Applications of Ruthenium Complexes Covalently Linked to Nucleic Acid Derivatives


1
Laboratory for Inorganic Chemical Biology, Chimie ParisTech, PSL University, F-75005 Paris, France
2
Laboratory for Bioorganic Chemistry of Nucleic Acids, Department of Structural Biology and Chemistry, Institute Pasteur, CNRS UMR3523, 28, rue du Docteur Roux, 75724 Paris CEDEX 15, France
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(7), 1515; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23071515
Submission received: 1 June 2018
/
Revised: 19 June 2018
/
Accepted: 20 June 2018
/
Published: 22 June 2018
(This article belongs to the Special Issue Oligonucleotides Application to Nano- and Biotechnology (DNA Origami, DNA Machine))
Abstract
:Oligonucleotides are biopolymers that can be easily modified at various locations. Thereby, the attachment of metal complexes to nucleic acid derivatives has emerged as a common pathway to improve the understanding of biological processes or to steer oligonucleotides towards novel applications such as electron transfer or the construction of nanomaterials. Among the different metal complexes coupled to oligonucleotides, ruthenium complexes, have been extensively studied due to their remarkable properties. The resulting DNA-ruthenium bioconjugates have already demonstrated their potency in numerous applications. Consequently, this review focuses on the recent synthetic methods developed for the preparation of ruthenium complexes covalently linked to oligonucleotides. In addition, the usefulness of such conjugates will be highlighted and their applications from nanotechnologies to therapeutic purposes will be discussed.
1. Introduction
The fantastic properties of DNA have made it a tool of choice for numerous applications. Due to its high fidelity of hybridization, selective, and programmable self-assembly properties and ease of synthesis, DNA has indeed emerged as a promising template for the construction of nanostructures [1,2,3,4,5,6]. Its double helical nature, consisting of a rigid, aromatic column of stacked base pairs, allows charge and electron transfer through DNA duplexes [7,8,9]. As carriers of genetic information and blueprints for protein synthesis, DNA and RNA, respectively, have also risen as targets for the inhibition of gene expression through the use of therapeutic antigene and antisense oligonucleotides [10,11,12,13,14] as well as catalytic nucleic acids [15,16,17,18,19,20,21].
Thereby, synthetic ways to chemically modify nucleic acids analogues have been developed over the past decades in order to obtain a better understanding of the role of DNA in various biological mechanisms or to add unexpected properties to natural processes [22]. For instance, oligonucleotides containing a peptide backbone (peptide nucleic acids, PNA) [23,24,25,26,27], unnatural bases [19,28,29,30,31], or altered phosphate groups [32,33] have been reported.
Similarly, a variety of groups have been attached at the 5′ or 3′ extremities of oligonucleotides, such as fluorophores [34], peptides [35], or metal complexes [36,37,38]. Among the latter, ruthenium complexes have received an increasing interest due to their remarkable redox and photophysical properties, which can easily be modulated by the nature of their ligands. The potential of these metal complexes is widely recognised and they have seen use in electron transfer studies [39,40], catalysis [41], photoprobes [42], or as optical devices [43]. Other chemical properties such as the rate of ligand exchange, the ability of ruthenium to bind biological molecules, the intercalative properties of its ligands and its capacity to interact with DNA in a variety of ways have made ruthenium complexes excellent candidates for cancer treatment and medical diagnosis [44,45,46]. Interestingly, several examples of correlation between different non covalent DNA binding modes of ruthenium complexes and their cytotoxic effects have been reviewed [47]. However, among these numerous applications of ruthenium complexes, only a few studies have been focusing on a covalent bonding interaction with nucleic acids.
In this context, this review will focus only on the elaboration of ruthenium complexes covalently linked to DNA oligonucleotides. We will not discuss the coupling of such complexes with PNA or other DNA analogues [48,49,50,51,52,53,54]. The different synthetic pathways to afford modified oligonucleotides will be discussed and a chronological overview of their most prominent applications in therapy, diagnostics, and as tools for chemical biology will be highlighted.
2. Electron Transfer Enhancement by Ruthenium-Nucleic Acid Complexes
The structural stability of double helical DNA is primarily due π-electron stacking interactions between adjacent base pairs. It is this π-system that enables electron transfer (ET) throughout DNA [55,56,57,58]. Electrons migrate across charge carrying bases in a multistep hopping process, where the transfer rate is dependent on base separation [59]. This primarily occurs through G:C pairs (G-hopping) but when the G-G distance exceeds four bases, A-hopping can be observed (over A:T sites) [60,61,62,63]. Long distance electron transfer is vital in biological processes such as DNA-binding or DNA damage and repair and it can also influence mutations [57,59,64]. Partially due to this, long-range ET through DNA has received considerable interest over the last few decades [64,65,66,67].
Extensive research on ruthenium-oligonucleotide conjugates has been carried out by the Barton group. It was first shown in 1992 that such complexes could function as sequence-specific molecular light switches for single-stranded DNA when [Ru(phen’)2dppz]2+ was tethered, via an amide bond, at the 5′-terminus of a 15-mer functionalised with hexylamine. This yielded a Ru-oligonucleotide complex (Ru-ODN) which only showed intense luminescence when in the presence of a complementary strand (Figure 1). This luminescence was positively correlated with intercalation of the complex into dsDNA [68,69]. Enhancement of the luminescence from the Ru-ODN was the result of the lowest excited state of [Ru(phen’)2dppz]2+, a metal-to-ligand charge transfer (MLCT) transition centred on the intercalating dppz-ligand [70,71,72]. No detectable luminescence was observed from the lone Ru-ODN or upon addition of a non-complementary strand due to the lack of phenazine ring protection from water [68,69]. Such characteristics are important for the sequence-specific targeting of single-stranded DNA.
Oligonucleotides are well-defined molecular templates which can be further modified with spectroscopically detectable and photochemically active metal-centres at specific locations [73]. These metal-oligonucleotide constructs are useful tools in understanding the mechanism of long-range ET. In this context, Barton et al. investigated in 1993 the long-range electron-donor abilities of a [Ru(phen’)2dppz]2+-oligonucleotide conjugate. To do so, [Ru(phen’)2dppz]2+ was tethered at the 5′-terminus of 15-mer functionalised with hexylamine via amide bond formation. Electron-accepting-[Rh(phi)2(phen’)]3+-oligonucleotides were also prepared [74]. Interestingly, the intense luminescence seen from the ruthenium-bearing 15-mer disappeared when the complementary strand was modified with [Rh(phi)2(phen’)]3+ (Table 1). Results showed that complete luminescence quenching is only observed when the acceptor is covalently bound within the same duplex as the donor, with the electron-transfer mechanisms occurring over distances as great as 40 Å. The lowest excited state of the rhodium complex, which results from a ligand-to-metal charge-transfer (LMCT), allows this direct photoinduced ET between the two metal centres in the same duplex at a rate of at least 3 × 109 s−1 which is comparable to the rate of charge transfer over long distances by the hopping mechanism (1010 s−1) [75]. Moreover, this is several magnitudes higher than the rates observed in natural DNA duplexes, which typically range from 104–106 s−1 [76]. The DNA helix is known to enhance ET processes, specifically ET occurring through π-stacks [77,78,79,80]. To confirm the role that ligand intercalation plays in this quenching, smaller oligonucleotides (5′-CGATTAGC-3′) were metallated with the complexes [Ru(phen)2phen’]2+ and [Rh(phen)2phen’]3+ [74]: it is known that the phenanthroline ligand has intercalation depths three times smaller than the dppz ligand [81,82]. Luminescence lifetime experiments yielded similar values independent of the presence of covalent binding between the ruthenium complex and the 8-mer (Table 2). Thus, the lack of intercalation capabilities implied the importance of this non-covalent interaction and the effect of metal-metal distance in the complete quenching of luminescence is demonstrated.
In 1995, Meade and Kayyem used ruthenium complexes as both donors and acceptors for ET through DNA. Two sets of complementary strands (8- and 14-mers) of ruthenium-containing oligonucleotides were synthesised to test the nucleic acid sequence dependency of the electron-transfer process (Figure 2). The acceptor ([Ru(bpy)2(im)]3+) and donor ([Ru(NH3)4(py)]2+) were covalently attached at the 2’ position of a 5′-terminal ribose following automated solid-phase DNA synthesis. Following this, the two ruthenium-modified oligonucleotides were annealed to yield the duplex DNA with a covalently bonded donor and acceptor. The rate constant for ET between metal centres (Ru-Ru distance = 21 Å) was found to be around 2.5 × 106 s−1, which is comparable to that of His39-modified cytochrome c (Fe-Ru distance = 20.3 Å), one of the most efficient protein systems known at this time [73].
Further long-range ET studies were completed by the group of Barton in 1997. A flash-quench strategy was employed to demonstrate ET through the DNA double helix [83]. Flash-quenching methods are popular for the study of electron-transfer reactions as they can be used to monitor the oxidation of guanine, the most readily-oxidised base [60,84,85,86]. In this experiment the DNA-bound intercalator [Ru(phen)(bpy’)(Me2dppz)]2+ was photolysed to enable the ET to an externally bound Ru(III) oxidative quencher, resulting in guanine oxidation in 5′-GG’-3′ sequences. Ru(II)-DNA was prepared by tethering this ruthenium complex to an oligonucleotide duplex containing 5′-GC-3′ or 5-GG-3′, allowing for oxidative damage comparisons [83]. An earlier part of this study showed that positions containing adjacent guanines, such as GG or GGG, are particularly susceptible to oxidation due to the electron-donor abilities of neighbouring guanines. The increased numbers of adjacent guanines was found to promote the oxidation of distant bases [80,83]. In the presence of the externally bound Ru(III), which was generated from Ru(NH3)63+, selective G-base damage at the 5′-G of a 5′-GG-3′ doublet was observed. In the [Ru(phen)(bpy’)(Me2dppz)]2+-oligonucleotide containing 5′-GC-3′, there was no preference and all guanine bases experienced equal oxidative damage. For the experiments in the absence of a 5′-GG-3′ guanine doublet, the lack of sequence specificity indicated equal energy sites across all G bases [83], providing further evidence of ET occurring through DNA base pair stacks [80,83].
It was also shown by the Barton group that long-range guanine oxidation was altered by the presence of a single base mismatch. Flash-quench experiments were carried out using [Ru(phen)(bpy’)(Me2dppz)]2+-DNA duplexes containing G·A or G·T mismatches approximately five base-pairs 3′-downstream from the Ru(II) binding site, as well as a single 5′-GG-3′ doublet around 11 base-pairs away. In the Ru-DNA duplex with a complete base-matched sequence, oxidative guanine damage occurred selectively at the 5′-GG-3′ doublet (lane 3–5 of Figure 3). In the sequence with the G·A mismatch, 16% guanine damage at the 5′-GG-3′ as well as damage of the mispaired guanine was seen (lanes 7–9 of Figure 3). 10% guanine damage at the 5′-GG-3′ site was recorded in the sequence with the more disruptive G·T mismatch, but only minor oxidative damage occurred to the mispaired guanine (lanes 11–13 of Figure 3). Among other things, base mismatches in DNA duplexes lead to localised helix disruption and the structural similarity of the G·A mismatch with the G·C Watson-Crick base pairing can be used to explain the observed difference in guanine oxidative damage between the G·A and G·T mispairings [83].
In 2001, Bhattacharya and Barton showcased the possibility of controlling long-range charge transport by manipulation of base-pairs dynamics. [Ru(bpy’)(dppz)(phen)]2+ was covalently bonded to the 5′-terminus of a series of 22-mers by a nine-carbon linker. Each sequence contained two 5′-GG-3′ sites in the complementary strand, between which a single base mismatch was introduced (Figure 4). All mispair-containing sequences were synthesised and the efficiency of electron transport through the base mismatches was determined by comparing the guanine oxidation of the 5′-G at the two 5′-GG-3′ sites: the ratio of distal/proximal damage. The value was found to be different for all base-pairings with the general trend of G·C ≈ G·G ≈ G·T ≈ G·A > A·A > C·C ≈ T·T ≈ C·A ≈ C·T, with the purine-purine mispairs yielding the highest ratios. The distal:proximal guanine oxidation trend was compared with various duplex properties, but the damage ratios correlated most closely with base-pair lifetimes. The lifetimes of the Ru-ODNs containing G·G, T·T, A·A, and C·C pairings were determined by monitoring the imino proton exchange by 1H NMR and comparing with that of the G·C Watson-Crick base pair (τex(G5NH) = 18 ms). The Ru-DNA duplex containing the G·G mismatch had the longest lifetime followed by the A·A, C·C, and T·T. The lifetime of the T·T mispair was four times shorter than that of G·G, yielding the lowest distal:proximal oxidation damage [87].
Hurley and Tor first reported the site-specific incorporation of metal complexes during the solid-phase synthesis oligonucleotides in 1998. This overcame restrictions associated with terminal-functionalization of oligonucleotides and removed the need of exposure to reactive metal precursors often associated with other approaches. In their approach, high-yield solid-phase phosphoramidite chemistry was utilised: the phosphoramidites were prepared by phosphitylation of their corresponding ruthenium and osmium containing protected nucleosides (Scheme 1). These modified 2’-deoxyuridine building blocks could then be used in automated solid-phase DNA synthesis [88].
A similar methodology was also applied to synthesise diasteromerically pure Ru(II)-ODNs. The absolute configuration of the Ru-oligonucleotide conjugates was controlled by using enantiomerically pure nucleoside phosphoramidites. [Ru(bpy)2(3-bromo-1,10-phenanthroline)]2+ was covalently attached to the 5-position of the nucleobase of 2’-deoxyuridine by a Sonogashira cross-coupling reaction, followed by the synthesis of the Ru(II)-nucleosides (Δ-1 and Λ-1) and the respective phosphoramidites (Δ-2 and Λ-2) (Figure 5) [89,90,91]. The CD spectra of the nucleosides showed that both configurations emitted at the same wavelength (632 nm) upon MLCT-band excitation and the lifetime of these excited-states was also the same (1.07 μs). It is not until incorporation into the respective oligonucleotides that the diastereomeric effects are visible. Specifically, the CD spectrum of Λ-Ru(II)-ODN is dominated by a large band at 300 nm which is the result of the polypyridine ligand and heterocyclic base transitions overlapping. There was also a minimal difference in thermal stability between the two isomers [91].
In a further study, the donor/acceptor interactions between the same ethynyl linked Ru(II)-nucleoside and its osmium analogue were investigated. [Ru(bpy)2(3-ethynyl-1,10-phenanthroline)]2+ was fixed at the 5′ terminus of a 19-mer and the position of the acceptor, [Os(bpy)2(3-ethynyl-1,10-phenanthroline)]2+, on the complementary strand was incrementally distanced by three base pairs, from 3 (16 Å) to 18 (60 Å) base pairs apart (Table 3 and Table 4). This was achieved using the corresponding epimeric phosphoramidites for solid-phase DNA synthesis. In quenching studies similar to those previously described in this review, a positive correlation between Os(II)-Ru(II) distance and average Ru(II) excited-state lifetime was seen.
The principal aim of this study was to determine the primary mechanism for the ET between these donor/acceptor metal centres. To do so, the ethynyl linked Ru(II)-donor was replaced with a saturated two-carbon linked complex to allow for increased flexibility (Figure 6). The same trend of distance dependent excited state lifetime for the Ru(II) donor was found to be true as for the more rigid nucleoside analogues. The energy transfer between the donor and acceptor was monitored using both steady-state and time-resolved techniques. This data was analysed in coherence with the Förster dipole-dipole and the Dexter electron exchange mechanisms. A significant discrepancy was observed between experimentally determined quenching data and theoretical Förster curves when the more rigid Λ-2 and Δ-2 building blocks were used. The rigidity of the linker arms was believed to affect the geometric factor κ2 used in the calculation of the critical Förster radius Ro in an unpredictable manner. When the more flexible linker was used, κ2 was closer to the typical value ascribed to this parameter and the correlation between calculated and experimental Förster curves was much higher. Therefore, the primary mechanism of the energy transfer was determined to be mainly based on Förster dipole-dipole interactions since efficiency decreased with increasing donor/acceptor distance. However, a minor contribution of the Dexter electron exchange mechanism, which operates at shorter distances, could not be entirely ruled out due to the variation from idealised Förster behaviour [92].
Modified nucleoside triphosphates can be used to incorporate functional groups and particularly metal complexes into DNA enzymatically (vide infra) [93,94]. This approach was used by Weizman and Tor in 2002 to develop metal-containing redox tags. Complexes of the general formula [Ru(bpyR2)L2]2+ were used due to the kinetic inertness, chemical stability, and ligand-based tunability of electrochemical potentials (E1/2) associated with ruthenium(II) polypyridyl complexes. There were three building blocks used in the synthesis: a substituted 2,4-pentadione or hydroxamic acid with a hydroxamate functionalised linker, a bis-substituted Ru2+ precursor and a modified nucleotide (Scheme 2 and Scheme 3). The Ru(II) complexes were bound by their succinimide ester to 5-propargylamino-dUTP (5-(3”-aminopropynyl)-2’-deoxyuridine-5′-triphosphate) via an amide bond (Scheme 2 and Scheme 3). Analysis of the primer extension reactions with the resulting modified triphosphate dRuTP into DNA clearly demonstrated that the modified triphosphate acted as a good substrate for the Klenow fragment of DNA polymerase I and that the Ru(II)-DNA had the expected lower electrophoretic mobility compared to unmodified strands (Figure 7) [95].
3. Nanomaterials Based on Ruthenium-Oligonucleotide Conjugates
The predictable behaviour of DNA has made it a useful scaffold to organize molecules into high-order 2- and 3-dimensional structures such as stars, disks, cubes, or even boxes [96,97]. In order to further expand the structural diversity accessible to nucleic acids, Sleiman et al. reported in 2001 the first synthesis of a Ru(II) complex linked to a branched oligonucleotide (17, Scheme 4), in which two DNA strands are covalently linked to the complex [98]. A cis-[Ru(bpy)2(imidazole)2]2+ complex is linked through hexyl spacers to two parallel dT10 oligonucleotides. The preparation of this molecule was made by a convergent solid phase strategy [99] described in Scheme 4. First, a stable ruthenium bis-phosphoramidite branching complex was synthesised which then could be appended on a dT10 oligonucleotide obtained by solid-phase synthesis. Due to the presence of two phosphoramidite moieties on the ruthenium complex, the 5′-termini of oligonucleotides needed to be in close proximity to each other on the solid support and therefore a high-density controlled pore glass (CPG) support was used.
The Ru-DNA conjugate 17, as a branched oligonucleotide, has the ability to specifically hybridize complementary single stranded DNA or RNA but could also be associated with analogues of molecule 17 containing mixed DNA sequences to create new DNA nanostructures [100]. Moreover, the presence of a ruthenium complex confers redox and luminescent properties to this DNA structure. In this context, the authors determined the hybridization efficiency of 17 with its complementary DNA dA10 by thermal denaturation experiments. When one equivalent of dA10 was added, the formation of a duplex with one of the complementary strands of the branched oligonucleotide was observed with a melting temperature (Tm value) of 21 °C, which is close to that of a natural Watson-Crick dA10/dT10 duplex (Tm = 22 °C). When two equivalents of dA10 were added, both strands of 17 were successfully hybridised (Tm = 26 °C). Despite these favourable assets, the presence of the two monodentate imidazole ligands along with the flexible hexyls spacers prevented control of the self-assembly process since no nanostructure containing two ruthenium complexes could be obtained.
