
Identification, Characterization and Quantification of Process-Related and Degradation Impurities in Lisdexamfetamine Dimesylate: Identifiction of Two New Compounds


Abstract
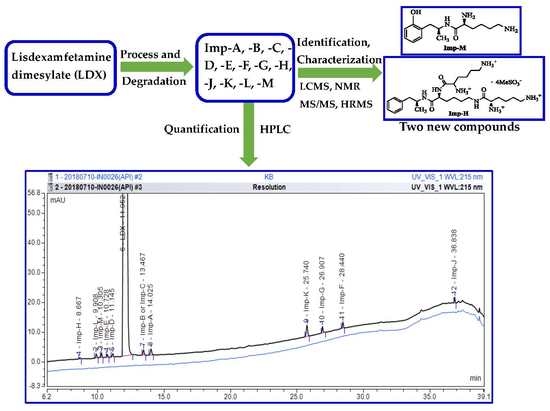
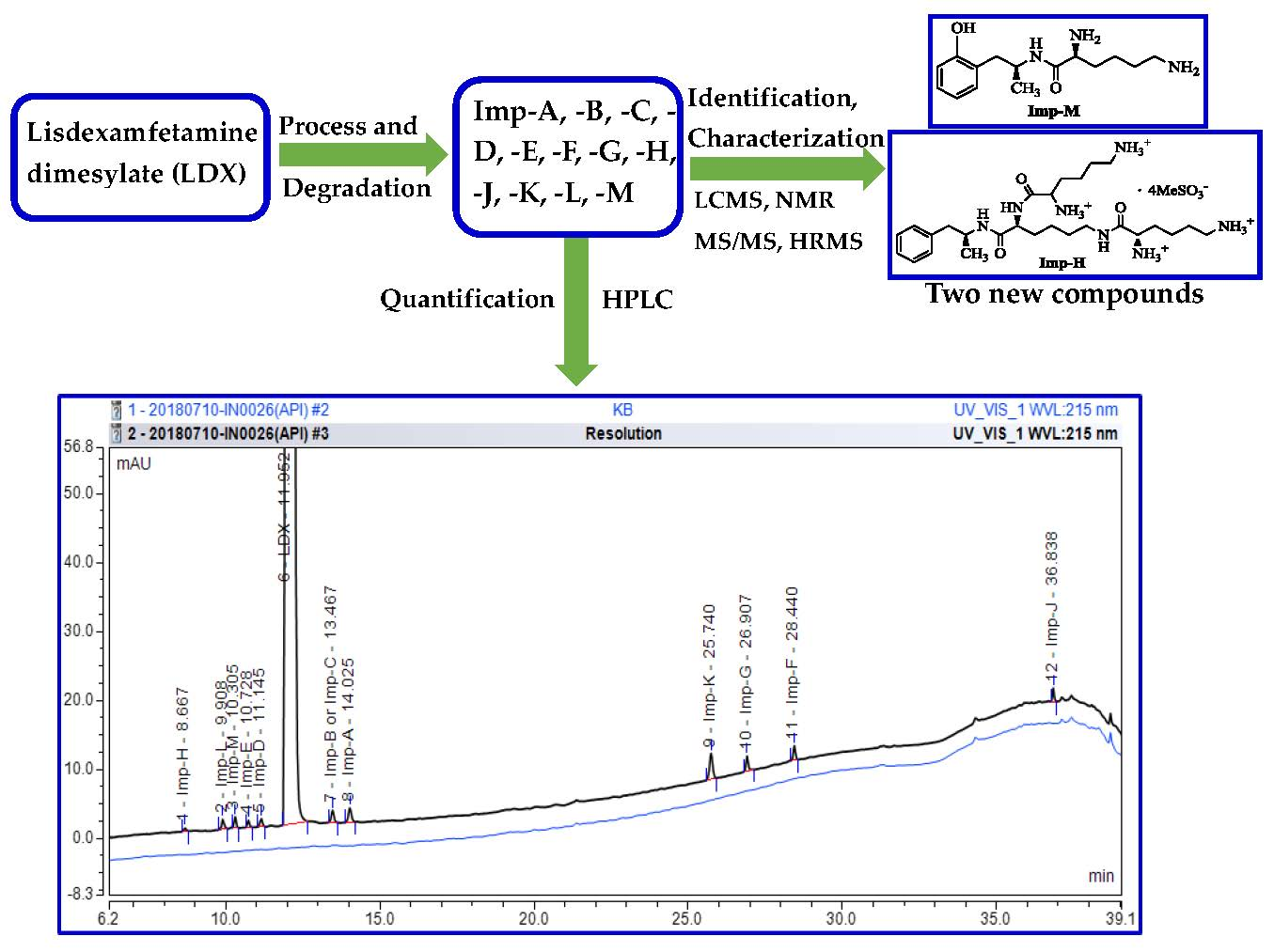
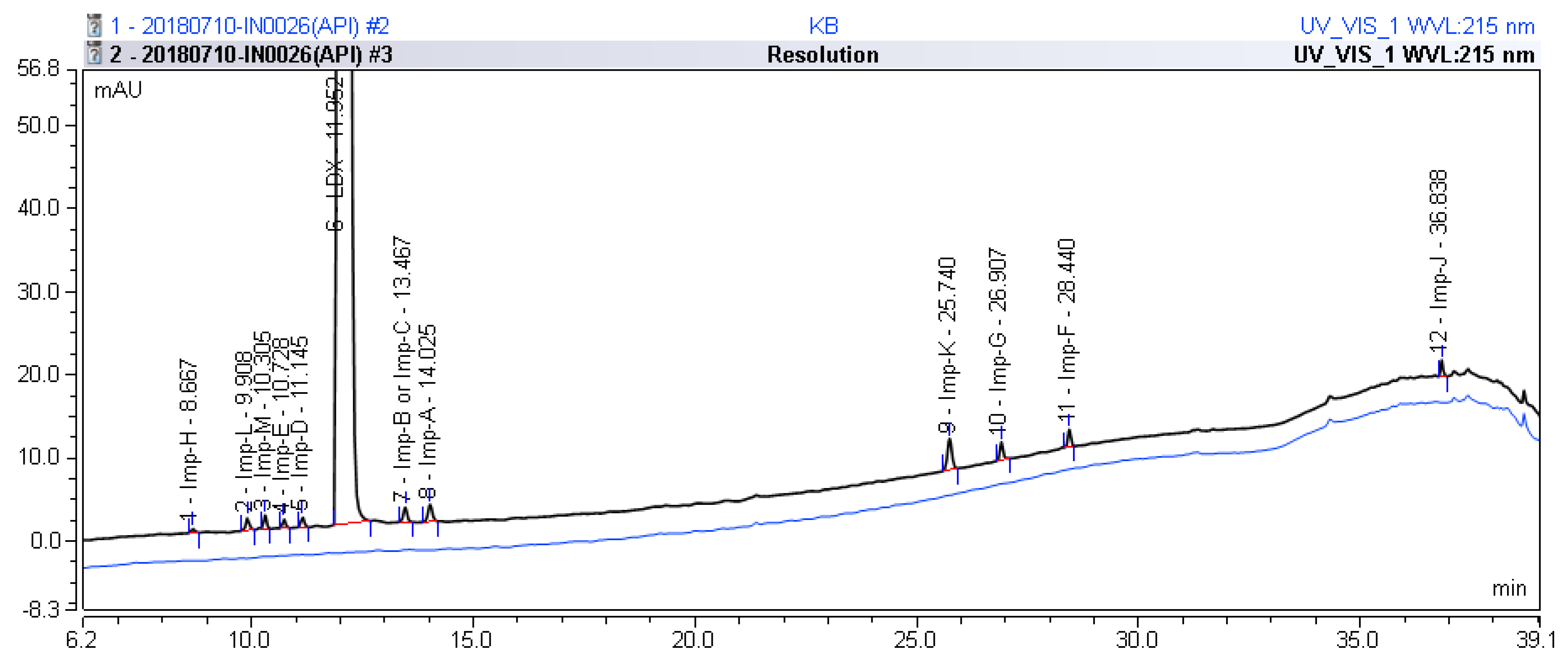
:
1. Introduction
2. Results and Discussion
2.1. Detection of Process-Related Impurities and Forced Degradation of LDX
2.2. Impurity Preparation and Structural Confirmation
2.2.1. Structural Elucidation and Control Strategy of Imp-B and Imp-C
2.2.2. Structural Elucidation and Control Strategy of Imp-F and Imp-G
2.2.3. Structural Elucidation and Control Strategy of Imp-D and Imp-E
2.2.4. Structural Elucidation and Control Strategy of Imp-H
2.2.5. Structural Elucidation and Control Strategy of Imp-L
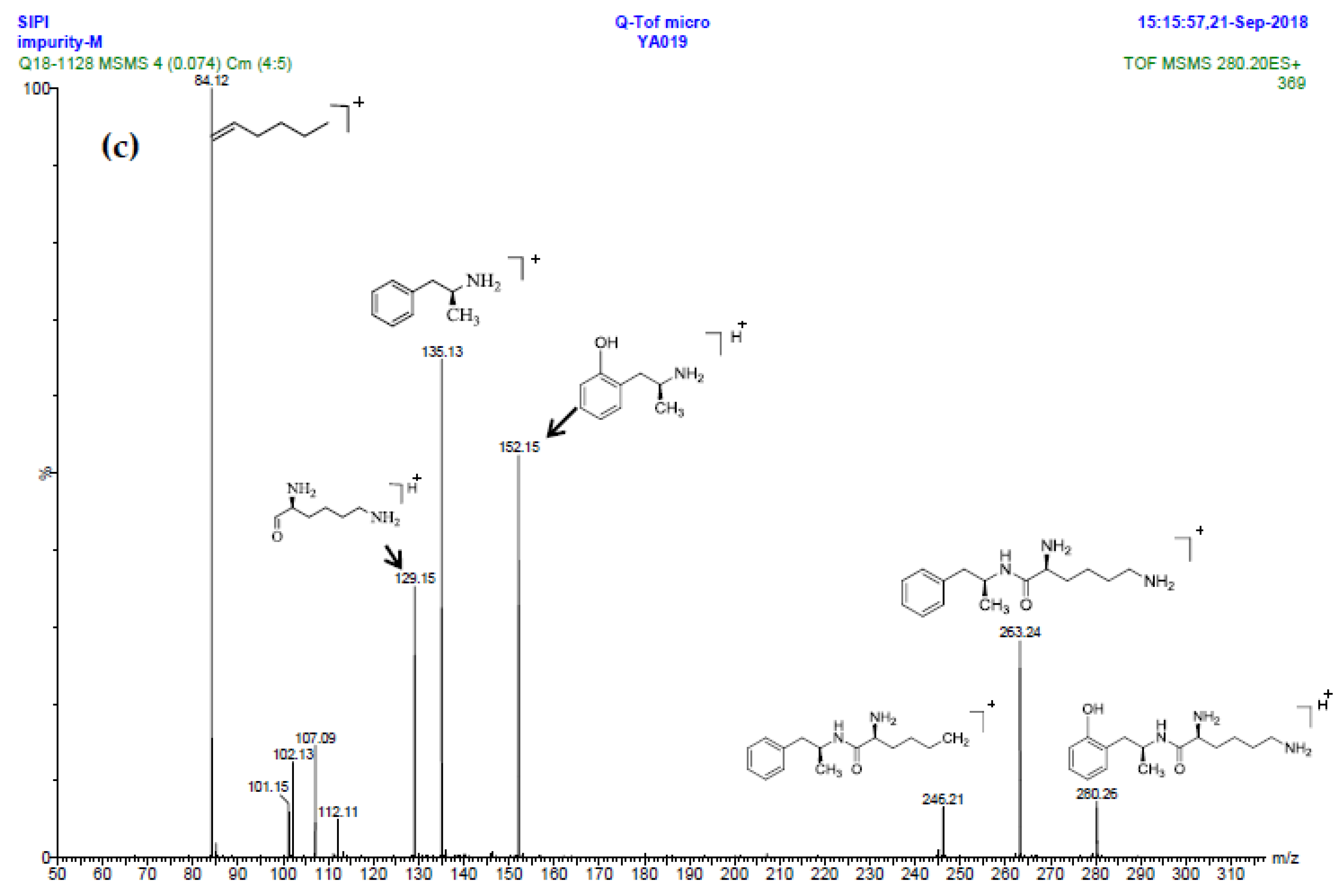
2.2.6. Structural Elucidation and Control Strategy of Imp-M
2.3. Possible Mechanisms for Formation of the Impurities
2.4. Optimization of the HPLC-UV Method
2.5. Validation of the HPLC-UV Method
2.5.1. System Suitability
2.5.2. Linearity, LOD, and LOQ
2.5.3. Accuracy, Precision, and Robustness
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Analytical HPLC Conditions
3.3. LC-MS Conditions
3.4. NMR Spectroscopy
3.5. FT-IR Spectroscopy
3.6. Preparation of Standard and Sample Solutions
3.7. Forced Degradation Study
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Biederman, J.; Krishnan, S.; Zhang, Y.X.; McGough, J.; Findling, R. Efficacy and tolerability of lisdexamfetamine dimesylate(NRP-104) in children with attention-deficit/hyperactivity disorder: A phase III, multicenter, randomized, double-blind, forced-dose, parallel-group study. Clin. Ther. 2007, 29, 450–463. [Google Scholar] [CrossRef]
- Michel, T.; Krishnan, S.; Bishop, B.; Lauderback, C.; Moncrief, J.; Oberlender, R.; Piccariello, T. Abuse-resistant Amphetamine Compounds. U.S. Patent 20050054561A1, 10 March 2005. [Google Scholar]
- Michel, T.; Krishnan, S.; Bishop, B.; Lauderback, C.; Moncrief, J.; Oberlender, R.; Piccariello, T. Abuse-resistant Amphetamine Prodrugs. U.S. Patent 20070042955A1, 22 January 2007. [Google Scholar]
