
Polyoxometalate-Based Catalysts for CO2 Conversion


1
State Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of LICP, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou 730000, China
2
University of Chinese Academy of Sciences, Beijing 100049, China
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(11), 2069; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112069
Submission received: 5 May 2019
/
Revised: 22 May 2019
/
Accepted: 23 May 2019
/
Published: 30 May 2019
(This article belongs to the Special Issue Polyoxometalates and Polyoxometalate-Based Systems)
Abstract
:Polyoxometalates (POMs) are a diverse class of anionic metal-oxo clusters with intriguing chemical and physical properties. Owing to unrivaled versatility and structural variation, POMs have been extensively utilized for catalysis for a plethora of reactions. In this focused review, the applications of POMs as promising catalysts or co-catalysts for CO2 conversion, including CO2 photo/electro reduction and CO2 as a carbonyl source for the carbonylation process are summarized. A brief perspective on the potentiality in this field is proposed.
1. Introduction
As the main greenhouse gas produced by human activity, about 10 ± 0.5 gigatons CO2 were released in 2018, bringing the atmospheric CO2 concentration level over the threshold of 400 ppm [1]. The large increase of global CO2 levels has led to serious environment problem. Meanwhile, it is also the most abundant and nontoxic carbon resource for preparing useful compounds [2]. From a practical point of view, its catalytic conversion is of significance to the supply of renewable energy, chemicals, and mitigation of global warming.
Polyoxometalates with {MOx} (x = 5, 6) as basic construction units have derived an enormous fraternity of inorganic molecular complexes [3,4,5,6]. To obtain various composite materials with specific function, the modification and decoration of POM can be attained by partially substituting {MOx} units with different transition metal moieties or attaching organometallic complexes onto POM. Based on geometrical morphology, most of POMs can be classified as Keggin, Wells-Dawson, Anderson-Evans, Silverton, Waugh, Strandberg, Lindqvist, and Peacock-Weakley type structures, which are named after their corresponding discoverers [7]. Among these types, Keggin-type POMs ([XM12O40]n−) are comparatively well-studied. POMs discussed in the following sections are mostly Keggin type [8]. The original Keggin structure is designated α-, which consists of a tetrahedron central ion, [XO4]n − 8, caged by twelve MO6 octahedron. The Keggin structure includes four additional isomers (β-, γ-, δ-, and ε-), each resulting from 60° rotations of the four {M3O13} units [9]. The lacunary species of Keggin-type POMs stem from removing a variable number of {MO6} octahedra from the plenary polyanion, resulting in metal-oxide clusters with vacant addenda metal sites. Such vacant sites can be treated as inorganic multidentate ligands (Figure 1) [10], For example, sandwich-type POMs are usually synthesized by the reaction of transition metal ions with appropriate lacunary POM precursors [5,8]. The extraordinary diversity and synthetic accessibility of POMs have led to a wide spectrum of applications, ranging from catalysis, biochemistry/medicinal chemistry, to materials science [11,12,13,14]. In the field of catalysis, the development of POMs as oxidation and acid catalysts was flourish in the past few decades, with molybdic and vanadomolybdic clusters being more employed in the former case and tungstic ones in the latter [12,15]. The explorations of POMs as catalysts (or co-catalysts) for many other industrially important transformations were sought-after.
As early as 1988, Kozik and co-workers first reported the coordination of CO2 with several POM derivatives [16]. Afterwards more researches about the interaction of POMs with CO2 emerged [17,18,19,20,21,22]. Since the interaction between CO2 and POM is one of the crucial steps for catalytic transformations of CO2, it is necessary to clarify the mode of such interaction. However, considering the facts that CO2 can be transformed to or with water and this transformation is both reversible and temperature-dependent, the explicit “real” form of dissolved CO2 to interact with POM catalyst, was challenging to be justified. POMs displayed variable interacting modes. 13C NMR, UV/vis, IR with isotope-labelled CO2 (13CO2 and C18O2), and X-ray crystallography are useful tools to solve this issue. As pointed in the seminal report of Kozik et al. [16], the IR spectra and thermochromic behavior of POM α-[SiW11O39Co]6− in the presence of either or was in contrast to the scenario with CO2. On 13C NMR spectra, the 13C chemical shifts of aqueous CO2, , and are respectively at 125, 160, and 162 ppm. After bubbling CO2 into hydrous toluene solution of α-[SiW11O39Co]6−, the appearance of two signals at 792 and 596 ppm demonstrated the presence of two different kinds of paramagnetic CO2 species. In their report, two patterns were suggested on the basis of all 13C NMR, IR observations: (i) the complexation between α-[SiW11O39Co]6− and CO2 was either via CO2 complexes with a direct η1 metal-carbon bond or bicarbonate complexes; (ii) the existence of H-bonding in the CO2 complexes was plausible. Other modes of POM-CO2 or POM- interaction were also discovered. Hill et al. reported the sandwich-type encapsulation of via forming [(YOH2)3(CO3)(A-α-PW9O34)2]11− [20]. [SiMo11CoO38(CO2)]n polymeric chains reported by Xu et al. displayed the μ-η1,η1-OCO linear coordination mode of POM-CO2 interaction [21]. Two kinds of POM-CO2 interaction modes were observed on [(n-C8H17)4N]8[α2-P2W17O61Zn-(CO2)] [21]. The stronger “side-on” binding of CO2 by is predominant at higher temperatures (room temperature down to ca. 250 K) and the more weakly coordinated “end-on” POM-Zn-O-C-O structures were observable at lower temperatures (Figure 2). One particular case was uptaking 30 CO2 molecules by one capsule of [{(MoVI) O21(H2O)6}12{ O4(CH3COO)}30]42−. In this case, CO2 reacted directly with the H2O ligands in { O4(H2O)2}2+ linkers to generate at pH 7. Then the uptake of 30 CO2 molecules was realized via the fast exchange of acetate by carbonate ligands [22]. Above cases indicated the abundance of POM-CO2 interaction mode.