In 2004, the same group applied their strategy to the elaboration of more rigid ruthenium branched oligonucleotides 18–22 (Figure 8) [101].
These molecules were able to form stable duplexes upon hybridization with their complementary strands, with a slight increase in their melting temperatures compared to the unmodified Watson-Crick duplexes (ΔTm of 1 to 4 °C). In terms of self-assembly properties, when an equimolar solution of 21 with its complementary ruthenium-containing oligonucleotide 22 was heated up to 90 °C, and then cooled down to 4 °C, a polymeric Ru-DNA species was detected. Under milder conditions (4 °C, 12 h), a dimer was created, which could be linear or cyclic (Scheme 5).
In order to characterize the nature of the dimer, an enzymatic digestion experiment was conducted. Mung Bean Nuclease has the ability to degrade selectively oligomeric species that contain single DNA strands. The dimer was found to remain unmodified upon this experiment, showing that the self-assembly of two ruthenium branched oligonucleotides leads to a cyclic nanostructure, addressable by means of light or electrical energy.
At the same period, Stewart and McLaughlin developed ruthenium bis(terpyridine) complexes linked to two identical 20-mer DNA sequences through triethylene glycol linkers (23, 24, Figure 9) [102].
A di(ethylene glycol) linker was added to a terpy ligand and the corresponding Ru(II) complex was synthesised [103]. The corresponding phosphoramidite molecules were then prepared and 23, 24 were synthesised through DNA automated synthesis. Due to the geometrical arrangement of these complexes, they are expected to form linear arrays under hybridisation. The complementary DNA sequences 23 and 24 were chosen so as to create a linear array in which ruthenium complexes are placed at a fixed and regular distance. With a 2:1 ratio or greater of 23:24 (or 24:23), a trimer 23=24=23 (or 24=23=24) was found as a major product. By reducing the number of equivalents, the length of the linear arrays was found to increase and products were analysed by non-denaturing PAGE experiments. The longest array was obtained for a 1:1 reaction and the procedure developed could be used to place ruthenium complexes at regular distances in linear arrays.
A few years later, Sartor et al. developed a new methodology to construct linear nanoassemblies of ruthenium-DNA conjugates in a programmable manner for applications such as symmetrical nanowires [104]. First, the synthesis of ruthenium-DNA conjugates is based on the coupling reaction of a Ru(II)(2,2’-bpy)2(4,4’-dicarboxy-2,2’-bpy) complex synthesised as described in [105] with commercially available oligonucleotides bearing an amino hexyl linker at the 5′ position. As shown on Figure 10, 8 mono Ru-DNA (m25–m32) and 8 bis Ru-DNA (b25–b32) molecules were prepared by this methodology. Sequences 25/26, 27/28, and 29/30 are complementary to each other.
With these Ru-DNA building blocks, a variety of linear assemblies could be created by preparing a solution of mono or bis Ru-DNA conjugates with their respective complementary complexes. The hybridization process promoted assemblies with one to seven ruthenium complexes (Figure 11). The distance between two ruthenium complexes could easily be modulated due to the different sizes of the oligonucleotide sequences. Interestingly, hybridisation of Ru-DNA 31b and 32b could be achieved with the use of single complementary DNA strands followed by a complementary mono-Ru-DNA to give a nanoassembly with three ruthenium complexes. This could also be achieved with only mono or bis Ru-DNA to increase the incorporation of ruthenium complexes. 31b was then hybridised with two equivalents of 25m and 27m (incorporation of 5 Ru) and 32b with two equivalents of 25m, 27m, and 29m (incorporation of 7 Ru).
This way of synthesis allows the programmable assembly of complementary Ru-DNA molecules in a linear manner. The number and position of ruthenium complexes incorporated in the DNA backbone can be easily controlled.
This work paved the way to the elaboration of other patterns. For instance, in 2013, DNA three-way junction-ruthenium complexes assemblies were created [106]. As a proof of concept, a ruthenium free three-branched DNA junction was first formed by hybridization of three complementary oligonucleotide sequences (1, 2, and 3) in which a dodecyl linker is present (Figure 12).
Oligonucleotides 1–8 were then used to incorporate different numbers of mono-Ru-DNA (m33–m35, Figure 12) at the periphery of the three-way junction (Figure 14). The introduction of ruthenium complexes at every end was believed to lock the constructs into three-point star motifs.
The authors successfully controlled the number of ruthenium complexes that could be incorporated at the periphery and were able to modulate the length of the binding arms.
The use of a bis-Ru-DNA construct permitted in a second step to connect two three-way junctions and to elaborate more complex structures (Figure 13). With a bis-Ru-DNA at the centre and no mono-Ru-DNA at the periphery of the three-arm star-shaped junctions, the efficiency formation of the assemblies decreased. On the other hand, other mono-Ru-DNA could be incorporated successfully at the periphery of the three-arm star-shaped junctions. However, when the structures were visualised by polyacrylamide gel, the size limit for the PAGE seemed to be reached suggesting the formation of rather large constructs. These studies demonstrate that the incorporation of ruthenium complexes at the periphery of a three-way junction can be used to study the thermal stability and the global structure of such assemblies.
4. Ru-DNA and the Antigene and Antisense Strategies
The use of the complementary properties of oligonucleotides has enabled the construction of complex assemblies at the nanoscale level. Yet, the specific binding of nucleic acid analogues can also be used to target DNA through the formation of a triple helix. In the antigene strategy, a triplex-forming oligonucleotide (TFO) blocks the transcription of DNA into mRNA and, therefore, acts directly on the regulation of gene expression, potentially knocking down the expression of disease-associated proteins [107,108].
In this context, Hélène et al. took an interest in the Δ and Λ enantiomers of the [Ru(phen)2dppz]2+ complex [109]. These molecules were previously reported as good double-stranded DNA binding agents, due to the intercalation of the ligands between DNA base pairs [71,72,110,111]. These interesting properties could be used for the stabilisation of DNA triple helices by conjugation of a Ru complex to the 5′-end of a TFO [112,113]. The presence of the ruthenium would also add photophysical properties, such as luminescence, long-distance electron transfer, or photocleavage to the resulting triple helices. Such Ru-oligonucleotides could be used in the context of the antigene therapy, but also as photosensitizers for directed photodamage of DNA, or for the study of DNA binding. Thus, two efficient synthetic approaches were developed to link Δ and Λ-[Ru(phen)2dppz]2+ at the 5′-end of oligonucleotides (Scheme 6) [114]. In the first approach, an alkylated ruthenium complex derivative is coupled to a thiol-modified oligonucleotide. The second synthetic route consists in the direct linkage of an amino-containing ruthenium complex with an oligonucleotide bearing an activated 5′-phosphate group. Even if the linkage of ODN to Ru(II) complex through an amine bond was effective, the thiol alkylation provided a better yield and the authors used 36 for further experiments.
The luminescence of the Δ and Λ enantiomers of the ruthenium complex linked to HIV-T oligonucleotide was investigated. Neither of the two moieties was luminescent in aqueous solution. Upon formation of the triple helix by addition of double-stranded DNA (composed of HIV-D1 and HIV-D2), the luminescence of the Δ enantiomer was increased by 6–10 times, unlike the Λ enantiomer, which showed no enhancement of luminescence. This phenomenon was previously reported for double-stranded DNA and can be explained by a difference in the geometry of intercalation. The geometry of the Λ enantiomer is more favourable to nonspecific inter- or intramolecular stacking interactions between the dppz ligand and the oligonucleotide bases [72,111,115].
The stability of triple-stranded DNA formed with HIV-D1, HIV-D2, and Λ-HIV-T-Ru was then investigated. Thermal denaturation experiments showed the stabilisation of the triple helix with the attachment of [Ru(phen)2dppz]2+, with a stabilisation reaching ΔTm = 12 °C. Unmodified HIV-T oligonucleotide was found to form less stable triplexes than HIV-T-Ru. Hence, competition studies revealed that the latter was able to displace the unmodified oligonucleotide from triplex structures. This study showed the effective intercalation of a Ru(II) complex between DNA base pairs, leading to a strong stabilisation of the triple helix, with an increase of its lifetime. The intense luminescence of the ruthenium complex upon formation of the triplex could be further used to study the kinetics of DNA binding or as a probe to study the hydrophobicity of an environment.
Another promising class of therapeutic agents are nucleic acids analogues binding directly to the encoding mRNA of a defined, disease-associated protein. Indeed, in the antisense strategy a chemically modified oligonucleotide (antisense oligonucleotide, ASO) recognises a stretch of mRNA through complementary base-pairing and the resulting steric blocks prevents translation [116]. Alternatively, ASOs can recruit RNase H which catalyses the cleavage of the RNA part of the ASO-mRNA duplex [117,118]. The development of molecules suited for the antisense strategy and gene silencing could serve as new anticancer agents [119]. Thereby, we will focus on complexes bearing a TAP ligand as non-intercalative agents. The excited states of these complexes are capable of oxidising free guanine or guanine nucleotides in DNA oligonucleotides [120] by a charge transfer process followed by a back electron transfer [121,122,123]. By measuring the reduction potential of several complexes, it was shown that at least two oxidising π–deficient TAP ligands were required in a complex to react with guanine [124]. In order to determine the nature of the reaction between guanine and these ruthenium complexes, the study of [Ru(TAP)3]2+ with guanosine monophosphate was carried out. This showed the formation of a covalent photoadduct between the exocyclic amine of the guanine nucleobase and one of the TAP ligands (Scheme 7) [125,126]. The mechanism involves the radical recombination of the protonated reduced ruthenium complex [127,128] and the deprotonated radical cation of guanine and rearomatisation of the nucleobase leads to the formation of the observed photoadduct. Interestingly, no bonding to the O6 centre of guanine was observed despite that radicals centred on O6 are usually the most stable.
In 2009, Kirsch-De Mesmaeker et al. applied this knowledge to gene silencing applications by synthesising ruthenium complexes bearing two TAP ligands [129]. Under light irradiation, and in the presence of an oligonucleotide duplex containing a guanine base on each strand, a photo-cross-linking reaction occurred. As a result, the exonuclease activity was inhibited and the DNA damaged [130].
Preliminary experiments showed that 37 and 38 (Figure 14) exhibited a moderate affinity for DNA (K = 3.9 × 104 M−1 and K = 1.5 × 106 M−1, respectively) [129]. Molecular modelling simulations showed an incomplete intercalation of the TPAC ligand into the base pairs of duplex DNA, revealing that 38 would be more constrained within the duplex. PAGE analysis of different duplexes containing a 32P-labeled strand in the presence of the ruthenium complexes and under irradiation was then carried out in order to study the photo-cross-linking process. Two photoadducts were found for complex 37: one corresponding to a photo-cross-linking reaction between the complex and the two strands of the duplex and the second within a single strand. For complex 38, no photo-cross-linking reaction could be detected under the same conditions. This phenomenon was ascribed to the intercalation of 38 into the stacked DNA base pairs which prevents the mobility of the complex inside the helix. The study of 38 showed that intercalative ligands do not possess the most favourable geometry to induce a photo-cross-linking reaction.
As 37 showed promising results for photo-cross-linking applications, Kirsch-De Mesmaeker and her group linked 37 to an oligonucleotide through an oxime bond. The oxyamino-aldehyde coupling reaction applied to oligonucleotides was first described by Lhomme and co-workers in 2000 to link an aldehyde containing oligonucleotide to a fluorophore bearing an oxyamine function, to give a stable conjugated oligonucleotide [133]. The oxime bond strategy was found to be a rapid, selective, and regiospecific manner to introduce functionalities at a preselected position inside the sequence of an oligonucleotide. This technique was then extended to the ligation of oligonucleotides with peptides, carbohydrates, glycopeptides, and PNA [134,135,136,137].
In 2003, Defrancq and co-workers applied this strategy to covalently couple an amino-modified phenanthroline derivative at the 5′ or the 3′-end of an oligonucleotide [138]. The oxyamino phenanthroline derivative synthetic route is presented in Scheme 8.
The oligonucleotide bearing an aldehyde at the 5′ or 3′-end was synthesised using standard automated solid-phase DNA synthesis. The conjugation of the two moieties was then carried out at pH 4.5 in ammonium acetate buffer, as slightly acidic conditions are needed to synthesise the oxime bond with efficiency through an addition-elimination mechanism. As two aldehyde-containing oligonucleotides can react with one aminooxy group [139], two equivalents of the phenanthroline derivative were used to minimise the formation of this by-product. This successful linkage of a phenanthroline derivative paved the way to the coupling of ruthenium complexes to oligonucleotides. In 2007, Defrancq et al. reported the first synthesis of Ru(II)-oligonucleotide complexes through the formation of an oxime bond [132]. [Ru(TAP)2phen’]2+ (39, Figure 14) and [Ru(TAP)2TAP’]2+ (40, Figure 14), where TAP’ or phen’ are the corresponding ligands bearing an aminooxy function were synthesised.
First, the phen and TAP ligands with Boc-protected aminooxy function were prepared by application of the synthetic pathway described above. The ligands were then reacted with [Ru(TAP)2(H2O)2]2+ [140] to give the corresponding complexes in 70% overall yield. The Boc function was then deprotected with a 1N HCl solution to avoid the use of TFA which, as an ambident reagent, could substitute a TAP ligand. Once the highly reactive aminooxy group was deprotected, 2 equivalents of the complexes were reacted with the aldehyde-containing oligonucleotides under acidic conditions. The expected products 41–44 (Figure 15) were obtained with an isolated yield of 40% for both a coupling at the 5′- or 3′-terminus of the oligonucleotide. Worthy of note, these conditions are compatible with the stability of the ruthenium complexes, as no degradation occurred during the reaction or the purification.
Following this methodology, Kirsch-De Mesmaeker and her group designed two polyazaaromatic ruthenium complexes to study the impact of a guanine base inside a ruthenium-oligonucleotide strand on photo-cross-linking reactions. A photoactive [Ru(TAP)2phen”]2+ Ru(T) and a non-photoactive [Ru(phen)2phen”]2+ Ru(P) were synthesised [141]. The phen” ligand corresponds to N-(2-(1,10-phenanthrolin-5-ylamino)-2-oxoethyl)-2-(aminooxy)acetamide and allows the linkage between the complexes and the 3′-end of modified oligonucleotides through an oxime bond. The attachment of the complexes at the 5′-end of ODNs was not considered as studies have shown that the efficiency of the photo-cross-linking process was higher on the 3′-end due to the suppression of the photodechelation process [142]. Three Ru-ODN sequences were created with two types of ODNs : (1) with a guanine base ODN(G); or (2) without a guanine base ODN(T) (Figure 16). ODN(T) was used as a negative control.
A similar intramolecular cyclic adduct, as described in Scheme 7, was formed when Ru(T)-ODN(G) was reacted alone or in the presence of non-complementary strands. Yet, when reacted in the presence of its complementary strand, and after 30 min illumination, an intermolecular photo-cross-linking process occurred between the TAP ligand of both Ru(T)-ODN(G) and Ru(T)-ODN(T) molecules and the guanine base of the complementary strand. No intramolecular process was detected in this case (Figure 17, Scheme 9) with no competition from the G nucleotide on the probe sequence is taking place. In addition, the intramolecular adduct, i.e., the reaction of the Ru complex with a G nucleotide in the probe sequence can be obtained, but it is occurring only in the absence of the target sequence. Therefore, this intramolecular adduct, named seppuku, prevents any side reaction from occurring [143].
This phenomenon was explained a few years later through a thorough biochemical analysis of the interaction of [Ru(TAP)2phen”]2+ with free guanine along with DNA oligonucleotides using molecular modelling simulations [144]. The formation of the intrastrand photoadduct requires a strong distortion of the oligonucleotide which cannot occur in the presence of the complementary strand containing a reachable guanine base, leading to a 100% interstrand photo-cross-linking process. Thereby, the formation of the intramolecular photoadduct enables the prevention of side reactions in the absence of the complementary strand or in the presence of non-complementary targets, without competing with the intrastrand process in the presence of the complementary strand. These results show the interest of ruthenium-based oligonucleotides for gene silencing methods but also as a way to prevent and detect mismatches in vivo.
In light of these results, [Ru(TAP)2phen”]2+ was tested in 2013 as a therapeutic agent against cervical cancer [145] through an antisense-based strategy. Cervical cancer is associated to human papillomavirus (HPV) infection [146]. The HPV oncogene E6 is able to form an inhibiting complex with the p53 nuclear phosphoprotein [147,148]. The latter, in its primal state, possesses antiproliferative properties by promoting cell cycle arrest and/or leading stressed cells to apoptosis [149]. The complexation of p53 by E6 leads to the abrogation of p53 functions, which cause the proliferation of cancer cells and tumour progression. Hence, antisense oligonucleotides targeting E6 could lead to a knockdown of this HPV oncogene and concomitantly restore the antiproliferative role of the p53 protein. In this context, [Ru(TAP)2phen”]2+ was first coupled to an oligonucleotide probe containing a guanine base and targeting a specific sequence of E6 at position 324 of the gene.
PAGE experiments showed that the ruthenium antisense oligonucleotide (Ru-ASO, Figure 18) in the absence of its complementary strand or with a non-complementary strand under irradiation gave, as expected, a cyclic adduct in 80% yield coming from an intramolecular reaction. Similarly, in the presence of its intended target, a photo-cross-linking process occurred also in 80% yield. An in vitro experiment with SiHa human cervical cancer cells revealed that Ru-ASO was able to inhibit, with specificity, the cell growth under light irradiation (45–50% of inhibition 24 h after irradiation). Western blot experiments attested that the inhibition was due to the decrease of E6 expression. The results showed that Ru-ASO reduces E6 expression with 60% efficiency, 24 h after irradiation while confocal microscopy studies revealed a restoration of p53 expression in these cells. Overall, this study demonstrated the efficiency and specificity of gene silencing therapy and gave preliminary results for the use of Ru-ASO in phototherapy.
5. Enzymatic Incorporation of Ru Complexes into DNA
The study of ruthenium complexes and more specifically of [Ru(bpy)3]2+ (bpy = 2,2’-bipyridine) [150] has shown that the photophysical properties of ruthenium complexes could be modulated by the nature of the ligand, making them good candidates as DNA probes. Thereby, in 2007, Hocek et al. took an interest in the synthesis of purine nucleosides (2’-deoxyadenosine [151] and 9-benzyladenine [152]) bearing in position 8 of the nucleobase phenanthroline or bipyridine ruthenium complexes. Their choice was motivated by the lack of reports concerning ruthenium probes conjugated to purines, in contrast with numerous reports of pyrimidine Ru-probes (vide supra). Unfortunately, due to the greater difficulties in preparation and chemical incorporation of this kind of molecules, the synthetic pathways developed gave low to moderate yields. As an alternative, Ru complexes were appended at position 8 of the nucleobase of purine triphosphates but these analogues revealed to be poor substrates for DNA polymerases [153,154]. As 7-substituted molecules were efficiently incorporated by the Pwo DNA polymerase [153,155,156,157], a new synthetic pathway was developed based on the Sonogashira reaction, to synthesise 7-deaza-2’-deoxyadenosines bearing bipyridine ligands and their ruthenium complexes. The complexes were connected to the nucleobase through an acetylene linker which ensured a certain rigidity often required for an efficient polymerase acceptance [158] as well as ease of synthesis. First, 7-iodo-7-deaza-2’-deoxyadenosine and two ruthenium-containing acetylenes (45, 46) were prepared according to literature protocols [152,159]. The coupling was performed through a Sonogashira reaction in a water/acetonitrile mixture (Figure 19). The choice of this solvent was made in order to be applicable for a further labelling of nucleoside triphosphates. Interestingly, the reaction gave much higher yields compared to 8-substituted-2’-deoxyadenosine analogues (16% for the first while the second could not be isolated).
