6.9 Hz, 1H), 8.51–8.47 (m, 1H), 8.40 (d, J = 6.9 Hz, 1H), 7.46 (dd, J = 15.2, 7.8 Hz, 2H), 7.37–7.31 (m, 1H), 7.12 (dd, J = 8.2, 2.9 Hz, 1H), 3.97 (s, 3H), 2.79 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.3 (Cq), 159.9 (Cq), 154.2 (Cq), 138.6 (Cq), 137.3 (Cq), 136.7 (Cq), 133.3 (Cq), 129.7 (CH), 128.8 (CH), 123.1 (CH), 123.0 (CH), 119.6 (CH), 119.3 (CH), 117.7 (CH), 114.8 (CH), 55.5 (OCH3), 14.9 (SCH3); HRMS: m/z [M + H]+ calcd for C17H15N4OS: 323.0960, found: 323.0961.
4-(2-Methoxyphenyl)-2-methylsulfanyl-pyrido[3,4]pyrazolo[4,3-d]pyrimidine (13): Using 2-methoxy-phenylboronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 4/6) to afford 13 as a yellow solid in a 20% yield. M.p. 194–196 °C; Rf = 0.3 (EtOAc/petroleum ether, 4/6); IR (ATR diamond cm−1): 3077, 2920, 1600, 1431, 1157, 878, 636; 1H-NMR (250 MHz, CDCl3): δ = 8.75 (d, J = 7.0 Hz, 1H), 8.40 (d, J = 7.0 Hz, 1H), 7.74 (d, J = 7.0 Hz, 1H), 7.52 (t, J = 7.0 Hz, 1H), 7.42 (t, J = 7.0 Hz, 1H), 7.29 (t, J = 7.0 Hz, 1H), 7.19–7.11 (m, 2H), 3.85 (s, 3H), 2.77 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.5 (Cq), 157.9 (Cq), 157.0 (Cq), 137.8 (Cq), 137.0 (Cq), 133.5 (Cq), 131.6 (CH), 131.4 (CH), 128.7 (CH), 125.5 (Cq), 122.6 (CH), 120.8 (CH), 119.5 (CH), 118.9 (CH), 112.2 (CH), 56.0 (OCH3), 14.8 (SCH3); HRMS: m/z [M + H]+ calcd for C17H15N4OS: 323.0959, found: 323.0961.
4-(4-Fluorophenyl)-2-methylsulfanyl-pyrido[3,4]pyrazolo[4,3-d]pyrimidine (14): Using 4-fluorophenyl-boronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 1/9) to afford 14 as a yellow solid in a 90% yield. M.p. 140–142 °C; Rf = 0.6 (EtOAc/petroleum ether, 1/9); IR (ATR diamond cm−1): 3095, 2920, 2849, 1635, 1556, 1420, 1210, 880, 749, 640, 524; 1H-NMR (250 MHz, CDCl3): δ = 8.97 (t, J = 6.8 Hz, 2H), 8.79 (d, J = 6.8 Hz, 1H), 8.39 (d, J = 6.8 Hz, 1H), 7.56–7.20 (m, 4H), 2.78 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 164.9 (d, J = 254.5 Hz, Cq), 162.4 (Cq), 153.2 (Cq), 138.5 (Cq), 138.5 (Cq), 136.6 (Cq), 133.4 (Cq), 132.4 (d, J = 8.7 Hz, 2 × CH), 128.7 (CH), 123.1 (CH), 119.5 (d, J = 33.8 Hz, 2 × CH), 115.8 (CH), 115.6 (CH), 14.6 (SCH3); HRMS: m/z [M + H]+ calcd for C16H12FN4S: 311.0762, found: 311.0761.