In this context, we have witnessed the leap in developing POMs-based catalysts for the photo- or electrocatalytic CO2 reduction and CO2 as C1 synthon in organic synthesis. Herein, in this review, we focused on the recent experimental and theoretical advances in CO2 transformation with POMs-based catalyst or co-catalyst. The key factors for high-efficiency POMs-catalyzed CO2 conversion are highlighted.
2. Photocatalytic CO2 Reduction
Only high-energy vacuum ultraviolet laser (<200 nm) is able to directly excite and split CO2 into CO and O fragments [23], therefore highly efficient photocatalyst is vital to motivate and accelerate photocatalytic CO2 reductions with low-energy common visible light (>380 nm). This reduction process usually involves multi-electron transfer and the final products are carbon monoxide (CO), formic acid (HCOOH), formaldehyde (HCHO), methanol (CH3OH), and methane (CH4) [24,25]. The corresponding redox potentials for the possible CO2 reduction at pH = 7 are listed in Table 1.
Since the pioneering application of Honda–Fujishima effect in TiO2-catalyzed CO2 photoreduction was reported [26], multitudinous different type of photocatalysts, including simple metal oxides [25], perovskite oxides [27,28], C3N4 [29], MOFs [30], conjugated polymers [31,32], and POMs [33,34] have been developed. Since the seminal study on the photochemistry of molybdates and tungstates for analytical purposes over half century ago [35,36], the photoredox chemistry of POMs has been verified for a long time. The metals in POMs are fully oxidized with d0 electron configuration. Light absorption is mainly attributed to O→M ligand to-metal charge transfer (LMCT) bands in the wide range of the electronic spectra. Consequently, an electron is promoted from a spin-paired, doubly occupied bonding orbital (HOMO) to an empty, antibonding orbital (LUMO), thus an oxo-centered radical is generated. This photo-excited POMs are more reactive both in oxidation and reduction than the non-excited species [37]. In the photocatalysis by POM with LMCT mode, forming lower-energy O→M LMCT transitions is vital to more efficiently absorb visible light. Substituting addenda centers with different metals has been well demonstrated as one of the effectual tactics to tune the photochemical properties of POMs and optimize their visible light absorption via narrowing the band gap between valence band and conduction band. For instance, Streb et al. discovered that the vanadium substitution effectively improved the visible light absorption and photocatalytic activity of molybdate [Mo6O19]2− [38]. TD-DFT calculations disclosed the corresponding contribution of O→V LMCT on absorption profile in the visible range. The progress of such strategy on POM-catalyzed selective photooxidation driven by visible light has been included in the recent review [15]. Besides altering the addenda centers on POMs, various other strategies have also been established in order to foster POMs as effective photocatalyst and most of their utilization were focused on photooxidation of water, photodegradation of organic dyes, as well as H2 evolution [37,39]. Here the progress of photoreduction of CO2 with POMs-based catalyst was summarized.
2.1. CO2 to CO
2.1.1. Homogeneous Catalysts
Neumann and co-workers reported that Ru-substituted POM ([RuIII(H2O)SiW11O39]5−) in combination with tertiary amines exhibited photoactivity for CO2 reduction Figure 3 [40]. Triethyl amine acted as the sacrificial agent. CO was the major product (~50 μmol after 20 h irradiation with ca. 2% quantum yield). None of HCOOH, MeOH, and CH4 was detected. UV/Vis, EPR, 13C NMR, and isotope labelling demonstrated that coordinated CO2 ([RuIII(CO2)SiW11O39]5−) was easily formed by substituting H2O in [RuIII(H2O)SiW11O39]5−. DFT results demonstrated that CO2 tended to coordinate to RuIII by forming a Ru-O bond in an “end-on” manner. Based on these experimental and computational results, the authors concluded that the RuIII site was responsible for activating CO2 via the coordination and the lacunary [SiW11O39]8− site acted as photocatalyst in this process.
By employing H2 instead of tertiary amines as the sacrificial agent, a viable CO2 reduction approach with Pt/C and ReI(L)(CO)3- O40 (L = 5,6-(15-crown-5)-1,10-phenanthroline) as synergistic catalysts was developed. The grafting of ReI(L)(CO)3 complexes onto POMs was achieved via the complexation of crown ether moiety on L with the sodium cation binding two PW12O40 moieties (Figure 4) [41]. H2 was oxidized with the facilitation of Pt(0) on the Pt/C surface to afford two protons. In the following step, protons and electrons retained by Pt were simultaneously transferred onto ReI(L)(CO)3(CH3CN)- O40 ( O40 + 2H+ + 2e− → MH3 O40) (Scheme 1). ReI(L)(CO)3-MH3 O40 possessed the photoreductive activity for converting CO2 to CO, because its transition to excited CO2-reduction active state was allowed by absorbing visible light. CO were almost exclusively produced with turnover of 22.6% and 1.1% quantum yield. Besides CO, only trace CH4 but no further CO reduction products (e.g., CH3OH, HCHO, and HCOOH) was detected on GC-MS after 14 h irradiation.