Photophysical and redox experiments showed a weak red luminescence with a low quantum yield for 45 and a moderate quantum yield of 0.03 for 46. Both molecules gave electrochemical oxidation (1.2 V), showing preliminary results for redox labelling of DNA. The use of this Sonogashira reaction enabled the incorporation of various metal complexes (i.e., Os, Fe, Ni) [161,162] on the nucleobase to yield both modified nucleoside triphosphates and DNA oligonucleotides. In 2009, [Ru(bpy)3]2+ was successfully linked to nucleoside triphosphates 47–50 (Scheme 10) [163]. The coupling reactions were conducted on the known analogues 5-iodo-2’-deoxyuridine 5′-triphosphate (5-I-dUTP), 5-iodo-2’-deoxycytidine 5′-triphosphate (5-I-dCTP), 7-iodo-7-deaza-2’-deoxyadenosine 5′-triphosphate (7-I-deaza-dATP), and 7-iodo-7-deaza-2’-deoxyguanosine 5′-triphosphate (7-I-deaza-dGTP) [154,164,165,166,167]. The Sonogashira reaction was ended in 1 h, even if total conversion was not achieved because of the partial decomposition and hydrolysis of the starting and final triphosphates. The authors also mentioned the quick hydrolysis (a few weeks) of the final products in water at −20 °C.
After HPLC purification, the efficiency of the polymerase incorporation of the modified triphosphates 47–50 was tested. Only Vent (exo−) and Pwo of the four polymerases that were tested gave full-length products under primer extension (PEX) reaction conditions. Other PEX experiments showed the successful incorporation of these modified dNTPs into diverse DNA sequences. However, the incorporation of two modified nucleoside triphosphates at adjacent positions led to an early termination of the polymerisation reaction.
Electrochemical studies were then carried out to ensure the potential of the modified dNTPs for DNA labelling. Unfortunately, the potential of the ruthenium (1.1–1.25 V) is overlapped by the potential of guanine bases (1.1 V), making 47–50 rather poor candidates for redox labelling of DNA. The luminescence of ruthenium complexes still allows the use of these molecules for luminescent spectroscopy detection.
6. Conclusions
Chemical modification of oligonucleotides is an alluring strategy to improve the potency of DNA and RNA to serve in a number of practical applications. For instance, the inclusion of modifications on the sugar and/or phosphate backbone of nucleotides increases the resistance of antisense oligonucleotides to nuclease degradation [116,168], the incorporation of fluorescent or electrochemical tags enables the localisation of biological targets both in vitro and in vivo [169,170], or the appendage of artificial nucleotides facilitates the immobilisation of oligonucleotides on solid supports [171,172]. The two main strategies for the synthesis of modified oligonucleotides are DNA automated synthesis and the polymerisation of modified nucleoside triphosphates. In this review, we describe several approaches for the covalent linkage of ruthenium complexes on various locations of DNA oligonucleotides.
The covalent linkage between nucleic acid derivatives and ruthenium complexes allows the direct application of the inherent properties of ruthenium complexes to probe DNA, detect mismatches in vivo, serve as gene silencing agents, or to stabilise DNA structures with the intercalation of ruthenium complexes. The elucidation of different mechanisms involved in DNA has also been possible, with the development of light switches, probes for the study of thermal stability or kinetics in DNA or the elucidation of several electron transfer mechanisms. The ease of functionalization of ruthenium complexes has allowed the incorporation of two different oligonucleotides for the elaboration of complex nanostructures. Depending on the coordination number of ruthenium, different shapes could be created and, among them, three-way DNA structures. As three-branched DNA structures are often found in the process of replication or recombination, this work could contribute to the better understanding of these biological processes.
Yet, among the numerous applications described in this review and to our surprise, few studies have been carried out to link the anticancer potency of ruthenium complexes to oligonucleotides. However, we are confident that more remarkable discoveries will be reported in the near future, highlighting the full potential of these compounds.
Author Contributions
Conceptualization and writing was done by all authors reviewing and editing was done by M.F., G.G., and M.H.
Acknowledgments
This work was financially supported by an ERC Consolidator Grant PhotoMedMet to G.G. (GA 681679) and has received support under the program “Investissements d’Avenir“ launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G.G.). M.F. gratefully acknowledges a fellowship from the doctoral school MTCI 5 University of Paris Descartes. M.H. acknowledges funding from Institut Pasteur. E.C. thanks Chimie ParisTech and the Institut Pasteur for a fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations were used in the manuscript:
| Bpy | 2,2’-Bipyridine |
| DMAP | 4-Dimethylaminopyrimidine |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| dNTP | Deoxyribose nucleoside triphosphate |
| Dppz | Dipyrido[3,2:a-2′,3′:c]phenazine |
| Im | Imidazole |
| LMCT | Ligand-to-metal charge transfer |
| Me2dppz | 9,10-Dimethyl-dipyridophenazine |
| MLCT | Metal-to-ligand charge transfer |
| ODN | OligoDeoxyriboNucleotide |
| Oligo | Oligonucleotide |
| PAGE | Polyacrylamide gel electrophoresis |
| Phen | 1,10-Phenanthroline |
| Phen’ | 5-(Glutaric acid monoamide)-1,10-phenanthroline |
| PNA | Peptide nucleic acid |
| Py | Pyridine |
| TAP | 1,4,5,8-Tetraazaphenanthrene |
| Terpy | Terpyridine |
| TFA | Trifluoroacetic acid |
| TPAC | Tetrapyridoacridine |
| TPPTS | Triphenylphosphine-3,3′,3′′-trisulfonic acid trisodium salt |
References
- Seeman, N.C.; Sleiman, H.F. DNA nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. [Google Scholar] [CrossRef]
- Aldaye, F.A.; Palmer, A.L.; Sleiman, H.F. Assembling materials with DNA as the guide. Science 2008, 321, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Liu, Y.; Yan, H. Designer DNA nanoarchitectures. Biochemistry 2009, 48, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. An overview of structural DNA nanotechnology. Mol. Biotechnol. 2007, 37, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Pitchiaya, S.; Krishnan, Y. First blueprint, now bricks: DNA as construction material on the nanoscale. Chem. Soc. Rev. 2006, 35, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Rothlisberger, P.; Gasse, C.; Hollenstein, M. Nucleic acid aptamers: Emerging applications in medical imaging, nanotechnology, neurosciences, and drug delivery. Int. J. Mol. Sci. 2017, 18, 2430. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.R.; Grodick, M.A.; Barton, J.K. DNA charge transport: From chemical principles to the cell. Cell Chem. Biol. 2016, 23, 183–197. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Silva, R.M.B.; Barton, J.K. Redox signaling through DNA. Isr. J. Chem. 2016, 56, 705–723. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Barton, J.K. DNA electrochemistry through the base pairs not the sugar–phosphate backbone. J. Am. Chem. Soc. 2005, 127, 10160–10161. [Google Scholar] [CrossRef] [PubMed]
- Buchini, S.; Leumann, C.J. Recent improvements in antigene technology. Curr. Opin. Chem. Biol. 2003, 7, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Oberbauer, R. Not nonsense but antisense—Applications of antisense oligonucleotides in different fields of medicine. Wien. Klin. Wochenschr. 1997, 109, 40–46. [Google Scholar] [PubMed]
- Miller, P.S. Development of antisense and antigene oligonucleotide analogs. Prog. Nucleic Acid Res. Mol. Biol. 1996, 52, 261–291. [Google Scholar] [PubMed]
- Saraswat, P.; Soni, R.R.; Bhandari, A.; Nagori, B.P. DNA as therapeutics; an update. Indian J. Pharm. Sci. 2009, 71, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Szabat, M.; Kierzek, R. Parallel-stranded DNA and RNA duplexes—Structural features and potential applications. FEBS J. 2017, 284, 3986–3998. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.A.; Moloney, F.J.; Cai, H.; Au-Yeung, A.; China, C.; Scolyer, R.A.; Yosufi, B.; Raftery, M.J.; Deng, J.Z.; Morton, S.W.; et al. Safety and tolerability of an intratumorally injected DNAzyme, dz13, in patients with nodular basal-cell carcinoma: A phase 1 first-in-human trial. Lancet 2013, 381, 1835–1843. [Google Scholar] [CrossRef]
- Hollenstein, M.; Hipolito, C.J.; Lam, C.H.; Perrin, D.M. Toward the combinatorial selection of chemically modified dnazyme RNase a mimics active against all-RNA substrates. ACS Comb. Sci. 2013, 15, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Krug, N.; Hohlfeld, J.M.; Kirsten, A.-M.; Kornmann, O.; Beeh, K.M.; Kappeler, D.; Korn, S.; Ignatenko, S.; Timmer, W.; Rogon, C.; et al. Allergen-induced asthmatic responses modified by a gata3-specific DNAzyme. N. Engl. J. Med. 2015, 372, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ding, J.; Liu, J. Theranostic dnazymes. Theranostics 2017, 7, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chan, K.M.; Kool, E.T. Fluorescent nucleobases as tools for studying DNA and RNA. Nat. Chem. 2017, 9, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, E.; Lam, C.H.; Perrin, D.M. A densely modified M2+-independent DNAzyme that cleaves RNA efficiently with multiple catalytic turnover. Chem. Sci. 2018, 9, 1813–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenstein, M. DNA catalysis: The chemical repertoire of DNAzymes. Molecules 2015, 20, 20777–20804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruthers, M.H. The chemical synthesis of DNA/RNA: Our gift to science. J. Biol. Chem. 2013, 288, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef] [PubMed]
- D'Agata, R.; Giuffrida, M.C.; Spoto, G. Peptide nucleic acid-based biosensors for cancer diagnosis. Molecules 2017, 22, 1951. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.M.; Mirhosseini, A.; Heiat, M.; Rad, H.S.; Mirnejad, R.; Choopani, A. Diagnostic and therapeutic applications of peptide nucleic acid in medical microbiology. Minerva Biotechnol. 2017, 29, 76–88. [Google Scholar]
- Wu, J.C.; Meng, Q.C.; Ren, H.M.; Wang, H.T.; Wu, J.; Wang, Q. Recent advances in peptide nucleic acid for cancer bionanotechnology. Acta Pharmacol. Sin. 2017, 38, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Vilaivan, T. Fluorogenic pna probes. Beilstein J. Org. Chem. 2018, 14, 253–281. [Google Scholar] [CrossRef] [PubMed]
- Hirao, I.; Kimoto, M.; Yamashige, R. Natural versus artificial creation of base pairs in DNA: Origin of nucleobases from the perspectives of unnatural base pair studies. Acc. Chem. Res. 2012, 45, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Cox, R.S.; Hirao, I. Unnatural base pair systems for sensing and diagnostic applications. Expert Rev. Mol. Diagn. 2011, 11, 321–331. [Google Scholar] [PubMed]
- Saito-Tarashima, N.; Minakawa, N. Unnatural base pairs for synthetic biology. Chem. Pharm. Bull. 2018, 66, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.W.; Romesberg, F.E. Expansion of the genetic alphabet: A chemist’s approach to synthetic biology. Acc. Chem. Res. 2018, 51, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, E.; Peyman, A. Antisense oligonucleotides—A new therapeutic principle. Chem. Rev. 1990, 90, 543–584. [Google Scholar] [CrossRef]
- Kurreck, J. Antisense technologies—Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M. Bioconjugation Protocols: Strategies and Methods; Springer Science & Business Media: Berlin, Germany, 2004; Volume 283. [Google Scholar]
- Taskova, M.; Mantsiou, A.; Astakhova, K. Synthetic nucleic acid analogues in gene therapy: An update for peptide-oligonucleotide conjugates. ChemBioChem 2017, 18, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Lesnikowski, Z.J. DNA as platform for new biomaterials. Metal-containing nucleic acids. Curr. Org. Chem. 2007, 11, 355–381. [Google Scholar] [CrossRef]
- Shao, F.; Barton, J.K. Long-range electron and hole transport through DNA with tethered cyclometalated iridium(III) complexes. J. Am. Chem. Soc. 2007, 129, 14733–14738. [Google Scholar] [CrossRef] [PubMed]
- Kudr, J.; Nejdl, L.; Skalickova, S.; Ruttkay-Nedecky, B.; Rodrigo, M.A.M.; Dostalova, S.; Jimenez, A.M.J.; Chudobova, D.; Cihalova, K.; Konecna, M.; et al. Plasmid HIV p24 gene detection on mercury film electrode using osmium labelling. Int. J. Electrochem. 2014, 9, 3409–3418. [Google Scholar]
- Barton, J.K.; Olmon, E.D.; Sontz, P.A. Metal complexes for DNA-mediated charge transport. Coord. Chem. Rev. 2011, 255, 619–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, B.; Kirsch-De Mesmaeker, A. Photo-reduction of polyazaaromatic ru(II) complexes by biomolecules and possible applications. Coord. Chem. Rev. 2006, 250, 1627–1641. [Google Scholar] [CrossRef]
- Noffke, A.L.; Habtemariam, A.; Pizarro, A.M.; Sadler, P.J. Designing organometallic compounds for catalysis and therapy. Chem. Commun. 2012, 48, 5219–5246. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.R.; Thomas, J.A. Ruthenium(II) polypyridyl complexes and DNA-from structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. [Google Scholar] [CrossRef] [PubMed]
- Newkome, G.R.; Cho, T.J.; Moorefield, C.N.; Baker, G.R.; Cush, R.; Russo, P.S. Self- and directed assembly of hexaruthenium macrocycles. Angew. Chem. Int. Ed. 1999, 38, 3717–3721. [Google Scholar] [CrossRef]
- Ang, W.H.; Casini, A.; Sava, G.; Dyson, P.J. Organometallic ruthenium-based antitumor compounds with novel modes of action. J. Organomet. Chem. 2011, 696, 989–998. [Google Scholar] [CrossRef]
- Moucheron, C. From cisplatin to photoreactive Ru complexes: Targeting DNA for biomedical applications. New J. Chem. 2009, 33, 235–245. [Google Scholar] [CrossRef]
- Ghesquiere, J.; Le Gac, S.; Marcelis, L.; Moucheron, C.; Kirsch-De Mesmaeker, A. What does the future hold for photo-oxidizing ru-ii complexes with polyazaaromatic ligands in medicinal chemistry? Curr. Top. Med. Chem. 2012, 12, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Novakova, O. DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resist. Updates 2006, 9, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Nickita, N.; Gasser, G.; Bond, A.M.; Spiccia, L. Synthesis, spectroscopic properties and electrochemical oxidation of Ru(II)-polypyridyl complexes attached to a peptide nucleic acid monomer backbone. Eur. J. Inorg. Chem. 2009, 2009, 2179–2186. [Google Scholar] [CrossRef]
- Joshi, T.; Patra, M.; Spiccia, L.; Gasser, G. Di-heterometalation of thiol-functionalized peptide nucleic acids. Artif. DNA PNA XNA 2013, 4, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, T.; Barbante, G.J.; Francis, P.S.; Hogan, C.F.; Bond, A.M.; Gasser, G.; Spiccia, L. Electrochemiluminescent monomers for solid support syntheses of Ru(II)-PNA bioconjugates: Multimodal biosensing tools with enhanced duplex stability. Inorg. Chem. 2012, 51, 3302–3315. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Barbante, G.J.; Francis, P.S.; Hogan, C.F.; Bond, A.M.; Spiccia, L. Electrochemiluminescent peptide nucleic acid-like monomers containing Ru(II)-dipyridoquinoxaline and Ru(II)-dipyridophenazine complexes. Inorg. Chem. 2011, 50, 12172–12183. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Gasser, G.; Martin, L.L.; Spiccia, L. Specific uptake and interactions of peptide nucleic acid derivatives with biomimetic membranes. RSC Adv. 2012, 2, 4703–4712. [Google Scholar] [CrossRef] [Green Version]
- Verheijen, J.C.; Van der Marel, G.A.; Van Boom, J.H.; Metzler-Nolte, N. Transition metal derivatives of peptide nucleic acid (PNA) oligomers: Synthesis, characterization, and DNA binding. Bioconj. Chem. 2000, 11, 741–743. [Google Scholar] [CrossRef]
- Gasser, G.; Sosniak, A.M.; Metzler-Nolte, N. Metal-containing peptide nucleic acid conjugates. Dalton Trans. 2011, 40, 7061–7076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubar, T.; Elstner, M. What governs the charge transfer in DNA? The role of DNA conformation and environment. J. Phys. Chem. B 2008, 112, 8788–8798. [Google Scholar] [CrossRef] [PubMed]
- Malyshev, D.A.; Romesberg, F.E. The expanded genetic alphabet. Angew. Chem. Int. Ed. 2015, 54, 11930–11944. [Google Scholar] [CrossRef] [PubMed]
- Giese, B. Electron transfer through DNA and peptides. Bioorg. Med. Chem. 2006, 14, 6139–6143. [Google Scholar] [CrossRef] [PubMed]
- Effi, B.; Yu, L.; Caroline, S.; Andreaseasd, U.; Hans-Achim, W. Acceleration of long-range photoinduced electron transfer through DNA by hydroxyquinolines as artificial base pairs. ChemPhysChem 2015, 16, 1607–1612. [Google Scholar]
- Giese, B. Electron transfer in DNA. Curr. Opin. Chem. Biol. 2002, 6, 612–618. [Google Scholar] [CrossRef]
- Boussicault, F.; Robert, M. Electron transfer in DNA and in DNA-related biological processes. Electrochemical insights. Chem. Rev. 2008, 108, 2622–2645. [Google Scholar] [CrossRef] [PubMed]
- Bixon, M.; Jortner, J. Incoherent charge hopping and conduction in DNA and long molecular chains. Chem. Phys. 2005, 319, 273–282. [Google Scholar] [CrossRef]
- Nitzan, A. Electron transmission through molecules and molecular interfaces. Annu. Rev. Phys. Chem. 2001, 52, 681–750. [Google Scholar] [CrossRef] [PubMed]
- Giese, B.; Amaudrut, J.; Köhler, A.-K.; Spormann, M.; Wessely, S. Direct observation of hole transfer through DNA by hopping between adenine bases and by tunnelling. Nature 2001, 412, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Siriwong, K.; Voityuk, A.A. Electron transfer in DNA. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 780–794. [Google Scholar] [CrossRef]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA charge transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [PubMed]
- Berlin, Y.A.; Kurnikov, I.V.; Beratan, D.; Ratner, M.A.; Burin, A.L. DNA electron transfer processes: Some theoretical notions. In Long-Range Charge Transfer in DNA II; Schuster, G.B., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 1–36. [Google Scholar]
- Venkatramani, R.; Keinan, S.; Balaeff, A.; Beratan, D.N. Nucleic acid charge transfer: Black, white and gray. Coord. Chem. Rev. 2011, 255, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, Y.; Barton, J.K. A sequence-specific molecular light switch tethering of an oligonucleotide to a dipyridophenazine complex of Ruthenium(II). J. Am. Chem. Soc. 1992, 114, 8136–8137. [Google Scholar] [CrossRef]
- Murphy, C.J.; Barton, J.K. Ruthenium complexes as luminescent reporters of DNA. Methods Enzymol. 1993, 226, 576–594. [Google Scholar] [PubMed]
- Olson, E.J.C.; Hu, D.; Hörmann, A.; Jonkman, A.M.; Arkin, M.R.; Stemp, E.D.A.; Barton, J.K.; Barbara, P.F. First observation of the key intermediate in the “light-switch” mechanism of [Ru(phen)2dppz]2+. J. Am. Chem. Soc. 1997, 119, 11458–11467. [Google Scholar] [CrossRef]
- Friedman, A.E.; Chambron, J.C.; Sauvage, J.P.; Turro, N.J.; Barton, J.K. Molecular light switch for DNA: Ru(bpy)2(dppz)2+. J. Am. Chem. Soc. 1990, 112, 4960–4962. [Google Scholar] [CrossRef]
- Lincoln, P.; Broo, A.; Norden, B. Diastereomeric DNA-binding geometries of intercalated ruthenium(II) trischelates probed by linear dichroism: [Ru(phen)2dppz]2+ and ru(phen)2bdppz2+. J. Am. Chem. Soc. 1996, 118, 2644–2653. [Google Scholar] [CrossRef]
- Meade, T.J.; Kayyem, J.F. Electron transfer through DNA: Site-specific modification of duplex DNA with ruthenium donors and acceptors. Angew. Chem. Int. Ed. 1995, 34, 352–354. [Google Scholar] [CrossRef]
- Murphy, C.J.; Arkin, M.R.; Jenkins, Y.; Ghatlia, N.D.; Bossmann, S.H.; Turro, N.J.; Barton, J.K. Long-range photoinduced electron transfer through a DNA helix. Science 1993, 262, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Fujitsuka, M.; Kawai, K.; Majima, T. Direct measurement of the dynamics of excess electron transfer through consecutive thymine sequence in DNA. J. Am. Chem. Soc. 2011, 133, 15320–15323. [Google Scholar] [CrossRef] [PubMed]
- Osakada, Y.; Kawai, K.; Fujitsuka, M.; Majima, T. Kinetics of charge transfer in DNA containing a mismatch. Nucleic Acids Res. 2008, 36, 5562–5570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromherz, P.; Rieger, B. Photoinduced electron transfer in DNA matrix from intercalated ethidium to condensed methylviologen. J. Am. Chem. Soc. 1986, 108, 5361–5362. [Google Scholar] [CrossRef]
- Purugganan, M.; Kumar, C.; Turro, N.; Barton, J. Accelerated electron transfer between metal complexes mediated by DNA. Science 1988, 241, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.M.; Harriman, A. Dynamics of electron transfer between intercalated polycyclic molecules: Effect of interspersed bases. J. Am. Chem. Soc. 1992, 114, 3656–3660. [Google Scholar] [CrossRef]
- Diederichsen, U. Charge transfer in DNA: A controversy. Angew. Chem. Int. Ed. 1997, 36, 2317–2319. [Google Scholar] [CrossRef]