- Lu, C.Y.; Feng, C.H. Identification of dimer impurities in ampicillin and amoxicillin by capillary LC and tandem mass spectrometry. J. Sep. Sci. 2007, 30, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.S.; Li, Y.; Qin, L. Isolation and identification of two unknown impurities from the raw material of clindamycin hydrochloride. J. Sep. Sci. 2014, 37, 2682–2687. [Google Scholar] [CrossRef] [PubMed]
- Carlos, G.; Comiran, E.; Herbstrith, M.; Limberger, R.; Bergold, A.; Froehlich, P. Development, validation and comparison of two stability-indicating RP-LC methods using charged aerosol and UV detectors for analysis of lisdexamfetamine dimesylate in capsules. Arab. J. Chem. 2016, 9, S1905–S1914. [Google Scholar] [CrossRef]
- Guideline, I.H.T. ICH Harmonised Tripartite Guideline: Impurities in new drug substances Q3A(R2). In Proceedings of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 25 October 2006. [Google Scholar]
- Yang, B.; Zhang, Y.J.; Zhang, S.S. Amidation of amines with eaters catalyzed by candida Antarctica lipase (CAL). Cheminform 2010, 36, 1312–1316. [Google Scholar]
- Routaboul, L.; Vanthuyne, N.; Gastaldi, S.; Gil, G.; Bertrand, M. Highly efficient photochemically induced thiyl radical-mediated racemization of aliphatic amines at 30 °C. J. Org. Chem. 2008, 73, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Goudriaan, P.E.; Kaiser, J.; Ibrahim, H.; Verspui, G.A.; Cox, D.P. Process for the preparation of lisdexamfetamine and related derivatives. U.S. Patent 20160376618A1, 29 December 2016. [Google Scholar]
- Enders, D.; Haertwig, A.; Raabe, G.; Runsink, J. Diastereo- and enantio-selective synthesis of vicinal amino alcohols by oxa-michael addition of N-formylnorephedrine to nitro alkenes. Eur. J. Org. Chem. 1998, 1771–1792. [Google Scholar] [CrossRef]
- Olah, G.A.; Fung, A.P.; Keumi, T. Oxyfunctionalization of hydrocarbons, hydroxylation of benzene and alkylbenzenes with hydrogen peroxide in hydrogen fluoride/boron trifluoride. J. Org. Chem. 1981, 46, 4305–4306. [Google Scholar] [CrossRef]
- Guideline, I.H.T. Validation of analytical procedures: text and methodology. In Proceedings of the International Conference on Harmonization, Geneva, Switzerland, November 2005; pp. 11–12. [Google Scholar]