The TD-DFT calculations disclosed a more detailed mechanism for the photoreduction of CO2 to CO with this POM-ReI complex catalyst [42]. Simulated absorption spectrum of ReI-POM complex was calculated at different functional level (i.e., X3LYP, M06X, B3LYP, and CAMB3LYP) and the computed results were found to be basically consistent with the experimental absorption spectrum (two major bands with maxima at 500 and 656 nm). In the ground state without any light irradiation, bonding CO2 with ReI center to afford the reactive POM-ReI- complex had to overcome a high energy barrier (≥38 kcal mol−1), therefore, the direct CO2 reduction with ReI-POM occurred less likely (Figure 5). In the conditions with photoexcitation, the excited ReI-POM was allowed to interact with CO2 to form POM-ReI-. The subsequently protonation and further electron reduction process were exothermic by more than 78 kcal mol−1. These energy profiles revealed that the role of POM as photosensitizer in the reduced state, electron “shuttles” as well as electron/proton reservoirs in this process. Plausible CO2 photoreduction mechanism consists of five steps: (1) photoexcitation and charge transfer; (2) disassociation of solvent molecule; (3) CO2 coordination; (4) proton and electron transfer; and (5) release of CO and recoordination of solvent.
Recently, by replacing crown ether-affiliated 1,10-phenanthroline with 6-hydrazinyl-2,2′-bipyridine on the Re complex, the Re2(CO)6Cl2L2-H3PW12O40 (L = 6-hydrazinyl-2,2′-bipyridine) hybrid pair and related relay process for CO2 reduction by photocatalysis and electrocatalysis was developed [43]. Initially, [ O40]3− was reduced to [ O40]5− by two electrons at −1.302 V (versus Fc/Fc+, Fc= ferrocene). Then the reduced POM undertook the functions of photoreductant and electron/proton acceptors. Photo irradiation promoted the generation of ReI(L−)(CO)3 via the electron transfer from [ O40]5− to ReI(L)(CO)3. This reductive ReI intermediate possessed reactivity for the selective CO2 reduction. Only CO was observed in this catalytic electro-photochemical reduction process. The result in the dark conditions indicated that light facilitated the CO2 reduction. The formation of dirhenium complex-H3PW12O40 hybrid pair via acid-base interaction was indispensable to this catalytic electro/photochemical transformation.
2.1.2. Heterogeneous Catalysts
Photocatalytic CO2 reduction by POMs-based heterogeneous catalyst has received intensive research interests. As a POM-stabilized multi–Co-oxide clusters, [Co4(PW9O34)2]10− is comprising a Co4O4 core stabilized by two oxidatively resistant polytungstate ligands (Figure 6a). The Na10[Co4(H2O)2(PW9O34)2 @graphitic carbon nitride hybrid material (Co4@g-C3N4) was reported for the efficient photocatalytic CO2 reduction [44]. It manifested the advantages of convenient recovery, steady reuse, simple preparation and flexible composition. The Co4@g-C3N4 photocatalyst with 43 wt% Co4 content showed high CO yield (107 μmol g−1 h−1) and excellent selectivity (94%) (Figure 6b). After 10 h reaction, the production of CO reached 896 μmol g−1 (Figure 6c). After 5 runs, the activity still remained (Figure 6d). Experimental and characterization results revealed that the Co4 unit both facilitated the charge transfer of g-C3N4 and significantly enhanced the surface catalytic oxidative activity.
NENU-10 and NENU-3 are respectively Ti-substituted Keggin-type POM [PTi2W10O40]7− (This Ti-disubstituted Keggin-type POM contains the Ti centers in relative 1,5 positions according to the IUPAC nomenclature. This anion is one of the few cases in the literature in which a salt of a disubstituted Keggin species displays a predominant isomer (ca. 75% of the α (1,5) isomer)) and Keggin-type POM [PW12O40]3− encaged into MOF Cu3(BTC)2 (BTC: benzene-1,3,5-tricarboxylate; Cu3(BTC)2 = HKUST-1) developed by Liu et al. Efficient photoreduction of CO2 catalyzed by hybrid material Au@NENU-10 (Both Au@NENU-10 and Au@NENU-3 were prepared by one-pot method. The Au NPs were in-situ deposited during the assembly of NENU-10 and NENU-3 as shown in Figure 7.) was recently achieved [45]. Table 2 illustrated the catalytic performance of as-prepared hybrid in the reductive CO2 transformation to CO and CH4 under visible-light irradiation. Compared to Ti-free Au@NENU-3, Au@NENU-10 presented both higher activity and selectivity in the CO2 photoreduction. Similarly, Au/K7(PTi2W10O40) also had better performance than Au/Na3(PW12O40). Results of control experiments implied the indispensability of Au nanoparticles to the CO reduction activity and precursors was inactive. Combing these patterns, authors deduced the following steps for the overall process: Firstly, Au nanoparticles produced electrons and holes where the holes oxidized H2O to generate two electrons and two protons. Then the electrons and protons directly transferred to [PTi2W10O40]7− to form [P(TiIII-H)2 O40]7−. Some of [P(TiIII-H)2 O40]7− species reduced CO2 to CO and H2, and others further obtained electrons and protons to form [P(TiIII-H2)2 O40]7− intermediates. Finally, [P(TiIII-H2)2 O40]7− reduced CO2 to CH4 and simultaneously returned to initial state. Ti-O-W in [PTi2W10O40]7− may responsible to absorb CO2. Compared with [PTi2W10O40]7− and [PW12O40]3−, [PTi2W10O40]7− had stronger electron-coupling protons ability.