- Kumar, C.V.; Barton, J.K.; Turro, N.J. Photophysics of ruthenium complexes bound to double helical DNA. J. Am. Chem. Soc. 1985, 107, 5518–5523. [Google Scholar] [CrossRef]
- Barton, J.K.; Goldberg, J.M.; Kumar, C.V.; Turro, N.J. Binding modes and base specificity of tris(phenanthroline)ruthenium(II) enantiomers with nucleic acids: Tuning the stereoselectivity. J. Am. Chem. Soc. 1986, 108, 2081–2088. [Google Scholar] [CrossRef]
- Arkin, M.R.; Stemp, E.D.; Pulver, S.C.; Barton, J.K. Long-range oxidation of guanine by Ru(III) in duplex DNA. Chem. Biol. 1997, 4, 389–400. [Google Scholar] [CrossRef]
- Stemp, E.D.A.; Barton, J.K. The flash-quench technique in protein-DNA electron transfer: Reduction of the guanine radical by ferrocytochrome c. Inorg. Chem. 2000, 39, 3868–3874. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Alshykhly, O.; Zhu, J.; Muller, J.G.; Burrows, C.J. Rates of chemical cleavage of DNA and RNA oligomers containing guanine oxidation products. Chem. Res. Toxicol. 2015, 28, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Langmaier, J.; Samec, Z.; Samcova, E.; Hobza, P.; Reha, D. Origin of difference between one-electron redox potentials of guanosine and guanine: Electrochemical and quantum chemical study. J. Phys. Chem. B 2004, 108, 15896–15899. [Google Scholar] [CrossRef]
- Bhattacharya, P.K.; Barton, J.K. Influence of intervening mismatches on long-range guanine oxidation in DNA duplexes. J. Am. Chem. Soc. 2001, 123, 8649–8656. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.J.; Tor, Y. Metal-containing oligonucleotides: Solid-phase synthesis and luminescence properties. J. Am. Chem. Soc. 1998, 120, 2194–2195. [Google Scholar] [CrossRef]
- Tzalis, D.; Tor, Y. Coordination compounds as building blocks: Single-step synthesis of multi-ruthenium(II) complexes. Chem. Commun. 1996, 1043–1044. [Google Scholar] [CrossRef]
- Connors, P.J.; Tzalis, D.; Dunnick, A.L.; Tor, Y. Coordination compounds as building blocks: Single-step synthesis of heteronuclear multimetallic complexes containing Ru(II) and Os(II). Inorg. Chem. 1998, 37, 1121–1123. [Google Scholar] [CrossRef]
- Hurley, D.J.; Tor, Y. Ru(II) and Os(II) nucleosides and oligonucleotides: Synthesis and properties. J. Am. Chem. Soc. 2002, 9, 3749–3762. [Google Scholar] [CrossRef]
- Hurley, D.J.; Tor, Y. Donor/acceptor interactions in systematically modified Ru(II)-Os(II) oligonucleotides. J. Am. Chem. Soc. 2002, 124, 13231–13241. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M. Nucleoside triphosphates—Building blocks for the modification of nucleic acids. Molecules 2012, 17, 13569–13591. [Google Scholar] [CrossRef] [PubMed]
- Hocek, M. Synthesis of base-modified 2′—Building beoside triphosphates and their use in enzymatic synthesis of modified DNA for applications in bioanalysis and chemical biology. J. Org. Chem. 2014, 79, 9914–9921. [Google Scholar] [CrossRef] [PubMed]
- Weizman, H.; Tor, Y. Redox-active metal-containing nucleotides: Synthesis, tunability, and enzymatic incorporation into DNA. J. Am. Chem. Soc. 2002, 124, 1568–1569. [Google Scholar] [CrossRef] [PubMed]
- Bujold, K.E.; Lacroix, A.; Sleiman, H.F. DNA nanostructures at the interface with biology. Chem 2018, 4, 495–521. [Google Scholar] [CrossRef]
- Chidchob, P.; Sleiman, H.F. Recent advances in DNA nanotechnology. Curr. Opin. Chem. Biol. 2018, 46, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Baca, I.; Mitra, D.; Zulyniak, H.J.; Banerjee, J.; Sleiman, H.F. Solid-phase synthesis of transition metal linked, branched oligonucleotides. Angew. Chem. Int. Ed. 2001, 40, 4629–4632. [Google Scholar] [CrossRef]
- Damha, M.J.; Ganeshan, K.; Hudson, R.H.E.; Zabarylo, S.V. Solid-phase synthesis of branched oligoribonucleotides related to messenger-RNA splicing intermediates. Nucleic Acids Res. 1992, 20, 6565–6573. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. DNA nanotechnology: Novel DNA constructions. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Di Cesare, N.; Sleiman, H.F. Self-assembly of cylclic metal—DNA nanostuctures using ruthenium tris(bipyridine)-branched oligonucleotides. Angew. Chem. Int. Ed. 2004, 43, 5804–5808. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.M.; McLaughlin, L.W. Design and synthesis of DNA-tethered ruthenium complexes that self-assemble into linear arrays. Chem. Commun. 2003, 2934–2935. [Google Scholar] [CrossRef]
- Constable, E.C.; Ward, M.D. Synthesis and coordination behavior of 6′,6″-bis(2-pyridyl)-2,2′-4,4″-2″,2″′-quaterpyridine—Back-to-back 2,2′-6′,2″-terpyridine. J. Chem. Soc. Dalton Trans. 1990, 1405–1409. [Google Scholar] [CrossRef]
- Irvoas, J.; Noirot, A.; Chouini-Lalanne, N.; Reynes, O.; Garrigues, J.C.; Sartor, V. Programmable multimetallic linear nanoassemblies of ruthenium-DNA conjugates. RSC Adv. 2012, 2, 9538–9542. [Google Scholar] [CrossRef] [Green Version]
- Terpetschnig, E.; Szmacinski, H.; Malak, H.; Lakowicz, J.R. Metal-ligand complexes as a new class of long-lived fluorophores for protein hydrodynamics. Biophys. J. 1995, 68, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Irvoas, J.; Noirot, A.; Chouini-Lalanne, N.; Reynes, O.; Sartor, V. DNA three-way junction-ruthenium complex assemblies. New J. Chem. 2013, 37, 2324–2329. [Google Scholar] [CrossRef] [Green Version]
- Calogero, A.; Hospers, G.A.P.; Mulder, N.H. Synthetic oligonucleotides: Useful molecules? A review. Pharm. World Sci. 1997, 19, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Hidenori, O.; Yosuke, T.; Shigeki, S. Aminopyridinyl-Pseudodeoxycytidine derivatives selectively stabilize antiparallel triplex DNA with multiple cg inversion sites. Angew. Chem. Int. Ed. 2016, 55, 12445–12449. [Google Scholar]
- Grimm, G.N.; Boutorine, A.S.; Lincoln, P.; Norden, B.; Helene, C. Formation of DNA triple helices by an oligonucleotide conjugated to a fluorescent ruthenium complex. ChemBioChem 2002, 3, 324–331. [Google Scholar] [CrossRef]
- Friedman, A.E.; Kumar, C.V.; Turro, N.J.; Barton, J.K. Luminescence of ruthenium(II) polypyridyls: Evidence for intercalative binding to z-DNA. Nucleic Acids Res. 1991, 19, 2595–2602. [Google Scholar] [PubMed]
- Hiort, C.; Lincoln, P.; Norden, B. DNA-binding of delta- ru(phen)2dppz2+ and lambda- ru(phen)2dppz2+. J. Am. Chem. Soc. 1993, 115, 3448–3454. [Google Scholar] [CrossRef]
- Silver, G.C.; Sun, J.S.; Nguyen, C.H.; Boutorine, A.S.; Bisagni, E.; Helene, C. Stable triple-helical DNA complexes formed by benzopyridoindole- and benzopyridoquinoxaline-oligonucleotide conjugates. J. Am. Chem. Soc. 1997, 119, 263–268. [Google Scholar] [CrossRef]
- Silver, G.C.; Nguyen, C.H.; Boutorine, A.S.; Bisagni, E.; Garestier, T.; Helene, C. Conjugates of oligonucleotides with triplex-specific intercalating agents. Stabilization of triple-helical DNA in the promoter region of the gene for the alpha-subunit of interleukin 2 (IL-2r alpha). Bioconj. Chem. 1997, 8, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Grimm, G.N.; Boutorine, A.S.; Helene, C. Rapid routes of synthesis of oligonucleotide conjugates from non-protected oligonucleotides and ligands possessing different nucleophilic or electrophilic functional groups. Nucleos. Nucleot. Nucl. 2000, 19, 1943–1965. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.D.; Kim, M.S.; Kim, S.K.; Lincoln, P.; Tuite, E.; Norden, B. Binding mode of [ruthenium(II) (1,10-phenanthroline)2L]2+ with poly(dT*dA-dT) triplex. Ligand size effect on third-strand stabilization. Biochemistry 1997, 36, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Istrate, A.; Katolik, A.; Istrate, A.; Leumann, C.J. 2′β-fluoro-tricyclo nucleic acids (2′F-tc-ANA): Thermal duplex stability, structural studies, and RNase H activation. Chem. Eur. J. 2017, 23, 10310–10318. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, P.E. (Ed.) Antisense DNA technology. In Vision Research Protocols; Humana Press: Totowa, NJ, USA, 2001; pp. 89–104. [Google Scholar]
- Tossi, A.B.; Kelly, J.M. A study of some polypyridylruthenium(II) complexes as DNA binders and photocleavage reagents. Photochem. Photobiol. 1989, 49, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, J.P.; Kirsch-De Mesmaeker, A.; Kelly, J.M.; Tossi, A.B.; Gorner, H. Photoinduced electron-transfer from nucleotides to ruthenium-tris-1,4,5,8-tetraazaphenanthrene—Model for photosensitized DNA oxidation. Photochem. Photobiol. 1992, 55, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Coates, C.G.; Callaghan, P.; McGarvey, J.J.; Kelly, J.M.; Jacquet, L.; Kirsch-De Mesmaeker, A. Spectroscopic studies of structurally similar DNA-binding ruthenium(II) complexes containing the dipyridophenazine ligand. J. Mol. Struct. 2001, 598, 15–25. [Google Scholar] [CrossRef]
- Kelly, J.M.; Feeney, M.M.; Tossi, A.B.; Lecomte, J.P.; Kirsch-De Mesmaeker, A. Interaction of tetra-azaphenanthrene ruthenium complexes with DNA and oligonucleotides—A photophysical and photochemical investigation. Anti-Cancer Drug Design 1990, 5, 69–75. [Google Scholar] [PubMed]
- Herman, L.; Ghosh, S.; Defrancq, E.; Kirsch-De Mesmaeker, A. Ru(II) complexes and light: Molecular tools for biomolecules. J. Phys. Org. Chem. 2008, 21, 670–681. [Google Scholar] [CrossRef]
- Jacquet, L.; Kelly, J.M.; Kirsch-De Mesmaeker, A. Photoadduct between tris(1,4,5,8-tetraazaphenanthrene)ruthenium(II) and guanosine-monophosphate—A model for a new mode of covalent binding of metal-complexes to DNA. J. Chem. Soc. Chem. Commun. 1995, 913–914. [Google Scholar] [CrossRef]
- Jacquet, L.; Davies, R.J.H.; Kirsch-De Mesmaeker, A.; Kelly, J.M. Photoaddition of Ru(tap)2(bpy)2+ to DNA: A new mode of covalent attachment of metal complexes to duplex DNA. J. Am. Chem. Soc. 1997, 119, 11763–11768. [Google Scholar] [CrossRef]
- Troian-Gautier, L.; Mugeniwabagara, E.; Fusaro, L.; Moucheron, C.; Kirsch-De Mesmaeker, A.; Luhmer, M. pH Dependence of photoinduced electron transfer with [ru(tap)32+]. Inorg. Chem. 2017, 56, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Troian-Gautier, L.; Mugeniwabagara, E.; Fusaro, L.; Cauet, E.; Kirsch-De Mesmaeker, A.; Luhmer, M. Photo-cidnp reveals different protonation sites depending on the primary step of the photoinduced electron-/proton-transfer process with Ru(II) polyazaaromatic complexes. J. Am. Chem. Soc. 2017, 139, 14909–14912. [Google Scholar] [CrossRef] [PubMed]
- Ghizdavu, L.; Pierard, F.; Rickling, S.; Aury, S.; Surin, M.; Beljonne, D.; Lazzaroni, R.; Murat, P.; Defrancq, E.; Moucheron, C.; et al. Oxidizing Ru(II) complexes as irreversible and specific photo-cross-linking agents of oligonucleotide duplexes. Inorg. Chem. 2009, 48, 10988–10994. [Google Scholar] [CrossRef] [PubMed]
- Lentzen, O.; Defrancq, E.; Constant, J.F.; Schumm, S.; Garcia-Fresnadillo, D.; Moucheron, C.; Dumy, P.; Kirsch-De Mesmaeker, A. Determination of DNA guanine sites forming photo-adducts with Ru(II)-labeled oligonucleotides; DNA polymerase inhibition by the resulting photo-crosslinking. J. Biol. Inorg. Chem. 2004, 9, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, J.P.; Kirsch-De Mesmaeker, A.; Demeunynck, M.; Lhomme, J. Synthesis and characterization of a new DNA-binding bifunctional Ruthenium(II) complex. J. Chem. Soc. Faraday Trans. 1993, 89, 3261–3269. [Google Scholar] [CrossRef]
- Villien, M.; Deroo, S.; Gicquel, E.; Defrancq, E.; Moucheron, C.; Kirsch-De Mesmaeker, A.; Dumy, P. The oxime bond formation as an efficient tool for the conjugation of ruthenium complexes to oligonucleotides and peptides. Tetrahedron 2007, 63, 11299–11306. [Google Scholar] [CrossRef]
- Trevisiol, E.; Renard, A.; Defrancq, E.; Lhomme, J. Fluorescent labelling of oligodeoxyribonucleotides by the oxyamino-aldehyde coupling reaction. Nucleos. Nucleot. Nucl. 2000, 19, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Forget, D.; Boturyn, D.; Defrancq, E.; Lhomme, J.; Dumy, P. Highly efficient synthesis of peptide-oligonucleotide conjugates: Chemoselective oxime and thiazolidine formation. Chem. Eur. J. 2001, 7, 3976–3984. [Google Scholar] [CrossRef]
- Singh, Y.; Spinelli, N.; Defrancq, E. Chemical strategies for oligonucleotide-conjugates synthesis. Curr. Org. Chem. 2008, 12, 263–290. [Google Scholar]
- Forget, D.; Renaudet, O.; Defrancq, E.; Dumy, P. Efficient preparation of carbohydrate-oligonucleotide conjugates (COCs) using oxime bond formation. Tetrahedron Lett. 2001, 42, 7829–7832. [Google Scholar] [CrossRef]
- Lee, E.J.; Mari, C.; Gel, M.; Gardiner, J.; Gasser, G.; Haylock, D. Immobilisation of multiple ligands using peptide nucleic acids: A strategy to prepare the microenvironment for cell culture. Chem. Select 2017, 2, 4028–4032. [Google Scholar] [CrossRef]
- Deroo, S.; Defrancq, E.; Moucheron, C.; Kirsch-De Mesmaeker, A.; Dumy, P. Synthesis of an oxyamino-containing phenanthroline derivative for the efficient preparation of phenanthroline oligonucleotide oxime conjugates. Tetrahedron Lett. 2003, 44, 8379–8382. [Google Scholar] [CrossRef]
- Tronchet, J.M.J.; Zosimolandolfo, G.; Balkadjian, M.; Ricca, A.; Zsely, M.; Barbalatrey, F.; Cabrini, D.; Lichtle, P.; Geoffroy, M. New types of spin-labeled sugar and nucleoside analogs—Pyrrolidine, morpholine and piperidine n-oxyls. Tetrahedron Lett. 1991, 32, 4129–4132. [Google Scholar] [CrossRef]
- Kirsch-De Mesmaeker, A.; Jacquet, L.; Nasielski, J. Ruthenium(II) complexes of 1,4,5,8-tetraazaphenanthrene (TAP) and 2,2′-bipyridine (bpy)—Ground-state and excited-state basicities of Ru2+(bpy)n(TAP)3-n (n = 0,1,2): Their luminescence quenching by organic buffers. Inorg. Chem. 1988, 27, 4451–4458. [Google Scholar] [CrossRef]
- Le Gac, S.; Rickling, S.; Gerbaux, P.; Defrancq, E.; Moucheron, C.; Kirsch-De Mesmaeker, A. A photoreactive Ruthenium(II) complex tethered to a guanine-containing oligonucleotide: A biomolecular tool that behaves as a “seppuku molecule”. Angew. Chem. Int. Ed. 2009, 48, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Deroo, S.; Le Gac, S.; Ghosh, S.; Villien, M.; Gerbaux, P.; Defrancq, E.; Moucheron, C.; Dumy, P.; Kirsch-De Mesmaeker, A. Oligonucleotide duplexes with tethered photoreactive Ruthenium(II) complexes: Influence of the ligands and their linker on the photoinduced electron transfer and crosslinking processes of the two strands. Eur. J. Inorg. Chem. 2009, 524–532. [Google Scholar] [CrossRef]
- Marcelis, L.; Ghesquiere, J.; Garnir, K.; Kirsch-De Mesmaeker, A.; Moucheron, C. Photo-oxidizing Ru-II complexes and light: Targeting biomolecules via photoadditions. Coord. Chem. Rev. 2012, 256, 1569–1582. [Google Scholar] [CrossRef]
- Marcelis, L.; Surin, M.; Lartia, R.; Moucheron, C.; Defrancq, E.; Kirsch-De Mesmaeker, A. Specificity of light-induced covalent adduct formation between Ru-II oligonucleotide conjugates and target sequences for gene silencing applications. Eur. J. Inorg. Chem. 2014, 3016–3022. [Google Scholar] [CrossRef]
- Reschner, A.; Bontems, S.; Le Gac, S.; Lambermont, J.; Marcelis, L.; Defrancq, E.; Hubert, P.; Moucheron, C.; Kirsch-De Mesmaeker, A.; Raes, M.; et al. Ruthenium oligonucleotides, targeting HPV16 E6 oncogene, inhibit the growth of cervical cancer cells under illumination by a mechanism involving p53. Gene Ther. 2013, 20, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.F.; Peto, J.; Meijer, C.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Lechner, M.S.; Mack, D.H.; Finicle, A.B.; Crook, T.; Vousden, K.H.; Laimins, L.A. Human papillomavirus e6 proteins bind p53 in vivo and abrogate p53-mediated repression of transcription. EMBO J. 1992, 11, 3045–3052. [Google Scholar] [PubMed]
- Thomas, M.; Pim, D.; Banks, L. The role of the e6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Durham, B.; Pinnick, D.V. Photochemistry of bis(2,2′-bipyridine) ruthenium(II) complexes. Abs. Am. Chem. Soc. 1982, 184, 190. [Google Scholar]
- Vrabel, M.; Pohl, R.; Klepetarova, B.; Votruba, I.; Hocek, M. Synthesis of 2′-deoxyadenosine nucleosides bearing bipyridine-type ligands and their Ru-complexes in position 8 through cross-coupling reactions. Org. Biomol. Chem. 2007, 5, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Vrabel, M.; Hocek, M.; Havran, L.; Fojta, M.; Votruba, I.; Klepetarova, B.; Pohl, R.; Rulisek, L.; Zendlova, L.; Hobza, P.; et al. Purines bearing phenanthroline or bipyridine ligands and their Ru-II complexes in position 8 as model compounds for electrochemical DNA labeling—Synthesis, crystal structure, electrochemistry, quantum chemical calculations, cytostatic and antiviral activity. Eur. J. Inorg. Chem. 2007, 2007, 1752–1769. [Google Scholar]
- Jager, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, O.; Famulok, M. A versatile toolbox for variable DNA functionalization at high density. J. Am. Chem. Soc. 2005, 127, 15071–15082. [Google Scholar] [CrossRef] [PubMed]
- Capek, P.; Cahova, H.; Pohl, R.; Hocek, M.; Gloeckner, C.; Marx, A. An efficient method for the construction of functionalized DNA bearing amino acid groups through cross-coupling reactions of nucleoside triphosphates followed by primer extension or PCR. Chem. Eur. J. 2007, 13, 6196–6203. [Google Scholar] [CrossRef]
- Olszewska, A.; Pohl, R.; Hocek, M. Trifluoroacetophenone-linked nucleotides and DNA for studying of DNA–protein interactions by 19F NMR spectroscopy. J. Org. Chem. 2017, 82, 11431–11439. [Google Scholar] [CrossRef]
- Hollenstein, M. Deoxynucleoside triphosphates bearing histamine, carboxylic acid, and hydroxyl residues—Synthesis and biochemical characterization. Org. Biomol. Chem. 2013, 11, 5162–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audrey, H.; Karin, B.; Kay, D.; Andreas, M. Structural basis for the KlenTaq DNA polymerase catalysed incorporation of alkenectural basis for tdified nucleotides. Chem. Eur. J. 2017, 23, 2109–2118. [Google Scholar]
- Hottin, A.; Marx, A. Structural insights into the processing of nucleobase-modified nucleotides by DNA polymerases. Acc. Chem. Res. 2016, 49, 418–427. [Google Scholar] [CrossRef]
- Seela, F.; Zulauf, M.; Rosemeyer, H.; Reuter, H. The DNA-stabilising nucleoside 7-iodo-2′-deoxytubercidin: Its structure in the solid state and in solution. J. Chem. Soc. Perkin Trans. 1996, 2, 2373–2376. [Google Scholar] [CrossRef]
- Vrabel, M.; Pohl, R.; Votruba, I.; Sajadi, M.; Kovalenko, S.A.; Ernsting, N.P.; Hocek, M. Synthesis and photophysical properties of 7-deaza-2′-deoxyadenosines bearing bipyridine ligands and their Ru(II)-complexes in position 7. Org. Biomol. Chem. 2008, 6, 2852–2860. [Google Scholar] [CrossRef]
- Kalachova, L.; Pohl, R.; Hocek, M. Synthesis of nucleoside mono- and triphosphates bearing oligopyridine ligands, their incorporation into DNA and complexation with transition metals. Org. Biomol. Chem. 2012, 10, 49–55. [Google Scholar] [CrossRef]
- Kalachova, L.; Pohl, R.; Bednarova, L.; Fanfrlik, J.; Hocek, M. Synthesis of nucleosides and dntps bearing oligopyridine ligands linked through an octadiyne tether, their incorporation into DNA and complexation with transition metal cations. Org. Biomol. Chem. 2013, 11, 78–89. [Google Scholar] [CrossRef]
- Vrabel, M.; Horakova, P.; Pivonkova, H.; Kalachova, L.; Cernocka, H.; Cahova, H.; Pohl, R.; Sebest, P.; Havran, L.; Hocek, M.; et al. Base-modified DNA labeled by [Ru(bpy)3]2+ and [Os(bpy)3]2+ complexes: Construction by polymerase incorporation of modified nucleoside triphosphates, electrochemical and luminescent properties, and applications. Chem. Eur. J. 2009, 15, 1144–1154. [Google Scholar] [CrossRef]
- Kovacs, T.; Otvos, L. Simple synthesis of 5-vinyl- and 5-ethynyl-2′-deoxyuridine-5′-triphosphates. Tetrahedron Lett. 1988, 29, 4525–4528. [Google Scholar] [CrossRef]
- Cahova, H.; Havran, L.; Brazdilova, P.; Pivonkova, H.; Pohl, R.; Fojta, M.; Hocek, M. Aminophenyl- and nitrophenyl-labeled nucleoside triphosphates: Synthesis, enzymatic incorporation, and electrochemical detection. Angew. Chem. Int. Ed. 2008, 47, 2059–2062. [Google Scholar] [CrossRef] [PubMed]
- McDougall, M.G.; Hosta, L.P.; Kumar, S.; Fuller, C.W. Analysis of DNA sequencing reaction products made with 7-halo-7-deaza-2′-deoxyguanosine-5′-triphosphate. Nucleos. Nucleot. 1999, 18, 1009–1011. [Google Scholar] [CrossRef]
- McDougall, M.G.; Sun, L.; Livshin, I.; Hosta, L.P.; McArdle, B.F.; Samols, S.B.; Fuller, C.W.; Kumar, S. Analogs of guanine nucleoside triphosphates for sequencing applications. Nucleos. Nucleot. Nucl. 2001, 20, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Kosuri, S.; Church, G.M. Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 2014, 11, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, D.; Jurkiewicz, P.; Cebecauer, M.; Hof, M.; Hocek, M. A rotational BODIPY nucleotide: An environment-sensitive fluorescence-lifetime probe for DNA interactions and applications in live-cell microscopy. Angew. Chem. Int. Ed. 2016, 55, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Neef, A.B.; Pernot, L.; Schreier, V.N.; Scapozza, L.; Luedtke, N.W. A bioorthogonal chemical reporter of viral infection. Angew. Chem. Int. Ed. 2015, 54, 7911–7914. [Google Scholar] [CrossRef] [PubMed]
- Bañuls, M.-J.; Jiménez-Meneses, P.; Meyer, A.; Vasseur, J.-J.; Morvan, F.; Escorihuela, J.; Puchades, R.; Maquieira, Á. Improved performance of DNA microarray multiplex hybridization using probes anchored at several points by thiol–ene or thiol–yne coupling chemistry. Bioconj. Chem. 2017, 28, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Rothlisberger, P.; Levi-Acobas, F.; Sarac, I.; Baron, B.; England, P.; Marliere, P.; Herdewijn, P.; Hollenstein, M. Facile immobilization of DNA using an enzymatic his-tag mimic. Chem. Commun. 2017, 53, 13031–13034. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram illustrating the occurrence of intercalation exclusively in the presence of a complementary strand [68,69].