- Guideline, I.H.T. Stability testing of new drug substances and products. Curr. Step 2003, 4, 1–24. [Google Scholar]
Sample Availability: Samples of the impurities A–M are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RRT | HRMS | Structure | Source | |
|---|---|---|---|---|---|
| [M + H]+ | Chemical Formula | ||||
| LDX | 1.00 | 264.2076 | C15H26N3O |  | Target compound |
| Imp-A | 1.17 | 136.1121 | C9H13N |  | Process and alkaline degradation |
| Imp-B | 1.12 | 264.2071 | C15H26N3O |  | Process and alkaline degradation |
| Imp-C | 1.12 | 264.2069 | C15H26N3O |  | Process and alkaline degradation |
| Imp-D | 0.93 | 392.3205 | C21H38N5O2 |  | Process |
| Imp-E | 0.89 | 392.3207 | C21H38N5O2 |  | Process |
| Imp-F | 2.38 | 364.2595 | C20H34N3O3 |  | Process |
| Imp-G | 2.25 | 364.2590 | C20H34N3O3 |  | Process |
| Imp-H | 0.72 | 520.3975 | C27H50N7O3 |  | Process |
| Imp-J | 3.08 | 464.3118 | C25H41N3O5 |  | Process |
| Imp-K | 2.15 | 186.0889 (M + Na+) | C10H13NO |  | Process |
| Imp-L | 0.82 | 250.1908 | C14H24N3O |  | Process |
| Imp-M | 0.86 | 280.2020 | C15H26N3O2 |  | Oxidative degradation |
| Position | LDX | Imp-B | Imp-C | Imp-D | Imp-E | Imp-F | Imp-G | Imp-H | Imp-L | Imp-M |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3.66–3.63 (t,1H) | 3.67–3.63 (t,1H) | 3.67–3.63 (t,1H) | 4.22–4.18 (t,1H) | 3.63–3.61 (t,1H) | 3.10–3.07 (t,1H) | 3.99–3.95 (t,1H) | 4.20–4.12 (dt,1H) | 4.93–4.88 (m,1H) | 3.65–3.63 (t,1H) |
| 3 | 1.48–1.40 (m (c),4H) | 1.76–1.67 (dd (d),2H | 1.76–1.68 (dd,2H) | 1.79–1.69 (m,2H) | 1.75–1.68 (m,2H) | 1.47,1.30 (m,2H) | 1.54–1.46 (m,2H) | 1.49–1.43 (m,2H) | 1.53–1.46 (m,2H) | 1.54–1.46 (dd,2H), |
| 4 | 1.03–0.92 (m,2H) | 1.35–1.26 (m,2H) | 1.35–1.26 (m,2H) | 1.39–1.31 (m,2H) | 1.38–1.27 (m,4H) | 1.24–1.17 (m,2H) | 1.28–1.23 (dd,2H) | 1.13–1.11 (d,2H) | 1.25–1.14 (m,2H) | 1.08–0.86 (m,2H). |
| 5 | 1.48–1.44 (m,4H) | 1.59–1.47 (m,2H) | 1.59–1.47 (m,2H) | 1.63–1.48 (m,6H) | 1.49–1.45 (m,2H) | 1.34–1.26 (m,2H) | 1.43–1.34 (m,2H) | 1.38–1.34 (m,6H) | 1.69–1.66 (m,2H) | 1.44–1.39 (m,2H), |