2.2. CO2 to HCOOH
HCOOH is one of the promising hydrogen carriers and the important C1 source for organic synthesis. The generation of HCOOH in CO2 photoreduction consumes the same amount of protons and electrons as that for CO formation, but it requires slightly higher reduction potential.
The POM microions are the molecules denoted as {POM} in dilute solutions, they tend to afford suprestructural macroions denoted as {POM}n (POM macroions). Three POM macroions catalysts, Na15[ O462H14(H2O)70]0.5[ O457H14(H2O)68]0.5 ({Mo154}1156), Na17[Mn6P3W24O94(H2O)2] ({Mn6P3W24}931) and (NH4)42[ O372(CH3COO)30(H2O)72]@RGO hybrid ({Mo132}1064@RGO; RGO = reduced graphene oxide) featured with peculiar structures, were respectively employed in the photocatalytic CO2 reduction coupling with water oxidation [46]. The enormous spherical superstructures in these three catalysts in dilute dispersion ({Mo154}1156, {Mn6P3W24}931, {Mo132}1064@RGO) are respectively made up by wheel-shaped {Mo154} rings, bent rod {Mn6P3W24} units and “Keplerate” {Mo132} spheres (Figure 8). The numbers of metal oxide cluster units in these superstructures were calculated by following equation: n = (4πR2)/(72.006σ2) × 60. (R, representing the hydrodynamic radius of POM vesicles formed in the dispersion, was obtained by dynamic light scattering analysis. σ, representing the diameter of isolated single cluster, was determined by the van der Waals radii of the constituent atoms.) The results showed that although HCOOH is the main product of CO2 reduction with these three catalysts, a considerable amount of HCHO was also obtained when {Mo154}1156 and {Mo132}1064@RGO catalysts were applied. In the case of {Mn6P3W24}931, CO2 was exclusively reduced to HCOOH. In terms of formic acid production, {Mo154}1156 and {Mo132}1064@RGO displayed better performance than {Mn6P3W24}931 (Table 3). This phenomenon can be rationally explained by that the simultaneous excitation of multitudinous photoactive clusters on the surface of {Mo154} and {Mo132} vesicles under light irradiation. For {Mo132}1064@RGO, good conductivity of RGO may help the facile electrons transfer.
Afterwards, POM macroions for the water oxidization-coupled CO2 photoreduction to HCOOH was extend to {Cu-PW12}n=1348−2024 ({Cu-PW12}n = [(K6.5Cu(OH)8.5(H2O)7.5)0.5@(K3PW12O40)]n) [47] and gigantic oxo-molybdate catalyst Na48[HxMo368O1032(H2O)240(SO4)48] ({Mo368}) [48]. In the former case, the max TON per mole was 613. Both FT-IR and Raman spectroscopies showed that the structure remained integral after reaction and the activity was kept after ten cycles. In the latter one, {Mo368}, which consisted of a central ball shaped unit {Mo288} and two {Mo40} capping units, showed excellent selectivity for HCOOH (95.73%) with impressive TON (27666) and TOF (4419 h−1). Its external quantum efficiency reached 0.6%. It is worth noting that in all above cases external photosensitizer was unessential. Because intervalence charge transfers (IVCT) of MoV to MoVI and WV to WVI have appropriate gap between conduction band and valence band, which can give rise to absorbance maxima in the region of visible light.
2.3. CO2 to CH4
Compared to the photoreductive conversion of CO2 to CO or HCOOH, photocatalytic methanation of CO2 involving an eight-electron transfer process was much more challenging. [PTi2W10O40]7− was reported as the first POM-based photocatalyst for reducing CO2 to CH4 with CH3OH as electron donator [49]. Although [PTi2W10O40]7− had poor activity and limited efficiency in photoreduction of CO2, this result suggested that POM could be an active catalyst in the CO2 photoreduction.
Since this time, few works focusing on photocatalytic CO2 reduction to CH4 during past decades. Recently, a remarkable achievement in photocatalytic CO2 reduction to CH4 with two hydrothermal-synthesized POM catalysts H[[Na2K4Mn4(PO4) (H2O)4]3[[Mo6O12(OH)3(HPO4)3(PO4)]4[Mn6(H2O)4]] (NENU-605) and H[[Na6CoMn3(PO4)(H2O)4]3[[Mo6O12(OH)3(HPO4)3(PO4)]4[Co1.5Mn4.5]] (NENU-606) was reported [50]. These two water-insoluble POMs showed good structural stability and extended solar spectrum absorption range in aqueous solutions.