Figure 2.
Structure of the duplex DNA (8- or 14-mer) containing ruthenium donor and acceptors [73].
Figure 2.
Structure of the duplex DNA (8- or 14-mer) containing ruthenium donor and acceptors [73].

Figure 3.
(a) PAGE analysis comparing the long-range oxidative damage on Ru(II)-modified DNA in duplexes with a G·A or G·T mismatch with damage in a complementary duplex. (b) Sequences of the duplexes used in the analysis. Arrows indicate the position of DNA damage as well as intensity of cleavage. Blue letters indicate the position of the mismatches as well as the GG doublet. Figure taken from [83] with permission.
Figure 3.
(a) PAGE analysis comparing the long-range oxidative damage on Ru(II)-modified DNA in duplexes with a G·A or G·T mismatch with damage in a complementary duplex. (b) Sequences of the duplexes used in the analysis. Arrows indicate the position of DNA damage as well as intensity of cleavage. Blue letters indicate the position of the mismatches as well as the GG doublet. Figure taken from [83] with permission.

Figure 4.
Structure of the Ru(II)-22 mer used in the mismatch experiments. The metal complex is linked, from the bpy’ ligand to the 5′ end of the duplex by a nine-carbon linker. All combinations (16) of base pairs and mismatches were synthesised [87].
Figure 4.
Structure of the Ru(II)-22 mer used in the mismatch experiments. The metal complex is linked, from the bpy’ ligand to the 5′ end of the duplex by a nine-carbon linker. All combinations (16) of base pairs and mismatches were synthesised [87].