| 6 | 2.78–2.73 (m,2H) | 2.78–2.71 (m,2H) | 2.78–2.70 (m,2H) | 2.82–2.76 (m,4H) | 3.03–2.98 (m,2H) | 2.89–2.84 (dd,2H) | 2.69–2.61 (m,2H) | 3.06–3.01 (m,2H) | 2.67–2.61 (dd,2H) | 2.68–2.61 (m,2H), |
| 7 | 4.16–4.04 (m,1H) | 3.97–3.93 (m,1H) | 3.97–3.93 (m,1H) | 3.97–3.92 (m,1H) | 4.15–4.07 (m,1H) | 3.93–4.03 (m,1H) | 4.32–4.32 (m,1H) | 4.02–3.95 (m,1H) | – | 4.20–4.09 (m,1H), |
| 8 | 1.13 (d (b),3H) | 1.03 (d,3H) | 1.02 (d,3H) | 1.01 (d,3H) | 1.14 (d,3H) | 1.03 (d,3H) | 1.15 (d,3H) | 1.07 (d,3H) | 1.38 (d,3H) | 1.11–1.10 (d,3H), |
| 9 | 2.65,2.75 (dd,2H) | 2.62,2.82 (dd,2H) | 2.62,2.82 (dd,2H) | 2.62,2.79 (dd,2H) | 2.68,2.76 (dd,2H) | 2.77,2.63 (dd,2H) | 2.84–2.71 (dd,2H) | 2.69–2.67 (d,2H) | 3.79 (t,1H) | 2.72,2.56 (dd,2H) |
| 11,13 | 7.24–7.18 (m,3H) | 7.23–7.17 (m,3H) | 7.23–7.16 (m,3H) | 7.23–7.17 (m,3H) | 7.23–7.18 (m,3H) | 7.20–7.17 (m,3H) | 7.24–7.18 (m,3H) | 7.20–7.19 (m,3H) | 7.34–7.28 (m,4H) | 7.04(d,1H), 7.01(t,H) |
| 12,14 | 7.31–7.26 (m,2H) | 7.32–7.26 (m,2H) | 7.32–7.25 (m,2H) | 7.31–7.26 (m,2H) | 7.30–7.26 (m,2H) | 7.29–7.26 (m,2H) | 7.31–7.28 (m,2H) | 7.30–7.23 (m,2H) | 7.34–7.28 (m,4H) | 6.69(d,1H), 6.78(t,H) |
| 15 | 7.24–7.18 (m,3H) | 7.23–7.17 (m,3H) | 7.23–7.16 (m,3H) | 7.23–7.17 (m,3H) | 7.23–7.18 (m,3H) | 7.20–7.17 (m,3H) | 7.24–7.18 (m,3H) | 7.20–7.19 (m,3H) | 7.34–7.28 (m,4H) | –– |
| 16 | 8.38–8.32 (d,1H) | 8.43–8.37 (d,1H) | 8.43–8.37 (d,1H) | 8.01–7.99 (d,1H) | 8.43–8.40 (t,1H) | 7.80–7.77 (d,1H) | 5.15 (d,1H) | 7.99–7.97 (d,1H) | 8.95–8.93 (d,1H) | 8.27 (d,1H) |
| 17 | 8.15–7.56 (s,6H) | 8.20–8.10 (s,3H) | 8.20–8.10 (s,3H) | 8.65–8.63 (d,1H) | 8.10–8.09 (d,3H) | – | 6.16 (s,1H) | 8.43–8.42 (d,2H) | 8.18–8.13 (m,3H) | 8.08 (s,2H) |
| 18 | 8.15–7.56 (s (a),6H) | 7.82–7.72 (s,3H) | 7.82–7.72 (s,3H) | 7.78 (s,3H) | 8.33–8.30 (d,1H) | 6.75–6.73 (t,1H) | – | 8.43–8.42 (d,2H) | 7.78 (s,3H) | 7.86 (s,2H) |
| 19 | 2.39 (s,6H) | 2.42 (s,6H) | 2.42 (s,6H) | 2.39 (s,9H) | 2.38 (s,9H) | – | – | 2.45 (s,12H) | 2.45 (s,6H) | 9.43 (s,1H) |
| 21,29 | 3.88–3.83 (t,1H) | 3.72–3.69 (t,1H) | 1.37 (s,9H) | 1.45 (s,9H) | 3.82–3.73 (t,1H) | |||||
| 22,31 | 1.63–1.48 (m,6H) | 1.60–1.52 (dd,2H) | 1.37 (s,9H) | 1.45 (s,9H) | 1.72–1.70 (d,4H) | |||||