Under CO2 atmosphere with triethanolamine (TEOA) as sacrificial agent and [Ru(bpy)3]Cl as photosensitizer, CH4 and CO were the main gaseous photoreduction products, only a trace amount of HCOOH was detected in the aqueous phase. The productivity of CH4 for NENU-605 and NENU-606 reached up to 170 nmol (894.7 nmol g−1 h−1) and 402 nmol (1747.8 nmol g−1 h−1) respectively. Good CH4 selectivity of 76.6% (NENU-605) and 85.5% (NENU-606) was achieved (Table 4, entry 1 and 2). Moreover, H2 evolution as the side reaction was not detected. Regarding to the higher CH4 selectivity of NENU-606 than NENU-605, the heterometallic MnII/CoII ions in NENU-606 might be more favorable to the adsorption and activation of CO2 than the homometallic MnII ions in NENU-605. In contrast, Mn[Mo6O12(OH)3(HPO4)3(PO4)]2 (NENU-607), the dimer containing only one MnII atom sandwiched between two P4Mo6 unit and displaying a similar connection mode to NENU-605 and NENU-606 (Figure 9), had much lower activity (Table 4, entry 3). Outcomes from control experiments demonstrated the necessities of both photosensitizer and light irradiation as well as the contribution from solvent effect (Table 4, entries 4–6). 13C-isotope labelling verified that CO2 was the carbon source of CO and CH4. Deduced from these patterns, a plausible multi-step mechanism for the photocatalytic CO2 methanation was proposed (Figure 10). Initially, the photosensitizer absorbs light to produce photo-excited electrons from its HOMO and then transfers electrons to the P4 unit through the matched LUMO positions (P4 + 6e− → P4 ). Simultaneously, the electron holes produced in the valence band of ruthenium complex was consumed by TEOA (TEOA + h+ → TEOA+). Subsequently, strongly reductive P4 unit further transfer electrons to the active metal center (MII + e− → MI). Then the adsorbed CO2 (MI → MI-CO2) obtains electrons from active metal sites (MI-CO2 → MII-) with the help of H2O as a proton source (MII- + H+ → MII-COOH). MII-CO was formed via the proton- and electron-assisted dehydroxylation (MII-CO2H + H+ + e− → MII-CO + H2O). CH4 is eventually produced with further six-electron transfer process (MII-CO + 6H+ + 6e− → MII + CH4 + H2O).
3. Electrocatalytic CO2 Reduction
Electrocatalytic CO2 reduction to produce various C1 and C2 molecules, for instance, CO, HCOOH, HCHO, CH3OH, CH4, oxalic acid, ethylene, and ethanol, is regarded as a promising proctol for the production of liquid fuels or bulk chemicals. The corresponding reactions and standard potentials were list in Table 5 [51].
Electrocatalytic reduction of CO2 with POM was first reported by Kozik et al. [52]. Cyclic voltammograms (CV) of several POMs with or without the presence of CO2 in various nonpolar solvents were compared. For α-[SiW11O39Co]6−, a large change on its CV was recorded after CO2 was bubbled. Meanwhile for α-[P2W18O62]6−, almost no change was observed before and after bubbling CO2. The evidence from CV hinted that α-[SiW11O39Co]6− might be active to CO2 reduction. Further investigations on reduced α-[SiW11O39Co]6− with CV indeed confirmed that α-[SiW11O39Co]6− showed the electrocatalytic for CO2 reduction. However, the final CO2 reduction product was unstated.
More than decade later, Proust et al. reinvestigated this reaction in more detail by using [α-SiW11O39Co]6− in the electro-assisted reduction of CO2 (Figure 11) [53]. The [α-SiW11O39Co]6− contained a CoII in place of a WVI. The square-pyramidal CoO5 with a vacant site was generated by losing a coordinated water molecule from CoO5(H2O) octahedral when the POM was extracted from aqueous to organic media. Except CO and HCHO, neither H2 nor other CO2 reduction products were detected. This indicated that the unique selectivity of [α-SiW11O39Co]6− POM catalyst in the electroreduction of CO2. The turnover of CO2 to CO reached to 3.7 with the faradic efficiency of 13%. HCHO was the other detectable product. Its amount varied from 2.1 × 10−7 to 2.2 × 10−6 mol with the faradic yield varying from 25% (high HCHO content with low electrolysis charge) to 0.8% (low HCHO content with high electrolysis charge).
Inspired by organometallic CO2 reduction catalyst [Cp*RhIII(bpy)Cl]+ (Cp* = pentamethyl-cyclopentadienyl anion; bpy = 2,2′-bipyridine), [α-H2PW11O39{RhIIICp*(OH2)}]3− with analogous coordination structure via grafting a {Cp*RhIII}2+ fragment on the monovacant [PW11O39]7− anion was prepared (Figure 12) and tested as the electrocatalyst for CO2 reduction [54]. Compared to previously reported [CoSiW11O39]6− catalyst, its electrochemical behavior in the presence of CO2 exhibited a clear improvement, strongly suggesting some interaction with the POM derivative despite the presence of a coordinating solvent. But H2 was still as major product (68% faradic yield) with HCOO− as minor reduction product (4.5% faradic yield).