Scheme 1.
Synthesis of phosphoramidites 6a and 6b [88].
Scheme 1.
Synthesis of phosphoramidites 6a and 6b [88].

Figure 5.
Chemical structures of the nucleosides containing the diastereomer Λ-1 and Δ-1 and their corresponding phosphoramidites (Λ-2 and Δ-2).
Figure 5.
Chemical structures of the nucleosides containing the diastereomer Λ-1 and Δ-1 and their corresponding phosphoramidites (Λ-2 and Δ-2).

Figure 6.
Structure of the Ru-containing-Uridine.

Scheme 2.
Synthesis and electrochemical data of acetyacetonate complexes (7–12) and hydroxamate complexes (13–15) [95].
Scheme 2.
Synthesis and electrochemical data of acetyacetonate complexes (7–12) and hydroxamate complexes (13–15) [95].

Scheme 3.
Synthesis of triphosphate 16 (dRuTP) [95].
Scheme 3.
Synthesis of triphosphate 16 (dRuTP) [95].

Figure 7.
Enzymatic incorporation of dRuTP. Primer extension experiments using the Klenow fragment of DNA polymerase I resolved on a 20% PAGE. Lanes 1 and 2: control reactions missing one natural triphosphate (dCTP or TTP); lane 3: control reaction with all four natural dNTPs; lane 4: four natural dNTPs and dRuTP; lane 5: dRuTP used instead of TTP; lane 6: control reactions without TTP and dRuTP. Figure taken from [95] with permission.
Figure 7.
Enzymatic incorporation of dRuTP. Primer extension experiments using the Klenow fragment of DNA polymerase I resolved on a 20% PAGE. Lanes 1 and 2: control reactions missing one natural triphosphate (dCTP or TTP); lane 3: control reaction with all four natural dNTPs; lane 4: four natural dNTPs and dRuTP; lane 5: dRuTP used instead of TTP; lane 6: control reactions without TTP and dRuTP. Figure taken from [95] with permission.

Scheme 4.
Synthetic pathway for the Ru-DNA conjugate 17.

Figure 8.
Structures of the Ru complex linked branched oligonucleotides 18–22.

Scheme 5.
Hybridization of 21 (blue) by the addition of its complementary Ru-DNA strand 22 (red). (a) 90 °C to 4 °C, overnight; (b) 4 °C, overnight [101].
Scheme 5.
Hybridization of 21 (blue) by the addition of its complementary Ru-DNA strand 22 (red). (a) 90 °C to 4 °C, overnight; (b) 4 °C, overnight [101].

Figure 9.
Structures of the Ru complex linked branched oligonucleotides 23, 24.

Figure 10.
Structures of m25–m32 mono Ru-DNA and b25–b32 bis Ru-DNA.

Figure 11.
Structures of the linear DNA nanoassemblies with one to seven ruthenium complexes [104].
Figure 11.
Structures of the linear DNA nanoassemblies with one to seven ruthenium complexes [104].

Figure 12.
Structures of m33–m35 mono Ru-DNA and b33–b35 bis Ru-DNA.

Figure 13.
Synthesis of the three-way junction by hybridization of DNA sequences 1, 2, and 3 (black lines represent the dodecyl linkers) and structures of three-way DNA junctions with mono Ru-DNA and double-star assemblies [106].
Figure 13.
Synthesis of the three-way junction by hybridization of DNA sequences 1, 2, and 3 (black lines represent the dodecyl linkers) and structures of three-way DNA junctions with mono Ru-DNA and double-star assemblies [106].

Scheme 6.
Preparation of oligonucleotide-Ru conjugates. 5′p = 5′-phosphate group, M = 5-methylcytosine [109].
Scheme 6.
Preparation of oligonucleotide-Ru conjugates. 5′p = 5′-phosphate group, M = 5-methylcytosine [109].

Scheme 7.
Formation of a covalent photoadduct between guanine and [Ru(TAP)3] under irradiation [125,126].

Figure 14.
Structures of the Ru complexes [Ru(TAP)2Phen]2+ 37, [Ru(TAP)2(TPAC)]2+ 38 [131], [Ru(TAP)2phen’]2+ 39, and [Ru(TAP)2TAP’]2+ 40 [132].

Scheme 8.
Preparation of the oxyamino phenanthroline derivative [138].
Scheme 8.
Preparation of the oxyamino phenanthroline derivative [138].

Figure 15.
Structures of the oligonucleotide-Ru complexes 41–44 [132].
Figure 15.
Structures of the oligonucleotide-Ru complexes 41–44 [132].

Figure 16.
Structures of the Ru-ODN molecules [141].
Figure 16.
Structures of the Ru-ODN molecules [141].

Figure 17.
PAGE analysis of the photochemical behaviour of Ru(T)-ODN(G) in the absence (lanes 1–6) and in the presence (lanes 7–12) of the complementary target strand. Lanes 13, 14, and 15 correspond to Ru(T)-ODN(G) that was irradiated for 30 min in the presence of non-complementary sequences. Figure taken from [141] with permission.
Figure 17.
PAGE analysis of the photochemical behaviour of Ru(T)-ODN(G) in the absence (lanes 1–6) and in the presence (lanes 7–12) of the complementary target strand. Lanes 13, 14, and 15 correspond to Ru(T)-ODN(G) that was irradiated for 30 min in the presence of non-complementary sequences. Figure taken from [141] with permission.

Scheme 9.
Mechanism of action of Ru(T)-ODN(G) molecules. (a) With a complementary target, interstrand photoreaction. (b) Without a target, or with a non-complementary strand, intramolecular photoreaction [141].
Scheme 9.
Mechanism of action of Ru(T)-ODN(G) molecules. (a) With a complementary target, interstrand photoreaction. (b) Without a target, or with a non-complementary strand, intramolecular photoreaction [141].

Figure 18.
Structure of Ru-ASO and its complementary sequence [145].
Figure 18.
Structure of Ru-ASO and its complementary sequence [145].

Figure 19.
Synthetic pathway for the preparation of nucleoside triphosphates 45, 46 [160].
Figure 19.
Synthetic pathway for the preparation of nucleoside triphosphates 45, 46 [160].

Scheme 10.
Synthetic pathway for the preparation of nucleotides 47–50 [163].
Scheme 10.
Synthetic pathway for the preparation of nucleotides 47–50 [163].