| 23,32 | 1.25 (d,2H) | 1.04,0.93 (m,2H). | 1.37 (s,9H) | 1.45 (s,9H) | 1.38–1.34 (m,6H) | |||||
| 24,33 | 1.63–1.48 (m,6H) | 1.38–1.27 (m,4H) | 1.62–1.53 (dd,4H) | |||||||
| 25,34 | 2.82–2.76 (m,4H) | 2.79–2.74 (dd,2H) | 2.84–2.71 (dd,4H) | |||||||
| 26,(35) | 7.78 (s,3H) | 7.70 (s,3H) | 7.75 (s,6H) | |||||||
| 27,(30) | 8.14 (s,3H) | 7.99 (d,3H) | 8.09 (s,6H) |
| Position | LDX | Imp–D | Imp–E | Imp–F | Imp–G | Imp–H | Imp–L | Imp–M |
|---|---|---|---|---|---|---|---|---|
| δC DEPT | δC DEPT | δC DEPT | δC DEPT | δC DEPT | δC DEPT | δC DEPT | δC DEPT | |
| 1 | 167.41– | 168.88– | 167.87– | 174.25– | 174.17– | 170.61– | 167.96– | 167.69– |
| 2 | 52.02 CH | 53.35 CH | 52.61 CH | 54.98 CH | 54.97 CH | 53.34 CH | 52.33 CH | 52.47 CH |
| 3 | 30.38 CH2 | 30.89 CH2 | 30.88 CH2 | 35.00 CH2 | 34.55 CH2 | 28.97 CH2 | 30.78 CH | 30.88 CH2 |
| 4 | 20.73 CH2 | 21.58 CH2 | 21.57 CH2 | 22.88 CH2 | 22.72 CH2 | 22.96 CH2 | 22.74 CH2 | 21.27 CH2 |
| 5 | 26.38 CH2 | 26.72 CH2 | 26.78 CH2 | 29.89 CH2 | 29.83 CH2 | 26.72 CH2 | 26.66 CH2 | 27.01 CH2 |
| 6 | 38.53 CH2 | 38.98 CH2 | 38.90 CH2 | 40.25 CH2 | 40.09 CH2 | 32.14 CH2 | 38.95 CH2 | 38.93 CH2 |
| 7 | 46.45 CH | 46.66 CH | 46.84 CH | 46.00 CH | 45.83 CH | 46.54 CH | – | 45.31 CH |
| 8 | 20.83 CH3 | 20.17 CH3 | 21.22 CH3 | 20.54 CH3 | 20.23 CH3 | 21.02 CH3 | 21.46 CH3 | 21.47 CH3 |
| 9 | 41.77 CH2 | 41.99 CH2 | 42.17 CH2 | 42.29 CH2 | 42.62 CH2 | 42.19 CH2 | 48.91 CH | 36.97 CH2 |
| 10 | 138.99 C | 139.43 C | 139.34 C | 139.43 C | 138.25 C | 139.51 C | 144.61 C | 125.37 C |
| 11 | 128.13 CH | 128.62 CH | 128.50 CH | 128.55 CH | 128.30 CH | 128.46 CH | 127.32 CH | 131.29 CH |
| 12 | 129.20 CH | 129.61 CH | 129.57 CH | 129.62 CH | 129.38 CH | 129.59 CH | 128.78 CH | 118.94 CH |
| 13 | 126.18 CH | 128.62 CH | 126.56 CH | 126.45 CH | 126.37 CH | 126.47 CH | 126.37 CH | 127.73 CH |
| 14 | 129.20 CH | 129.61 CH | 129.57 CH | 129.62 CH | 129.38 CH | 129.55 CH | 128.78 CH | 115.28 CH |
| 15 | 128.13 CH | 128.62 CH | 128.50 CH | 128.55 CH | 128.30 CH | 128.49 CH | 127.32 CH | 155.85 C |
| 19 | 39.80 CH3 | 40.07 CH3 | 40.13 CH3 | 156.03– | 156.10– | 40.16 CH3 | 40.11 CH3 | – |