2-Methylsulfanyl-4-[4-(trifluoromethyl)phenyl]pyrido[3,4]pyrazolo[4,3-d]pyrimidine (15): Using 4-trifluoromethylphenyl boronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 0.5/9.5) to afford 15 as a yellow solid in a 70% yield. M.p. 184–186 °C; Rf = 0.6 (EtOAc/petroleum ether, 0.5/9.5); IR (ATR diamond cm−1): 3076, 1643, 1541, 1466, 1255, 1230, 1086, 857, 753, 620; 1H-NMR (400 MHz, CDCl3): δ = 9.04 (d, J = 8.2 Hz, 2H), 8.82 (d, J = 6.9 Hz, 1H), 8.42 (t, J = 6.9 Hz, 1H), 7.84 (d, J = 8.2 Hz, 2H), 7.48 (t, J = 6.9 Hz, 1H), 7.38 (t, J = 6.9 Hz, 1H), 2.79 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.4 (Cq), 152.6 (Cq), 139.1 (q, J = 1.4 Hz, Cq), 139.0 (Cq), 136.7 (Cq), 133.4 (Cq), 132.7 (q, J = 33.0 Hz, Cq), 132.5 (Cq), 130.4 (2 × CH), 128.8(CH), 125.4 (q, J = 3.7 Hz, 2 × CH), 123.4 (CH), 119.7 (CH), 119.6 (CH), 14.7 (SCH3); HRMS: m/z [M + H]+ calcd for C17H12F3N4S: 361.0730, found: 361.0729.
4-(2-Methylsulfanylpyrido[3,4]pyrazolo[4,3-d]pyrimidin-4-yl)benzonitrile (16): Using 4-cyanophenyl-boronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 2/8) to afford 16 as a yellow solid in a 58% yield. M.p. 230–232 °C; Rf = 0.76 (EtOAc/petroleum ether, 2/8); IR (ATR diamond cm−1): 2921, 2223, 1731, 1548, 1494, 1255, 1275, 1018, 877, 748, 631; 1H-NMR (400 MHz, CDCl3): δ = 9.10 (d, J = 8.5 Hz, 1H), 8.85 (d, J = 6.9 Hz, 1H), 8.46 (d, J = 6.9 Hz, 1H), 7.89 (d, J = 8.5 Hz, 1H), 7.54 (t, J = 6.9 Hz, 1H), 7.43 (t, J = 6.9 Hz, 1H), 2.81 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.4 (Cq), 151.7 (Cq), 139.9 (Cq), 139.3 (Cq), 136.5 (Cq), 133.5(Cq), 132.3 (2 × CH), 130.5 (2 × CH), 128.8 (CH), 123.6 (CH), 119.9 (CH), 119.8 (CH), 118.7 (Cq), 114.4 (Cq), 14.8 (SCH3); HRMS: m/z [M + H]+ calcd for C17H12N5S: 318.0808, found: 318.0807.
4-(2-Methylsulfanylpyrido[3,4]pyrazolo[4,3-d]pyrimidin-4-yl)phenol (17): Using 4-hydroxyphenylboronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 3/7) to afford 17 as a yellow solid in a 45% yield. M.p. 254–256 °C; Rf = 0.4 (EtOAc/petroleum ether, 2/8); IR (ATR diamond cm−1): 3100, 2928, 2214, 1607, 1550, 1494, 1274, 1128, 1021, 877, 748, 631; 1H-NMR (250 MHz, CDCl3): δ = 9.08 (d, J = 8.6 Hz, 2H), 8.83 (d, J = 6.9 Hz, 1H), 8.44 (d, J = 6.9 Hz, 1H), 7.87 (d, J = 8.6 Hz, 1H ), 7.51 (t, J = 6.9 Hz, 1H), 7.42 (t, J = 6.9 Hz, 1H), 2.79 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.5 (Cq), 151.8 (Cq), 139.9 (Cq), 136.5 (Cq), 133.9 (Cq), 132.3 (2 × CH), 130.5 (2 × CH), 128.8 (CH), 123.6 (CH), 119.9 (CH), 119.8 (CH), 118.7 (Cq), 114.6 (Cq), 14.8 (SCH3); HRMS: m/z [M + H]+ calcd for C16H13N4OS: 309.0805, measured: 309.0804.