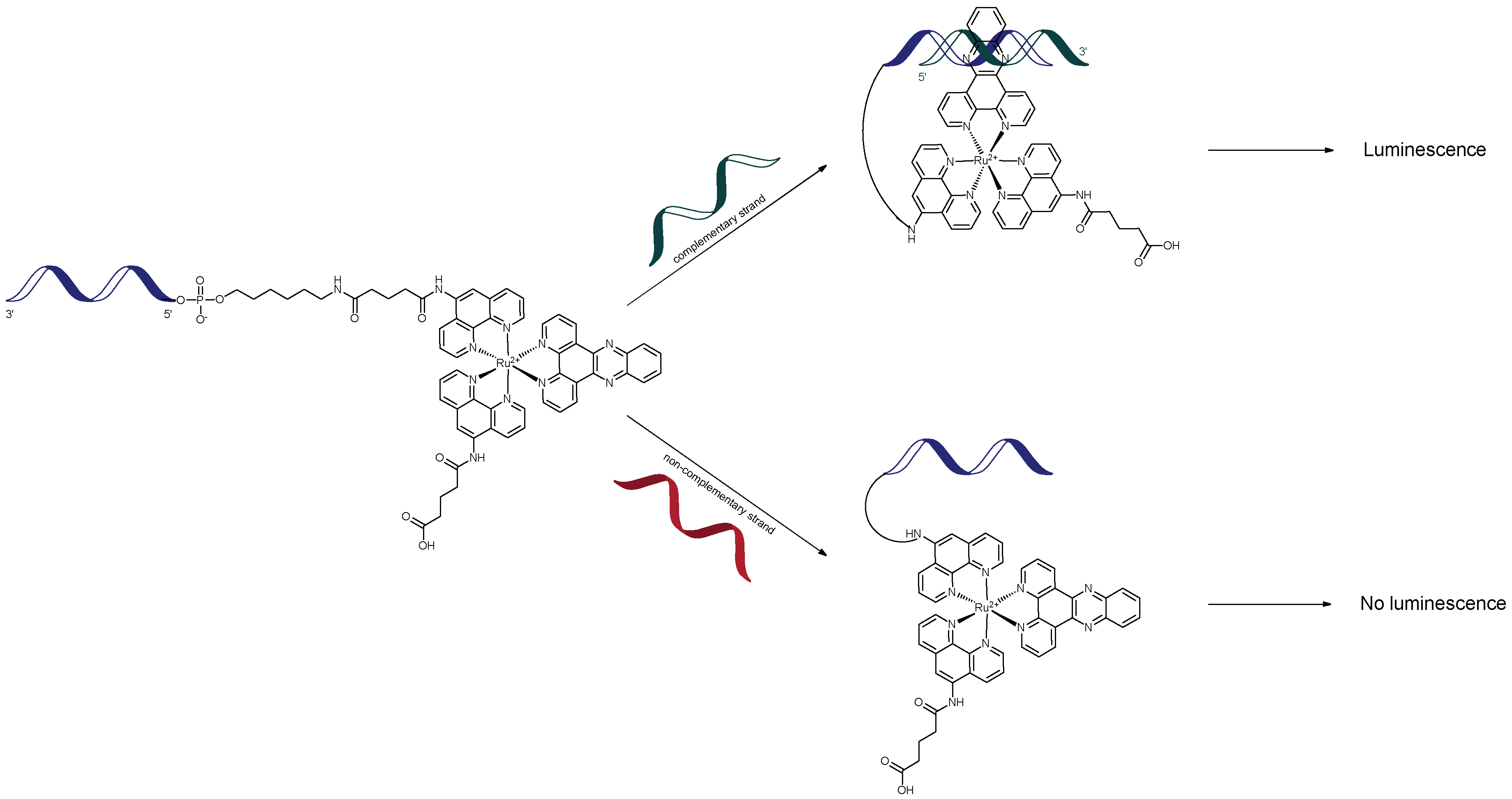{kind=link}
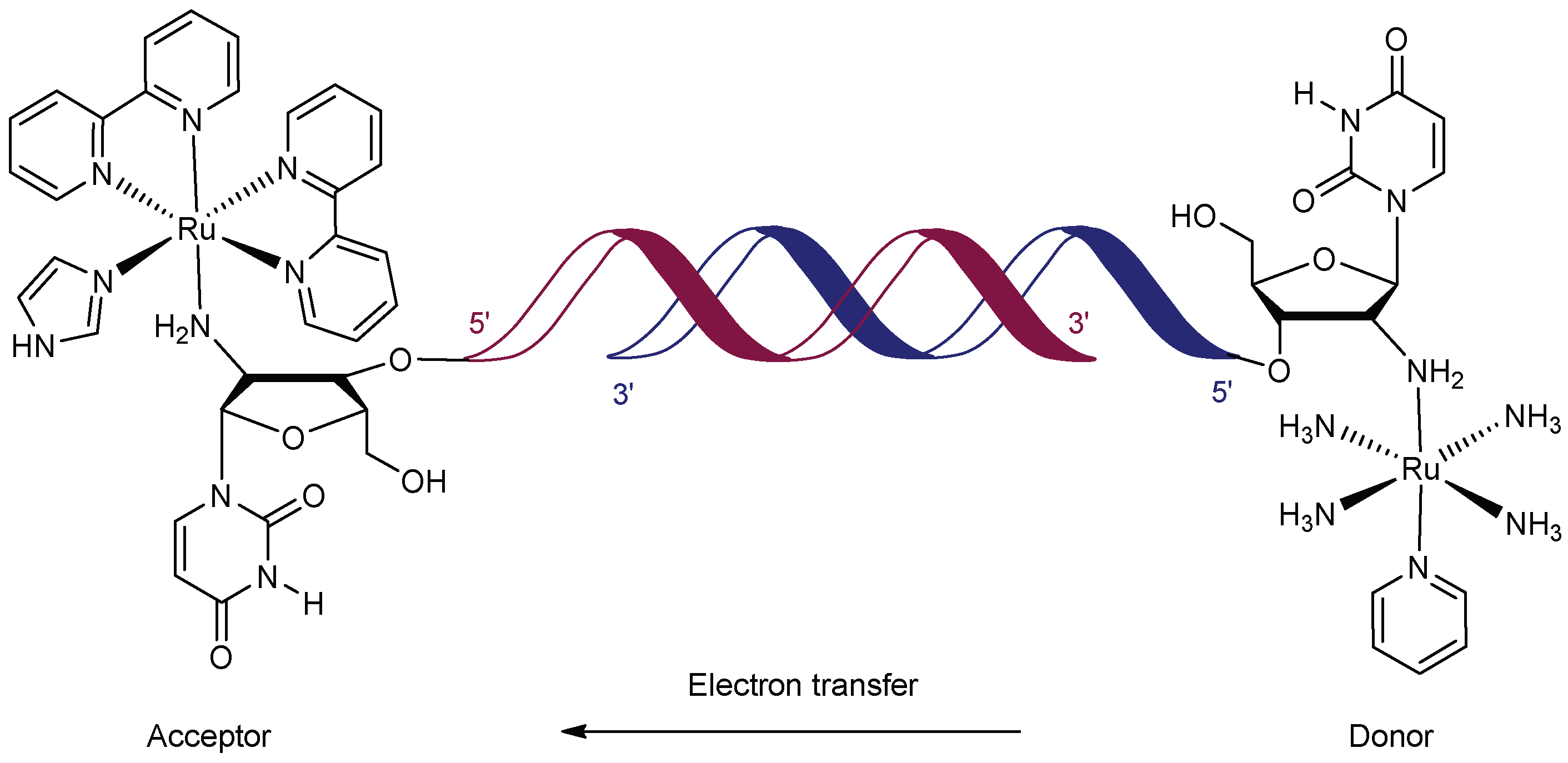{kind=link}
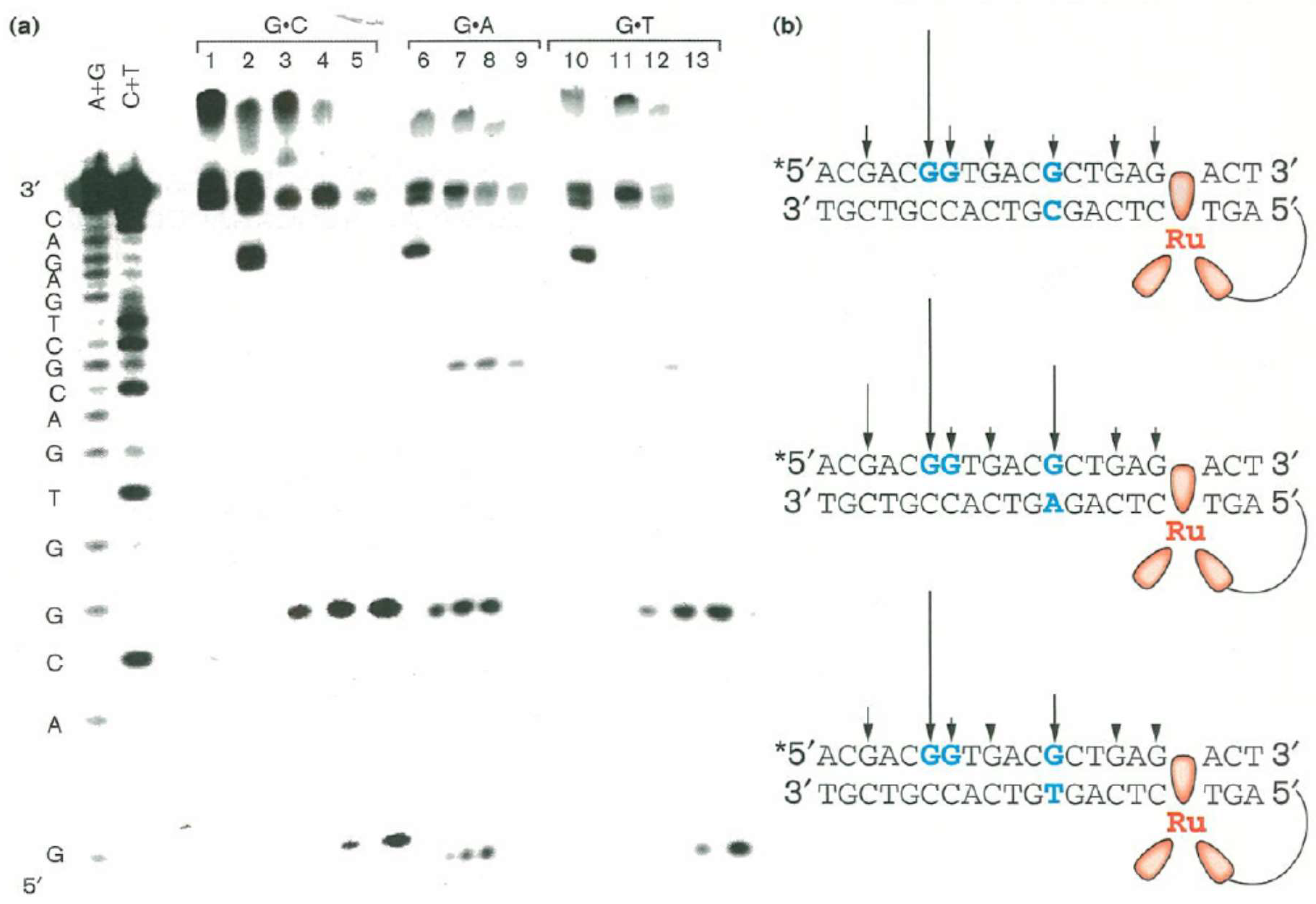{kind=link}
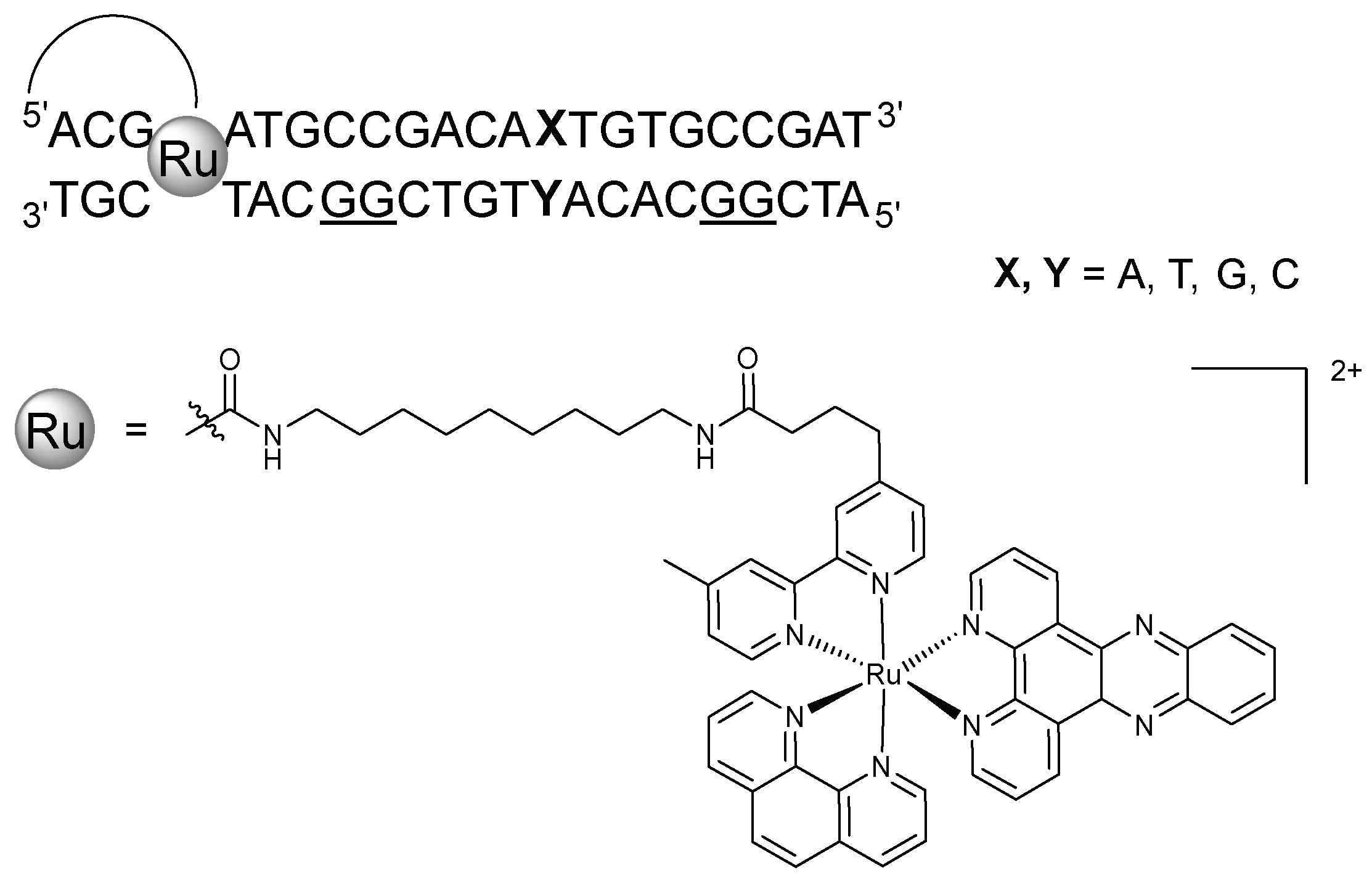{kind=link}
{kind=link}
{kind=link}
{kind=link}
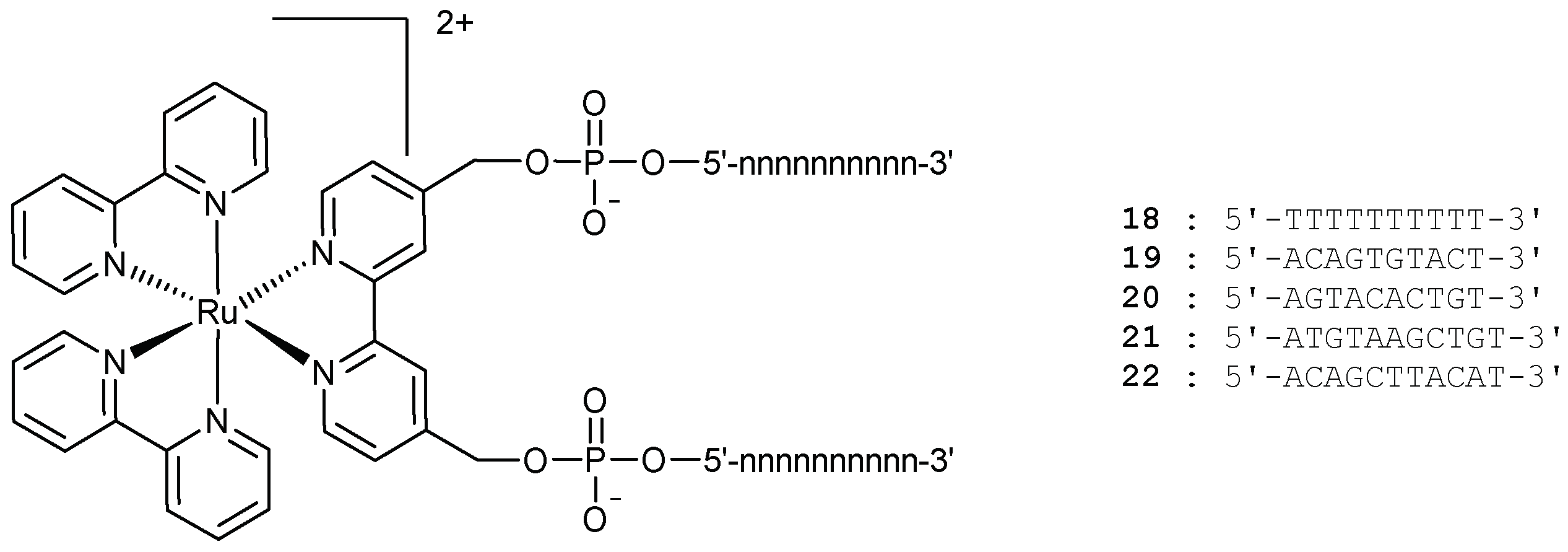{kind=link}
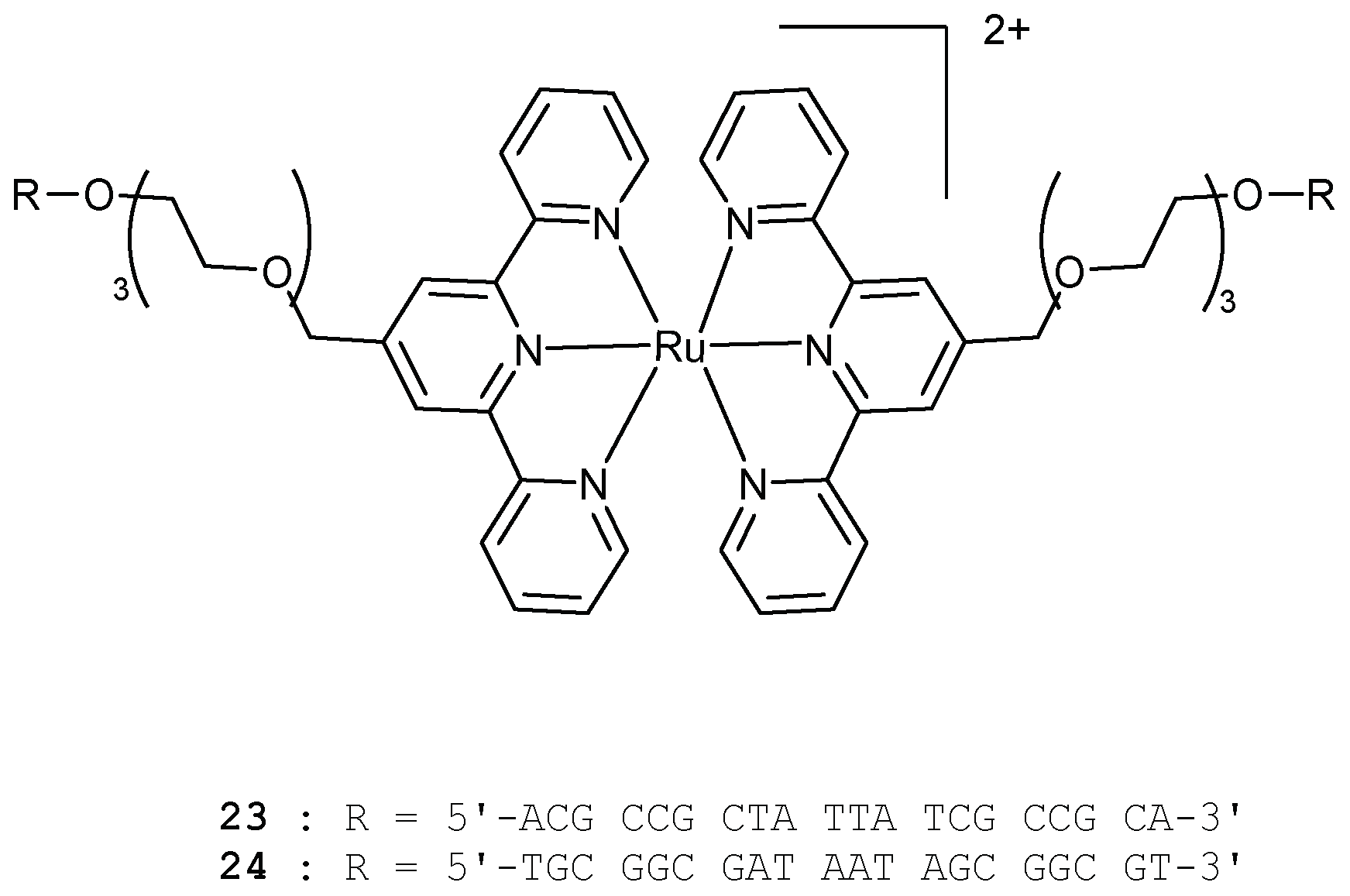{kind=link}
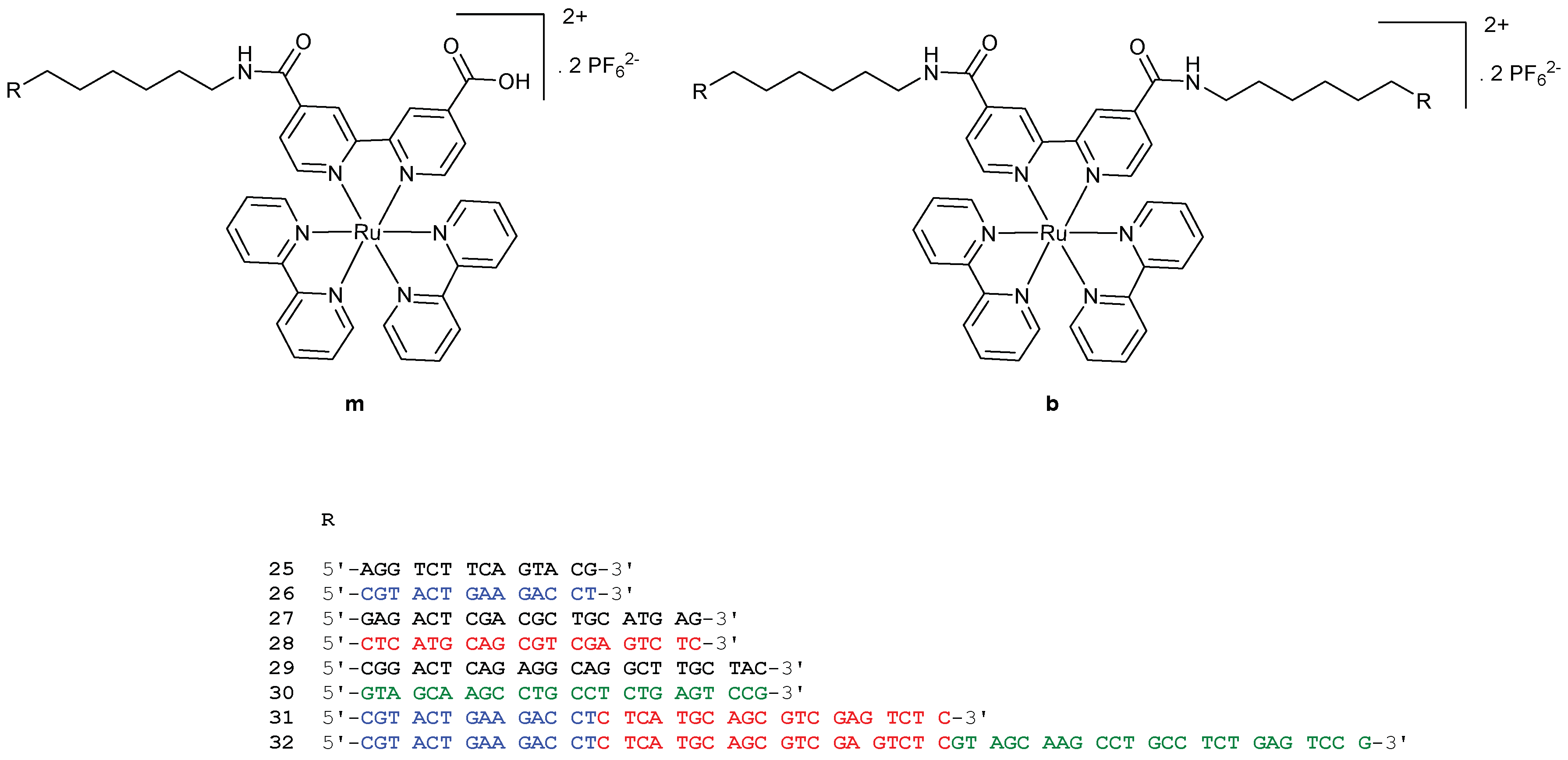{kind=link}
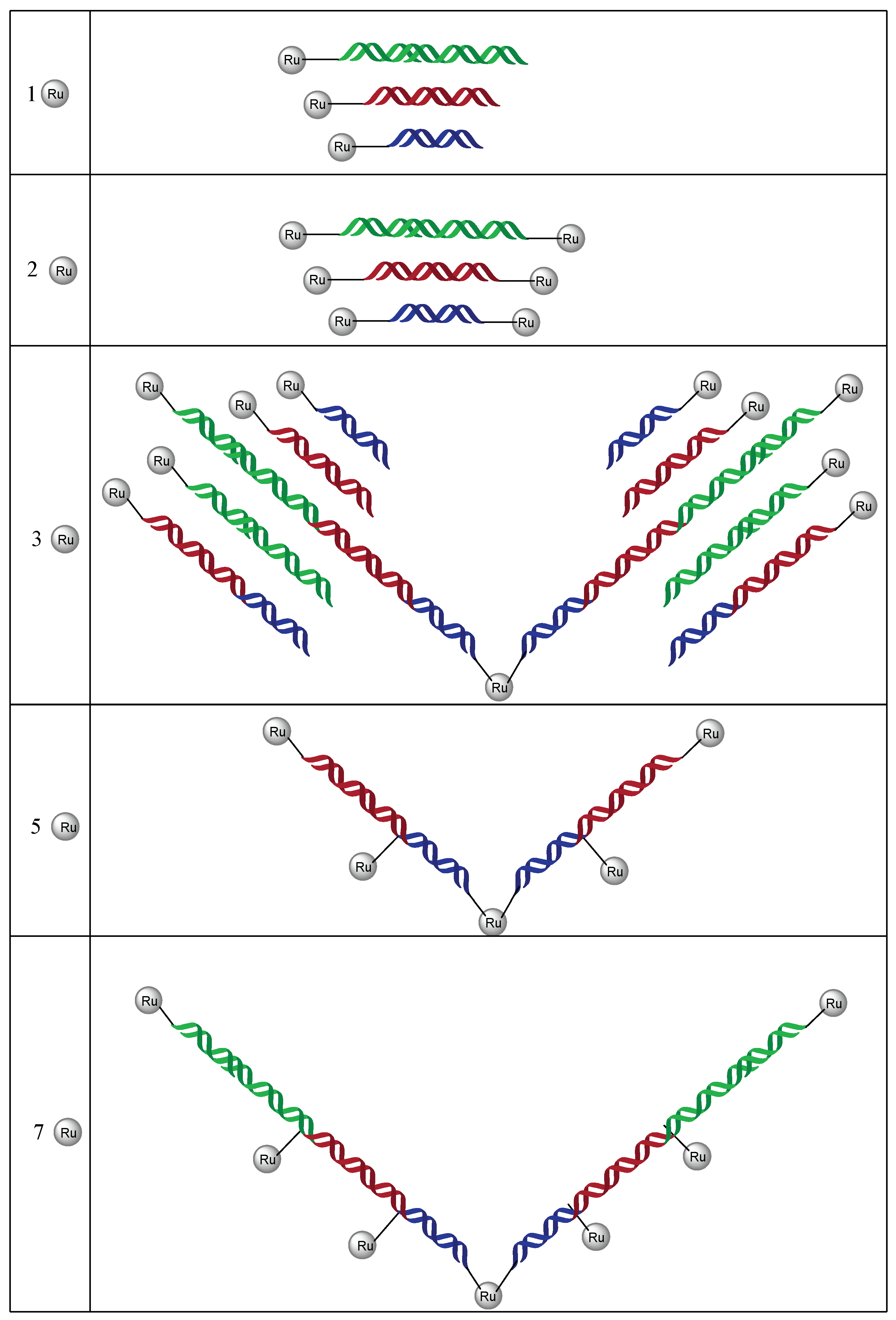{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
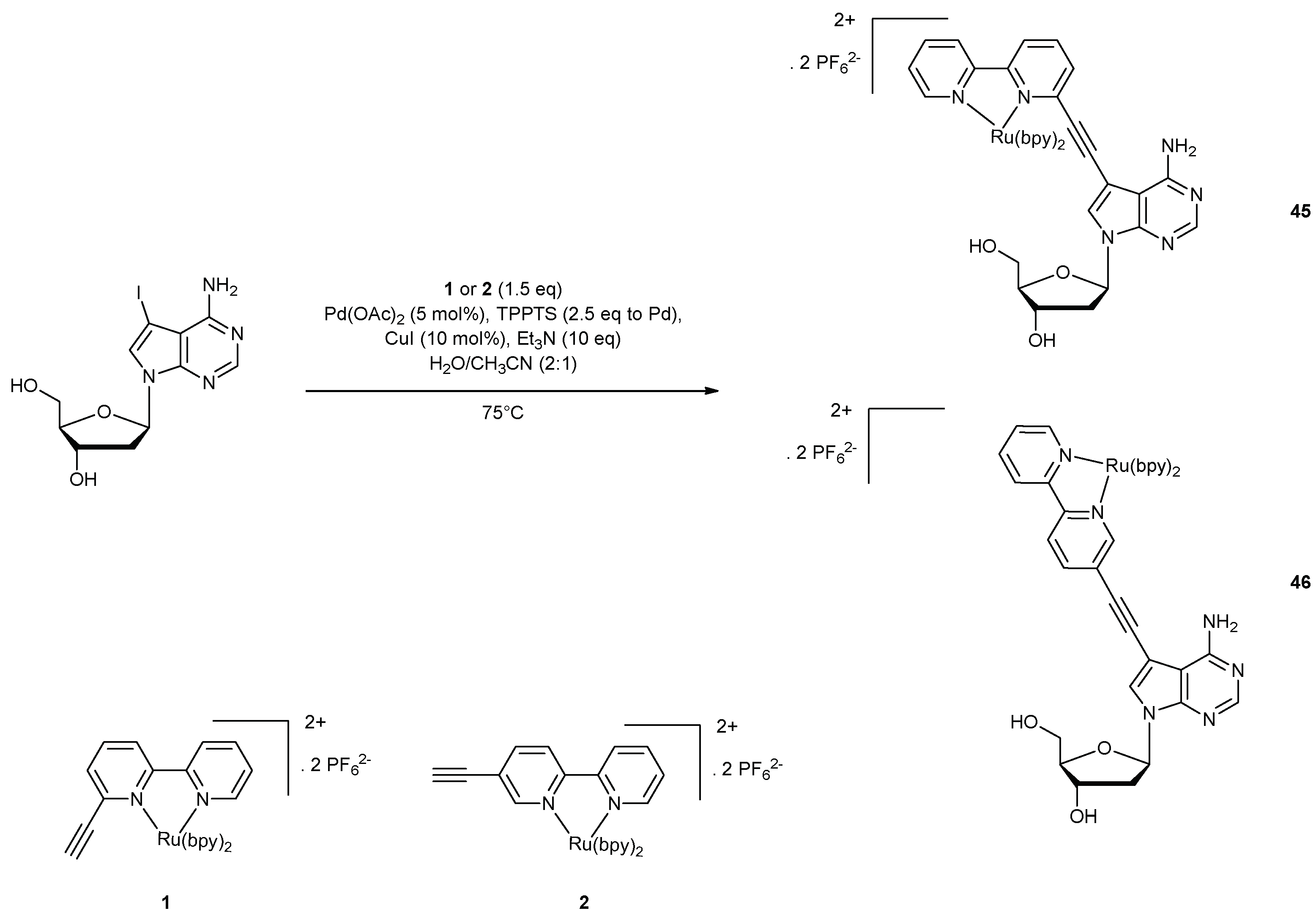{kind=link}
{kind=link}
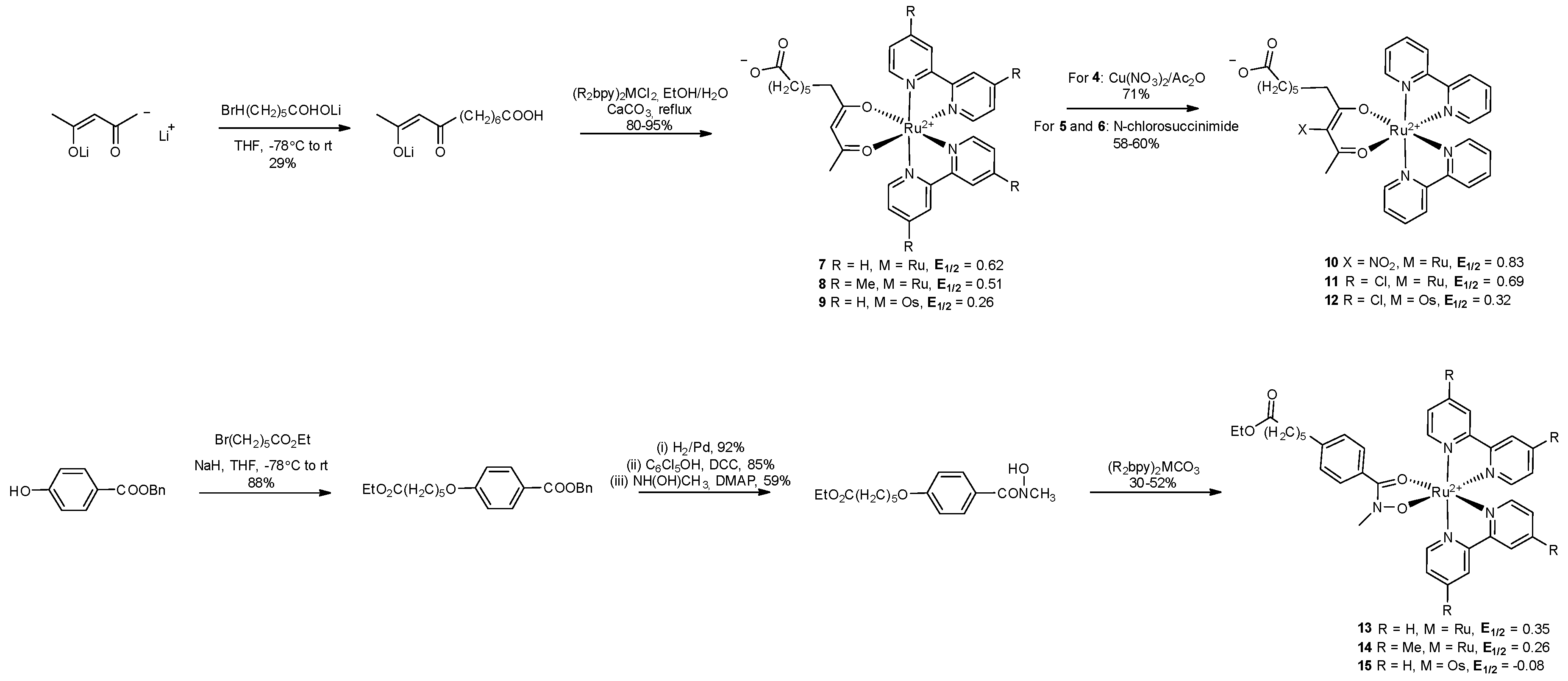{kind=link}
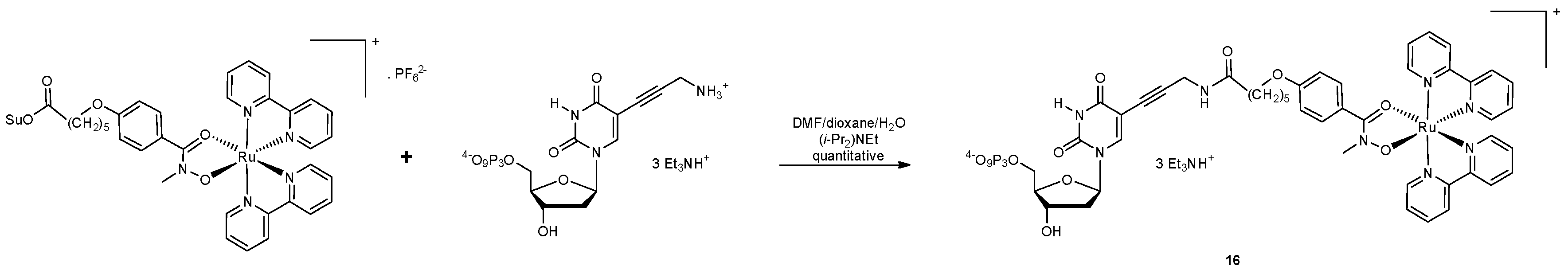{kind=link}
{kind=link}
{kind=link}
{kind=link}
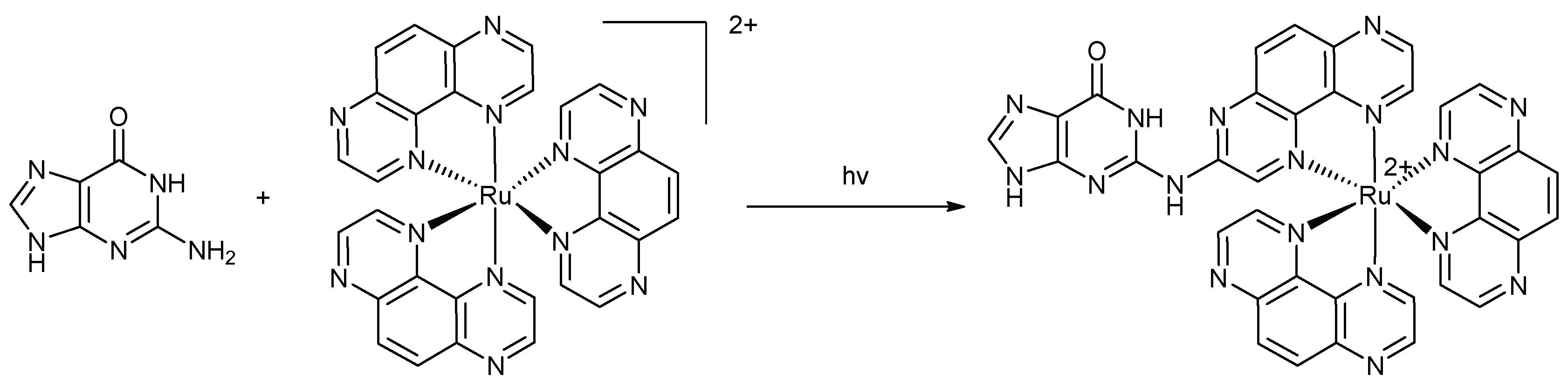{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Luminescent intensity of intramolecular metal-DNA complexes relative to that of 5′-Ru(phen’)2dppz-AGRGCCAAGCTTGCA-3′, integrated from 500 to 800 nm [74].
Table 1.
Luminescent intensity of intramolecular metal-DNA complexes relative to that of 5′-Ru(phen’)2dppz-AGRGCCAAGCTTGCA-3′, integrated from 500 to 800 nm [74].
| Sample | Relative Intensity |
|---|---|
 | 1.00 |
 | 0.00 |
 | 1.15 |
 | 0.08 |
 | 0.43 |