| 20 | 170.61– | 168.67– | 77.73 C | 79.07 C | 168.63– | |||
| 21 | 52.47 CH | 52.53 CH | 28.74 CH3 | 28.42 CH3 | 52.53 CH | |||
| 22 | 31.98 CH2 | 31.07 CH2 | 28.74 CH3 | 28.42 CH3 | 30.90 CH2 | |||
| 23 | 22.63 CH2 | 21.64 CH2 | 28.74 CH3 | 28.42 CH3 | 21.65 CH2 | |||
| 24,33 | 26.86 CH2 | 28.83 CH2 | 26.78 CH2 | |||||
| 25,34 | 39.10 CH2 | 38.96 CH2 | 38.97 CH2 | |||||
| 28 | 168.69– | |||||||
| 29 | 52.25 CH | |||||||
| 31 | 30.84 CH2 | |||||||
| 32 | 21.33 CH2 |
| Compound | System Suitability | Linearity | Sensitivity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RRT a | PC b | SF c | R d | Range (μg/mL) | R e | Slope | Intercept | CF f | LOD g (μg/mL) | LOQ h (μg/mL) | |
| LDX | - | 19755 | 1.28 | 6.09 | 0.5100–20.4000 | 0.9999 | 0.1461 | −0.0055 | - | 0.3060 | 0.5100 |
| Imp-A | 1.17 | 105353 | 1.15 | 66.28 | 0.5025–20.1000 | 1.0000 | 0.2271 | −0.0276 | 0.61 | 0.3028 | 0.5025 |
| Imp-B | 1.12 | 121354 | 1.02 | 3.50 | 0.5110–20.4400 | 1.0000 | 0.1262 | −0.0141 | 1.10 | 0.3010 | 0.5105 |
| Imp-C | 1.12 | 125659 | 1.03 | 3.50 | 0.5047–20.1900 | 1.0000 | 0.1119 | −0.0010 | 1.31 | 0.3026 | 0.5105 |
| −Imp-D | 0.93 | 98972 | 1.09 | 3.33 | 0.5160–20.6400 | 1.0000 | 0.1112 | −0.0079 | 1.04 | 0.3035 | 0.5070 |
| Imp-E | 0.89 | 118365 | 1.14 | 3.19 | 0.5135–20.5400 | 1.0000 | 0.0836 | −0.0013 | 1.66 | 0.3041 | 0.5051 |
| Imp-F | 2.38 | 749255 | 1.63 | 12.44 | 0.5022–20.0900 | 1.0000 | 0.1700 | −0.0061 | 0.86 | 0.3013 | 0.5023 |
| Imp-G | 2.25 | 864920 | 1.16 | 80.95 | 0.5070–20.2800 | 1.0000 | 0.1641 | −0.0119 | 0.89 | 0.3042 | 0.5070 |
| Imp-H | 0.725 | 46840 | 1.28 | 8.53 | 0.5028–20.1900 | 1.0000 | 0.1524 | −0.0085 | 0.92 | 0.3028 | 0.5062 |
| Imp-J | 3.08 | 28465 | 1.14 | - | 0.5080–20.3200 | 0.9998 | 0.1269 | 0.0110 | 1.15 | 0.3028 | 0.5041 |
| Imp-K | 2.15 | 322224 | 1.12 | 7.00 | 0.5105–20.256 | 0.9999 | 0.1389 | 0.0105 | 1.32 | 0.3036 | 0.5075 |
| Imp-L | 0.83 | 90222 | 1.02 | 8.53 | 0.5180–20.7200 | 1.0000 | 0.1159 | −0.0056 | 1.19 | 0.3041 | 0.5180 |
| Imp-M | 0.86 | 114119 | 1.17 | 3.40 | 0.5240–20.9592 | 1.0000 | 0.1614 | −0.0011 | 0.91 | 0.3060 | 0.5140 |