4-(2-Furyl)-2-methylsulfanyl-pyrido[3,4]pyrazolo[4,3-d]pyrimidine (18): Using 2-furanylboronic acid as coupling reagent and following the general procedure A, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 4/6) to afford 18 as a green solid in a 74% yield. M.p. 190–192 °C; Rf = 0.6 (EtOAc/petroleum ether, 3/7); IR (ATR diamond cm−1): 2917, 2849, 1737, 1523, 1497, 1233, 1165, 1018, 885, 747, 516; 1H-NMR (400 MHz, CDCl3): 1H-NMR (400 MHz, CDCl3): δ = 8.83 (d, J = 7.0 Hz, 1H), 8.38 (d, J = 7.0 Hz, 1H), 8.06 (d, J = 4.4 Hz, 1H), 7.85 (d, J = 2.8 Hz, 1H), 7.46 (t, J = 7.0 Hz, 1H), 7.35 (t, J = 7.0 Hz, 1H), 6.73 (dd, J = 4.4, 2.8 Hz, 1H), 2.78 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 162.6 (Cq), 149.7 (Cq), 146.6 (CH), 145.2 (Cq), 137.7 (Cq), 134.4 (Cq), 133.6 (Cq), 128.9 (CH), 123.2 (CH), 119.5 (CH), 119.1 (2CH), 112.8 (CH), 14.7 (SCH3); HRMS: m/z [M + H]+ calcd for C14H11N4OS: 283.0649, found: 283.0648.
General procedure B for C-2 Liebeskind–Srogl cross-coupling reaction: Compound 10, 11 or 14 (0.15 mmol), aryl boronic acid (1.5 equiv.), Cu-(I) thiophene-2-carboxylate (CuTc) (3.0 equiv.), and Pd(PPh3)4 (10 mol%) were dissolved in anhydrous THF (5 mL) under argon in a microwave sealed vial. The reaction mixture was irradiated under microwave at 100 °C for 1h30. After cooling, the solvent was evaporated under reduced pressure and the residue was diluted in an aq. satd. NaHCO3 solution (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with water (5 mL), dried with anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the desired compounds.
2,4-Bis(p-tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (20): Starting from 10, using p-tolylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 1/9) to afford 20 as a yellow solid in a 78% yield. M.p. 176–178 °C; Rf = 0.7 (EtOAc/petroleum ether, 2/8); IR (ATR diamond, cm−1): 2922, 2851, 1704, 1524, 1495, 1134, 1017, 834, 746; 1H-NMR (400 MHz, CDCl3): δ = 8.94 (d, J = 7.9 Hz, 2H), 8.83 (d, J = 7.0 Hz, 1H), 8.60 (d, J = 7.9 Hz, 2H), 8.49 (d, J = 7.0 Hz, 1H), 7.48–7.38 (m, 3H), 7.37–7.29 (m, 3H), 2.47 (s, 3H), 2.43 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 156.5 (Cq), 153.9 (Cq), 141.5 (Cq), 139.5 (Cq), 138.2 (Cq), 137.5 (Cq), 136.3 (Cq), 134.8 (Cq), 134.0 (Cq), 130.0 (2 × CH), 129.4 (2 × CH), 129.3 (2 × CH), 128.9 (CH), 128.0 (2 × CH), 123.2 (CH), 119.7 (CH), 119.2 (CH), 21.7 (CH3), 21.5 (CH3); HRMS: m/z [M + H]+ calcd for C23H19N4: 351.1525, found: 351.1524.
2-(4-Methoxyphenyl)-4-(p-tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (21): Starting from 10, using 4-methoxyphenylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 1/9) to afford 21 as an orange solid in a 75% yield. M.p. 202–204 °C; Rf = 0.6 (EtOAc/petroleum ether, 1.5/8.5); IR (ATR diamond, cm−1): 2921, 2851, 1608, 1524, 1495, 1234, 1024, 750; 1H-NMR (400 MHz, CDCl3): δ = 8.98 (d, J = 8.5 Hz, 2H), 8.88 (d, J = 7.0 Hz, 1H), 8.71 (d, J = 8.5 Hz, 2H), 8.53 (d, J = 7.0 Hz, 1H), 7.50 (t, J = 7.0 Hz, 1H), 7.46 (d, J = 8.5 Hz, 2H) 7.38 (t, J = 7.0 Hz, 1H), 7.10 (d, J = 8.5 Hz, 2H), 3.94 (s, 3H), 2.52 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 161.0 (Cq), 156.4 (Cq), 154.0 (Cq), 141.5 (Cq), 138.2 (Cq), 137.4 (Cq), 134.7 (Cq), 134.0 (Cq), 131.9 (Cq), 130.0 (2 × CH), 129.6 (2 × CH), 129.4 (2 × CH), 128.9 (CH), 123.1 (CH), 119.7 (CH), 119.2 (CH), 113.9 (2 × CH), 55.4 (OCH3), 21.7 (CH3); HRMS: m/z [M + H]+ calcd for C23H19N4O: 367.1554, found: 367.1553.