Table 2.
Luminescent lifetimes of tris(phenanthroline)metal-DNA complexes. Samples were dissolved in buffer (5 mM tris, 50 mM NaCl, pH 7.2) to ≤5 μM and experiments performed at 10 °C to ensure duplex stability [74].
Table 2.
Luminescent lifetimes of tris(phenanthroline)metal-DNA complexes. Samples were dissolved in buffer (5 mM tris, 50 mM NaCl, pH 7.2) to ≤5 μM and experiments performed at 10 °C to ensure duplex stability [74].
| Sample | Lifetime (ns) |
|---|---|
 | 820 |
 | 820 |
 | 830 |
 | 830 |

Table 3.
Table showing the position of the osmium acceptor, [Os(bpy)2(3-ethynyl-1,10-phenanthroline)]2+, in various 19-mer duplexes where the ruthenium donor position, [Ru(bpy)2(3-ethynyl-1,10-phenanthroline)]2+, remains the same (U-19). The duplex containing strand 1 and 2 is used as a control.
Table 3.
Table showing the position of the osmium acceptor, [Os(bpy)2(3-ethynyl-1,10-phenanthroline)]2+, in various 19-mer duplexes where the ruthenium donor position, [Ru(bpy)2(3-ethynyl-1,10-phenanthroline)]2+, remains the same (U-19). The duplex containing strand 1 and 2 is used as a control.
| Strand | Sequence | Os Position | Weighted Average Time (μs) |
|---|---|---|---|
| 1 2 |  | ||
| 3 2 |  | 1.31 ± 0.01 | |
| 3 4 |  | U-4 | 0.20 ± 0.06 |
| 3 5 |  | U-7 | 0.27 ± 0.01 |
| 3 6 |  | U-10 | 0.63 ± 0.01 |
| 3 7 |  | U-13 | 1.02 ± 0.12 |
| 3 8 |  | U-16 | 1.24 ± 0.02 |
| 3 9 |  | U-19 | 1.27 ± 0.01 |
Table 4.
Showing the position of the osmium acceptor, [Os(bpy)2(3-ethynyl-1,10-phenanthroline)]2+, in various 19-mer duplexes where the ruthenium donor position, 1, remains the same (U-19) [92].
Table 4.
Showing the position of the osmium acceptor, [Os(bpy)2(3-ethynyl-1,10-phenanthroline)]2+, in various 19-mer duplexes where the ruthenium donor position, 1, remains the same (U-19) [92].
| Strand | Sequence | Os Position | Weighted Average Time (μs) |
|---|---|---|---|
| 10 2 |  | 0.59 ± 0.01 | |
| 10 4 |  | U-4 | 0.07 ± 0.01 |
| 10 5 |  | U-7 | 0.18 ± 0.01 |
| 10 6 |  | U-10 | 0.37 ± 0.01 |
| 10 7 |  | U-13 | 0.52 ± 0.01 |
| 10 8 |  | U-16 | 0.58 ± 0.01 |
| 10 9 |  | U-19 | 0.59 ± 0.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Flamme, M.; Clarke, E.; Gasser, G.; Hollenstein, M. Applications of Ruthenium Complexes Covalently Linked to Nucleic Acid Derivatives. Molecules 2018, 23, 1515. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23071515
AMA Style
Flamme M, Clarke E, Gasser G, Hollenstein M. Applications of Ruthenium Complexes Covalently Linked to Nucleic Acid Derivatives. Molecules. 2018; 23(7):1515. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23071515
Chicago/Turabian StyleFlamme, Marie, Emma Clarke, Gilles Gasser, and Marcel Hollenstein. 2018. "Applications of Ruthenium Complexes Covalently Linked to Nucleic Acid Derivatives" Molecules 23, no. 7: 1515. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23071515