| Impurity | 0.05%–1% | 0.05%–2% | 0.05%–3% | 0.10%–1% | 0.10%–2% | 0.10%–3% | 0.15%–1% | 0.15%–2% | 0.15%–3% | Mean | RSD (n = 9) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Imp-A | 91.8 | 90.2 | 92.6 | 89.6 | 92.7 | 94.1 | 92.0 | 92.5 | 94.1 | 92.0 | 1.83 |
| Imp-B | 102.6 | 105.1 | 100.2 | 100.9 | 91.3 | 97.3 | 92.6 | 90.8 | 90.5 | 96.8 | 5.82 |
| Imp-C | 92.8 | 90.4 | 91.2 | 100.6 | 98.7 | 99.5 | 97.4 | 102.4 | 98.5 | 96.8 | 4.44 |
| Imp-D | 99.7 | 96.3 | 95.8 | 97.7 | 96.3 | 97.6 | 94.1 | 97.0 | 93.1 | 96.4 | 2.04 |
| Imp-E | 99.1 | 101.0 | 102.7 | 101.6 | 96.5 | 95.7 | 103.5 | 103.4 | 102.6 | 100.6 | 2.91 |
| Imp-F | 103.9 | 96.6 | 97.1 | 103.3 | 105.6 | 99.3 | 98.8 | 100.3 | 98.1 | 100.3 | 3.19 |
| Imp-G | 101.5 | 102.4 | 100.3 | 103.3 | 96.6 | 102.6 | 95.8 | 100.9 | 100.5 | 100.4 | 2.59 |
| Imp-H | 99.5 | 98.7 | 102.5 | 101.8 | 98.9 | 103.5 | 96.9 | 99.5 | 102.1 | 100.3 | 2.16 |
| Imp-J | 92.7 | 95.4 | 93.9 | 95.0 | 96.2 | 93.5 | 91.0 | 99.4 | 94.0 | 94.5 | 2.50 |
| Imp-K | 99.7 | 103.5 | 101.6 | 98.7 | 99.5 | 96.4 | 105.1 | 103.6 | 104.2 | 101.3 | 2.90 |
| Imp-L | 91.4 | 90.6 | 101.2 | 95.7 | 95.5 | 94.8 | 96.7 | 90.0 | 94.9 | 94.5 | 3.69 |
| Imp-M | 97.8 | 98.5 | 93.2 | 95.1 | 94.4 | 97.6 | 91.3 | 95.5 | 92.7 | 95.1 | 2.62 |
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | RSD (n = 6) |
|---|---|---|---|---|---|---|---|
| C (mg/mL) | |||||||
| 1.0080 | 1.0225 | 1.0180 | 1.0290 | 1.0130 | 1.0095 | ||
| Imp-A | 0.10 | 0.10 | 0.09 | 0.10 | 0.10 | 0.10 | 4.22 |
| Imp-B | 0.10 | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 | 2.19 |
| Imp-C | 0.12 | 0.12 | 0.12 | 0.12 | 0.12 | 0.13 | 3.24 |
| Imp-D | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.08 | 1.67 |
| Imp-E | 0.10 | 0.10 | 0.10 | 0.11 | 0.10 | 0.10 | 1.83 |
| Imp-F | 0.12 | 0.12 | 0.12 | 0.12 | 0.11 | 0.11 | 1.20 |
| Imp-G | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 1.30 |
| Imp-H | 0.10 | 0.11 | 0.11 | 0.10 | 0.10 | 0.10 | 1.51 |
| Imp-J | 0.11 | 0.11 | 0.10 | 0.10 | 0.10 | 0.10 | 2.14 |
| Imp-K | 0.11 | 0.11 | 0.11 | 0.10 | 0.10 | 0.11 | 1.63 |
| Imp-L | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 | 1.32 |
| Imp-M | 0.12 | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 | 1.87 |