2-(3-Methoxyphenyl)-4-(p-tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (22): Starting from 10, using 3-methoxyphenylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 3/7) to afford 22 as a yellow solid in a 48% yield. M.p. 130–132 °C; Rf = 0.5 (EtOAc/petroleum ether, 5/5); IR (ATR diamond, cm−1): 2922, 2801, 1722, 1584, 1486, 1240, 1036, 745; 1H-NMR (250 MHz, CDCl3): δ = 8.97 (d, J = 8.5 Hz, 2H), 8.87 (d, J = 7.0 Hz, 1H), 8.53 (d, J = 8.5 Hz, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.31 (s, 1H), 7.53–7.34 (m, 5H), 7.04 (d, J = 7.0 Hz, 1H), 3.99 (s, 3H), 2.50 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 159.9 (Cq), 156.1 (Cq), 153.9 (Cq), 141.6 (Cq), 140.5 (Cq), 138.2 (Cq), 137.6 (Cq), 137.6 (Cq), 134.9 (Cq), 130.0 (2 × CH), 129.5 (CH), 129.4 (2 × CH), 129.0 (CH), 123.4 (CH), 120.7 (CH), 119.7 (CH), 119.3 (CH), 115.6 (CH), 113.1 (CH), 55.5 (OCH3), 21.7 (CH3); HRMS: m/z [M + H]+ calcd for C23H19N4O: 367.1554, found: 367.1553.
2-(2-Methoxyphenyl)-4-(p-tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (23): Starting from 10, using 3-methoxyphenylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 4/6) to afford 23 as a yellow solid in a 25% yield. M.p. 160–162 °C; Rf = 0.5 (EtOAc/petroleum ether, 6/4); IR (ATR diamond, cm−1): 3068, 2923, 2836, 2211, 1636, 1543, 1188, 935, 736; 1H-NMR (250 MHz, CDCl3): δ = 8.89 (d, J = 8.3 Hz, 3H), 8.56 (d, J = 8.3 Hz, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.52–7.35 (m, 5H), 7.17–7.08 (m, 2H), 3.93 (s, 3H), 2.47 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 157.9 (Cq), 157.3 (Cq), 154.2 (Cq), 141.5 (Cq), 137.8 (Cq), 137.1 (Cq), 134.8 (Cq), 133.9 (Cq), 132.1 (Cq), 130.1 (CH), 130.1 (2 × CH), 129.9 (CH), 129.4 (2 × CH), 128.9 (CH), 123.3 (CH), 120.8 (CH), 119.8 (CH), 119.2 (CH), 112.5 (CH), 56.2 (OCH3), 21.7 (CH3); HRMS: m/z [M + H]+ calcd for C23H19N4O: 367.1554, found: 367.1553.
2-(4-Fluorophenyl)-4-(p-tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (24): Starting from 10, using 4-fluorophenylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 1/9) to afford 24 as a yellow solid in a 85% yield. M.p. 156–158 °C; Rf = 0.6 (EtOAc/petroleum ether, 1.5/8.5); IR (ATR diamond, cm−1): 3061, 2920, 1524, 1493, 1427, 1375, 1208, 1135, 748; 1H-NMR (400 MHz, CDCl3): δ = 8.94 (d, J = 8.2 Hz, 2H), 8.84 (d, J = 6.9 Hz, 1H), 8.71 (d, J = 8.2 Hz, 2H), 8.48 (d, J = 6.9 Hz, 1H), 7.48 (t, J = 6.9 Hz, 1H), 7.43 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 6.9 Hz, 1H), 7.22 (d, J = 8.2 Hz, 2H), 2.50 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 164.0 (d, J = 251.8 Hz, Cq), 155.5 (Cq), 153.9 (Cq), 141.7 (Cq), 138.1 (Cq), 137.4 (Cq), 135.2 (d, J = 2.9 Hz, Cq), 134.7 (Cq), 133.8 (Cq), 130.0 (d, J = 8.4 Hz, 2 × CH), 130.0 (2 × CH), 129.4 (2 × CH), 129.0 (CH), 123.3 (CH), 119.5 (d, J = 28.9 Hz, 2 × CH), 115.4 (CH), 115.2 (CH), 21.7 (CH3); HRMS: m/z [M + H]+ calcd for C22H16FN4: 355.1355, found: 355.1353.