The remarkable capability of storing and donating electrons endows POMs the feature of reversibly transferring multi-electrons. Hence, selecting proper co-catalyst to establish synergistic catalytic systems containing POM catalyst is reasonable to achieve the efficient CO2 reduction. On one side, by taking the advantage of POMs on reversible transfer of multi-electrons or protons, co-catalysts can efficiently overcome the barrier of CO2 activation. On the other side, co-catalysts with good conductivity and adsorbing CO2 capability can facilitate the electroreduction and improve the activity of catalytic system. Poor electrical conductivity and electron-donating capability are the major drawbacks for MOFs being as efficient electrocatalysts. Therefore, the integrated use of POM and MOF would combine the strengths of each other and generate electrocatalysts with excellent activity [55,56,57].
Following the above intention, Lan et al. prepared various Polyoxometalate-Metalloporphyrin Organic Frameworks (PMOFs) by assembling Zn-ε-Keggin cluster ε- O40Zn4 with M-TCPP (M-TCPP = tetrakis[4-carboxyphenyl]-porphyrinato-M; M = Co, Fe, Ni, Zn) via the links between Zn2+ ions of POM and carboxylates of porphyrin in hydrothermal conditions (Figure 13) and measured their catalytic performances (Table 6) [58]. In the skeleton of these M-PMOFs, the moieties of Zn-ε-Keggin cluster, as electron reservoirs, potently helped the electron transfer to CO2 reduction catalyst M-TCPP and improve its performance of CO2 reduction. The best results were obtained on Co-PMOF with lower onset potential, tafel slope, and electrochemical impedance as well as higher partial CO current density (jCO), electrochemical active surface area (ECSA), faradic efficiency for CO (FECO) and turnover frequency (TOF). Co-PMOF converted CO2 to CO with a superior faradic efficiency of 99%. The value of TOF was elevated to 1656 h−1 at −0.8 V. No evident activity attenuation was observed during the 36 h stability test, which indicated the robustness of Co-PMOF.
DFT calculations was employed to explicitly understand the excellent performance of Co-MPOF and synergism between Zn-ε-Keggin cluster and Co-TCPP. Owing to rather high Gibbs free energies of 0.96 eV (ΔG1 in Figure 14a), the formation of adsorbed intermediates * COOH was regarded as the rate-determining step (RDS) for CO2 reduction on Zn-ε-Keggin cluster. On the contrary, the formation of adsorbed intermediates * CO on Co-TCPP was RDS with high Gibbs free energies of 0.53 eV (ΔG2 in Figure 14a). In accordance with expectation, remarkable drops of both ΔG1 and ΔG2, particularly the much lower ΔG1 = 0.34 eV, were revealed. For Co-PMOF, the more favorable CO2 reduction active site is Co on Co-TCPP instead of Zn on POM. In fact, this Zn POM was inactive in CO2 reduction, and the synergistic effect was derived from the intramolecular electron transfer between the electron mediator of POM and Co-TCPP. The effect of different metal center in porphyrin was also computed (Figure 14b). The energy profile of reaction on different metal centers was well consistent with the outcomes of experiments. Based on the experimental results and theoretical calculations, the possible mechanism about reducing CO2 to CO on Co-PMOF was suggested. Firstly, the POM captures and transfers electrons from the electrode to the CoII center. Subsequently, CoII centers was reduced to CoI. Then CoI interacts with CO2 to afford CoII-* COO−, which was converted to CoII-* CO via the proton-coupled electron transfer. Finally, CO is desorbed and released (Figure 14c,d).
The design and application of [α-SiW12O40]4−-modified AgNC@BSA catalyst (AgNC@BSA = silver nanoclusters capped with bovine serum albumin; BSA is both the stabilizer and reducing reagent for the synthesis of AgNC) in the electroreduction of CO2 to CO also exhibited the same concept of synergism [59]. Similar to the case of Co-PMOF, although [α-SiW12O40]4− had little CO2 reduction activity, it played the role of electron transfer mediator and assisted the CO2 reduction through its strong interaction with CO2 on the surface of AgNC. It had excellent faradic efficiency (>75%) in DMF containing 1% (v/v) H2O. The overpotential of [α-SiW12O40]4−-modified AgNC@BSA electrode was about 0.7 V. The onset potential for this POM-decorated AgNC electrocatalyst was about 400 mV more positive than that on bulk Ag.
Metal nanoparticles stabilized by POMs was extensively researched [60,61]. By replacing BSA with POM as the stabilizer of AgNC, three Ag-POM nanocomposites respectively with [PMo12O40]3−, [α-SiW12O40]4− and [PW12O40]3− were synthesized by electrodeposition method [62]. POM is the promoter for the CO2 reduction. Due to the higher charge density and stronger basicity associated with [PMo12O40]3−, the activity of Ag-[PMo12O40]3− nanocomposite was better than Ag-[PW12O40]3− and Ag-[α-SiW12O40]4−. This nanocomposite exhibited efficient and sustained CO2 reduction at a wide potential range with faradic efficiency of 90 ± 5%. The Tafel slope, an inherent property of electrocatalyst determined by the rate-limiting step (formation of or protonation of ), calculated for Ag-[PMo12O40]3− (60 mV dec−1) was very close to the theoretical value (59 mV dec−1), which indicated the faster formation of on the Ag-[PMo12O40]3− nanocomposite-modified electrode
4. Electromicrobial Conversion of CO2
Since the introduction of lithoautotrophic bacterium Ralstonia eutropha H16 as the production host for biological formate conversion as well as the coupling between this biological formate conversion method and the electroreductive conversion of CO2 to formate on indium cathode was employed to produce fuels for internal combustion engines from CO2 [63], the integration of electrocatalytic reduction with microbial conversion represents an edge-cutting strategy for the direct but non-photosynthetic CO2 conversion to important molecules.