| RRT = 1.32 | 0.01 | 0.01 | 0.01 | 0.02 | 0.02 | 0.01 | 2.85 |
| Compound | Column Temperature | Flow Rate | UV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 27 °C | 30 °C | 33 °C | 0.9 mL/min | 1.0 mL/min | 1.1 mL/min | 213 nm | 215 nm | 217 nm | |
| Imp-A | 0.10 | 0.10 | 0.09 | 0.10 | 0.10 | 0.09 | 0.11 | 0.10 | 0.10 |
| Imp-B | 0.13 | 0.12 | 0.13 | 0.12 | 0.13 | 0.12 | 0.12 | 0.13 | 0.13 |
| Imp-C | 0.10 | 0.11 | 0.10 | 0.13 | 0.12 | 0.13 | 0.11 | 0.10 | 0.12 |
| Imp-D | 0.09 | 0.09 | 0.08 | 0.09 | 0.09 | 0.10 | 0.09 | 0.09 | 0.08 |
| Imp-E | 0.11 | 0.10 | 0.11 | 0.11 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 |
| Imp-F | 0.10 | 0.09 | 0.09 | 0.09 | 0.10 | 0.10 | 0.09 | 0.10 | 0.09 |
| Imp-G | 0.09 | 0.09 | 0.09 | 0.10 | 0.09 | 0.10 | 0.09 | 0.09 | 0.10 |
| Imp-H | 0.09 | 0.09 | 0.08 | 0.09 | 0.09 | 0.08 | 0.09 | 0.09 | 0.09 |
| Imp-J | 0.08 | 0.09 | 0.08 | 0.07 | 0.08 | 0.07 | 0.07 | 0.07 | 0.07 |
| Imp-K | 0.10 | 0.10 | 0.11 | 0.10 | 0.10 | 0.11 | 0.11 | 0.10 | 0.10 |
| Imp-L | 0.11 | 0.12 | 0.12 | 0.11 | 0.11 | 0.12 | 0.12 | 0.12 | 0.12 |
| Imp-M | 0.08 | 0.08 | 0.09 | 0.08 | 0.09 | 0.08 | 0.09 | 0.09 | 0.09 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; Meng, L.; Zhao, C.; Zhang, T.; Qiu, P.; Zhang, F. Identification, Characterization and Quantification of Process-Related and Degradation Impurities in Lisdexamfetamine Dimesylate: Identifiction of Two New Compounds. Molecules 2018, 23, 3125. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23123125
Gao S, Meng L, Zhao C, Zhang T, Qiu P, Zhang F. Identification, Characterization and Quantification of Process-Related and Degradation Impurities in Lisdexamfetamine Dimesylate: Identifiction of Two New Compounds. Molecules. 2018; 23(12):3125. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23123125
Chicago/Turabian StyleGao, Shenghua, Lili Meng, Chunjie Zhao, Tao Zhang, Pengcheng Qiu, and Fuli Zhang. 2018. "Identification, Characterization and Quantification of Process-Related and Degradation Impurities in Lisdexamfetamine Dimesylate: Identifiction of Two New Compounds" Molecules 23, no. 12: 3125. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23123125