4-(p-Tolyl)-2-[4-(trifluoromethyl)phenyl]pyrido[3,4]pyrazolo[4,3-d]pyrimidine (25): Starting from 10, using 4-trifluoromethylphenylboronic acid as coupling reagent and following the general procedure B, compound was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 1/9) to afford 25 as a yellow solid in a 60% yield. M.p. 174–176 °C; Rf = 0.7 (EtOAc/petroleum ether, 2/8); IR (ATR diamond, cm−1): 2953, 2924, 2852, 1615, 1527, 1508, 1429, 1355, 1295, 1177, 797; 736; 1H-NMR (400 MHz, CDCl3): δ = 8.91 (d, J = 8.0 Hz, 2H), 8.83 (d, J = 7.0 Hz, 1H), 8.79 (d, J = 8.0 Hz, 2H), 8.46 (d, J = 7.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.49 (t, J = 7.0 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.36 (t, J = 7.0 Hz, 1H), 2.49 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 154.6 (Cq), 153.8 (Cq), 142.3 (q, J = 1.3 Hz, Cq) 141.8 (Cq), 138.1 (Cq), 137.5 (Cq), 134.9 (Cq), 133.6 (Cq), 130.8 ( Cq), 130.0 (2 × CH), 129.3 (q, J = 251.3, CF3),129.4 (2 × CH), 129.0 (CH), 128.2 (2 × CH), 125.3 (q, J = 3.8 Hz, 2 × CH), 123.7 (CH), 119.6 (CH), 119.5 (CH), 21.7 (CH3); HRMS: m/z [M + H]+ calcd for C23H16F3N4: 405.1320, found: 405.1321.
4-[4-(p-Tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidin-2-yl]phenol (26): Starting from 10, using 4-hydroxyphenylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 2.5/7.5) to afford 26 as a yellow solid in a 40% yield. M.p. 182–184 °C; Rf = 0.4 (EtOAc/petroleum ether, 3/7); IR (ATR diamond, cm−1): 3108, 2923, 2215, 1615, 1547, 1508, 1429, 1355, 1293, 1157, 787, 746; 1H-NMR (250 MHz, Acetone-d6): δ = 9.08 (t, J = 7.8 Hz, 3H), 8.63 (d, J = 8.7 Hz, 2H), 8.54 (d, J = 7.8 Hz, 1H), 7.75–7.59 (m, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 2.48 (s, 3H); 13C-NMR (101 MHz, Acetone-d6): δ = 159.2 (Cq), 155.9 (Cq), 153.0 (Cq), 141.6 (Cq), 138.2 (Cq), 136.9 (Cq), 134.5 (Cq), 134.1 (Cq), 130.6 (Cq), 129.9 (2 × CH), 129.5 (2 × CH), 129.3 (CH), 129.2 (2 × CH), 124.0 (CH), 120.0 (CH), 119.1 (CH), 115.2 (2 × CH), 20.7 (CH3); HRMS: m/z [M + H]+ calculated for C22H17N4O: 353.1397, found: 353.1396.
2-(2-Furyl)-4-(p-tolyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (27): Starting from 10, using 2-furanylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 2/8) to afford 27 as a yellow solid in a 60% yield. M.p. 146–148 °C; Rf = 0.6 (EtOAc/petroleum ether, 3/7); IR (ATR diamond, cm−1): 3542, 3100, 2922, 2852, 1715, 1525, 1508, 1356, 1275, 1182, 783, 753,612; 1H-NMR (400 MHz, CDCl3) δ 8.93 (d, J = 8.2 Hz, 2H), 8.88 (d, J = 6.9 Hz, 1H), 8.60 (d, J = 6.9 Hz, 1H), 7.71 (d, J = 3.6 Hz, 1H), 7.54–7.48 (m, 2H), 7.45 (d, J = 8.2 Hz, 2H), 7.39 (t, J = 6.9 Hz, 1H), 6.65 (dd, J = 3.6, 1.7 Hz, 1H), 2.51 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 154.6 (Cq), 153.6 (Cq), 150.1 (Cq), 144.2 (CH), 141.8 (Cq), 137.4 (Cq), 137.3 (Cq), 134.6 (Cq), 133.4 (Cq), 130.0 (2 × CH), 129.4 (2 × CH), 128.9 (CH), 123.4 (CH), 119.8 (CH), 119.3 (CH), 112.1 (CH), 112.0 (CH), 21.7 (CH3); HRMS: m/z [M + H]+ calcd for C20H15N4O: 327.1241, found: 327.1240.