Microorganisms, represented by R. eutropha and Clostridium spp, have been well harnessed to transform CO2 and H2 to energy-dense liquid fuels [64]. As potent hydrogen-oxidizing bacterium, R. eutropha has exhibited its versatility in synthesize poly[R-(−)-3-hydroxybutyrate], which is one of the ingredients to manufacture biodegradable plastics [65]. On the other aspect, many POMs-based catalysts have presented excellent activity for hydrogen evolution reaction (HER) [12,66]. Searching biocompatible POM-based HER catalysts and utilizing suitable microorganism to catalytically reduce CO2 by consuming H2 generated from HER can effectually accomplish the electromicrobial conversion of CO2.
Recently, Zhang and co-workers demonstrated the feasibility of such reckoning. The combination of a heterometallic Co/Cu-containing polyoxometalate/carbon cloth (Cu6Co7/CC; Cu6Co7 = [Cu(en)2]6[((PW9O34)Co3(OH)(H2O)2(O3PC(O)(C3H6NH3)PO3))2Co]2−, en = ethanediamine; see the structure of Cu6Co7 POM in Figure 15; CC = carbon cloth) precatalyst with bacterium R. eutropha H16 was developed to achieved the electricity-driven bioconversion of CO2 to biomass in neutral water [67]. Cu6Co7/CC hybrid material possessed good biocompatibility in this CO2 conversion system. It was evidenced that most of the H2 produced on the Cu6Co7/CC cathode was consumed by the bacteria for living and growth. In excess of half input electrical energy was transported into biomass. The authors assumed that the electricity would be supplied by a photovoltaic device with an efficiency of 18%, the expected overall solar-to-biomass efficiency could reach 10%, which would be nearly 10 times higher than the natural photosynthesis. This Cu6Co7/CC-R. eutropha hybrid system exemplified the potential to explore non-photosynthetic but highly efficient CO2-fixation methods by using POMs-based catalysts and solar electricity.
5. Non-Reductive CO2 Conversion to Carbonyl-Contained Organic Chemicals
Non-reductive chemical CO2 conversion provides diverse alternatives to achieve both the synthesis of practical chemicals and CO2 fixation at the same time [68]. Currently, the industrial manufacture of several chemicals demands CO2 as starting material [69]. Predominantly, due to the basic properties of POM-based materials, the capture and transformation of CO2 with these materials has been widely explored. Therefore, the versatility of POMs-based materials in non-reductive chemical CO2 fixation has been fully presented. Various useful chemicals, including cyclic carbonate [19], dimethyl carbonate [70,71,72,73,74], urea [74], quinazoline-2,4-(1H,3H)-diones [74,75], 2-benzimidazolone [74], 2-oxazolidinones [18], α-methylene cyclic carbonate [74], and methacrylic acid [76,77] were able to be synthesized with POM-based catalyst (Scheme 2). Considering that several reviews have already given comprehensive summaries of representative progress on non-reductive CO2 fixation by POM catalysts [69,78,79], herein we briefly introduced the recent advances about non-reductive CO2 conversion to organic chemicals, which were not collected in the very recent review published in 2018 [79].
Various important applications of cyclic carbonates make the research on POM or other material-catalyzed synthesis of cyclic carbonates always receive long-lasting interests [80]. By integrating POM components, chiral organic catalysts and CO2 activator in the skeleton of a single MOF, the polyoxometallate-organocatalyst-metal organic framework (POMOF) was designed by Duan group [81]. Chiral cyclic carbonates were produced efficiently from olefins and CO2 on the POMOFs tandem catalyst. By ingeniously connecting Keggin-type POM anions α-[ZnW12O40]6−, l-proline-derived asymmetric organocatalysts pyrrolidine-2-yl-imidazole (PYI) and NH2-functionalized bridging links 2-amino-4,4′-bipyridine via the six-coordinated ZnII nodes in these POMOFs (α-[ZnW12O40]6−-PYIs), a clear division of catalytic working for oxidation-coupled CO2 conversion was established that POM as an oxidation catalyst, pyrrolidine moiety as a chiral organocatalyst, ZnII ions as Lewis acid catalyst to activate the epoxide intermediate and amino groups on 4,4′-bipyridines as renewable CO2 absorption reagent in this tandem process (Figure 16). The multi-catalytic sites were orderly distributed and spatially matched in the framework. The captured CO2 molecules are synergistically fixed and activated by well-positioned pyrrolidine and amine groups, providing further compatibility with the terminal W=O activated epoxidation intermediate and driving the tandem catalytic process in a single workup stage and an asymmetric fashion.