2-(2-Tolyl)-4-(p-methoxyphenyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (29): Starting from 11, using p-tolylboronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 3/7) to afford 29 in a 86% yield. M.p. 210–212 °C; Rf = 0.4 (EtOAc/petroleum ether, 3/7); IR (ATR diamond, cm−1): 3012, 1633, 1531, 1432, 1254, 1114, 1021, 804, 744; 1H-NMR (400 MHz, CDCl3): δ = 8.90 (d, J = 6.8 Hz, 1H), 8.74 (d, J = 8.8 Hz, 2H), 8.10 (d, J = 8.8 Hz, 1H), 7.94 (d, J = 8.8 Hz, 2H), 7.55–7.39 (m, 3H), 7.38–7.26 (m, 1H), 7.03 (d, J = 8.8 Hz, 2H), 3.90 (s, 3H), 2.52 (s, 3H) ppm; 13C-NMR (101 MHz, CDCl3): δ =164.4 (Cq), 163.9 (Cq), 162.4 (Cq), 141.6 (Cq), 136.4 (Cq), 136.1 (Cq), 131.8 (Cq), 131.3 (2 × CH), 130.2 (CH), 130.2 (2 × CH), 129.5 (2 × CH), 126.6 (CH), 120.3 (CH), 118.4 (CH), 114.3 (2 × CH), 103.3 (Cq), 91.0 (Cq), 56.0 (OCH3), 22.2 (CH3); HRMS: m/z [M + H]+ calcd for C23H19N4O [M + H]+ 367.1553, found 367.1556.
2-(2-Tolyl)-4-(p-fluorophenyl)pyrido[3,4]pyrazolo[4,3-d]pyrimidine (30): Starting from 14, using p-tolyl-boronic acid as coupling reagent and following the general procedure B, the crude product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, 2/8) to afford 30 in a 53% yield. M.p. 160–162 °C; Rf = 0.6 (EtOAc/petroleum ether, 2/8); IR (ATR diamond, cm−1): 3061, 2920, 1524, 1493, 1427, 1375, 1208, 1135, 748; 1H-NMR (400 MHz, CDCl3) δ = 9.13 (d, J = 8.7, Hz, 2H), 8.87 (d, J = 6.9 Hz, 1H), 8.63 (d, J = 7.8 Hz, 2H), 8.54 (d, J = 6.9 Hz, 1H), 7.52 (t, J = 6.9 Hz, 1H), 7.43–7.30 (m, 5H), 2.49 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 164.8 (d, J = 251.8 Hz, Cq), 156.5 (Cq), 152.5 (Cq), 139.6 (Cq), 138.4 (Cq), 137.3 (Cq), 136.1 (Cq), 134.8 (Cq), 132.9 (d, J = 3.0 Hz, Cq), 132.3 (d, J = 8.6 Hz, 2 × CH), 129.3 (2 × CH), 128.9 (CH), 127.9 (2 × CH), 123.4 (CH), 119.6 (d, J = 32.1 Hz, 2 × CH), 115.8 (CH), 115.5 (CH), 21.4 (CH3); HRMS: m/z [M + H]+ calcd for C22H16FN4: 355.1354, found: 355.1352.

 and
and 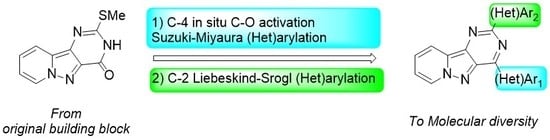




{kind=link}
{kind=link}
{kind=link}
{kind=link}






