In conjunction with ionic liquid co-catalyst, two kinds of POMs attached with metal carbonyl {P2W15O56Co3(H2O)3(OH)3Mn(CO)3}8− [82] and {(Se2W11O43)(Mn(CO)3)4}8− [83] were reported as efficient catalysts for the cycloaddition of CO2 with epoxides under mild conditions. A rare three-dimensional CO2-linked POM polymer {PMo12O40Zn4(CO2)}2− exhibited superior performance in the cycloaddition of CO2 with epoxides [19]. The structural feature of catalyst is that the CO2 ligand connects with two Zn-ε-Keggin cores in a linear and symmetrical μ2-η2o,o coordination pattern(Figure 17). Polyoxoniobates (DBUH)3(NbO5), (TBA)6[Nb10O28] and Na16[SiNb12O40] were applied as Lewis base-type catalysts for the cycloaddition of CO2 with epoxides under halide-free conditions [84,85,86]. Carbon nanotubes-supported Fe1.5PMo12O40 (theoretical formulas) obtained 57.7% propylene oxide conversion and 99.0% propylene carbonate selectivity, both activity and selectivity were higher than Fe1.5PMo12O40, Co1.5PMo12O40, Cu1.5PMo12O40, and Zn1.5PMo12O40 (theoretical formulas). The good activity can be attributed to the well dispersion of the Fe1.5PMo12O40 on the CNTs [87].
6. Outlook
POMs are regarded as discrete metal oxide clusters with much smaller nanometric size than bulk metal oxides, therefore most of the distinctions on chemical properties, especially CO2 reductive conversion-related redox property, between bulk metal oxides and POMs were originated from quantum size effect. Quantum size effect leads to the changes of electronic configurations. These changes arise through systematic transformations in the density of electronic energy levels as a function of the size. By plotting the correlation between redox potential and electron density per volume (Figure 18a) as well as the diagram of band structure (Figure 18b), the quantum size effect-resulted distinction on redox property among bulk WO3, colloid WO3 and [SiW12O40]4− has been clearly illustrated [88]. Despite the prevalence of both POMs and bulk oxides as photocatalysts of harvesting light in recent years, the smaller size endows POMs with wider gap and lower reduction potentials. The case given by Hill and Geletii et al. involving water oxidation catalyst [Co4(H2O)2(PW9O34)2]10− (Co4POM) also manifested the distinction between POM and bulk metal oxide on redox property. In their report, (PW9O34)9−-sandwiched Co4 unit in Co4POM was unambiguously identified as the dominant active center for water splitting and not CoOx. [89]
The prospering arising of new POM structures provides vast opportunities to excavate latent catalysts for chemical CO2 conversion. Titanium oxo-clusters [90] and polyoxo-noble-metalates [91] are exemplified as two promising representatives among these new POM structures. Regarding titanium oxo-clusters (also named polyoxotitanates), there are diverse variants including titanium oxoalkoxides clusters TinOm(OR)4n–2m, titanium oxo-carboxo-alkoxides clusters TinO2n–x/2–y/2(OR)x(OOCR’)y, and titanium oxocarboxo clusters [TinOm(OOCR)p] (2m + p = 4n) [90]. The tunable wide polynuclearity (the number of titanium atoms n varies from 2 to 28) and facile fabrication of hybrid architectures with organic components by covalent bonding are the two major features of these clusters. MOF MIL-125 (MIL stands for Material from Insitut Lavoisier) consisting of 1,4-benzenedicarboxylate-connected titanium-oxo-hydroxo clusters has been reported to possess possible photocatalytic property when alcohols are adsorbed inside its framework [92]. Based on this indication and as the extensive applications of titanium oxide in CO2 photocatalytic reduction [93,94,95], there are likely to be extensive catalytic application of hybrid materials containing titanium oxo-cluster cores in CO2 reductive conversion. The emergance of various novel polyoxo-noble-metalates including polyoxopalladates, -platinates, and -aurates [91] also provides different prospects for catalytic CO2 reductive conversion. On these polyoxo-noble-metalates, the noble metal atoms Pd, Pt, and Au act as “addenda” atoms rather than as heteroatoms [91]. Despite that there are no direct accessible active sites on noble metal centers with saturated coordination environments, the very recent report about the catalytic Suzuki-Miyaura cross-coupling application of polyoxopalladate-based MOF materials has implied that these polyoxo-noble-metalates may acquire catalytic activity via in situ partial reduction [96]. For polyoxoplatinate [Pt12O8(SO4)12]4− [97], although it is insoluble in any media, its six dumbbell-shaped [Pt2]6+ anionic units perhaps will attract electrocatalytic research interests, because they may be in-situ partially reduced to molecular mixed-valent metal clusters or platinum black-type “suboxides” [98]. Then these plausible in-situ generated species may be applied as HER catalysts for electromicrobial conversion of CO2.
More than the mode of LMCT, developing POM-based catalysts with the intrinsic function of metal-to-metal charge transfer (MMCT) will also be beneficial to look for more efficient POM-based approach to harvest visible-near infrared emission of sunlight with higher quantum yield. The MMCT transition occurs when two metal centers with different valence states are coupled by bridging ligands (Ma+-O-M’b+). The two metal centers are respectively reduced and oxidized (Ma+-O-M’b+ → M(a−1)+-O-M’(b+1)+) during this transition. The MMCT transition often takes place in polynuclear complexes [99]. The oxo-bridged and all-inorganic heterobinuclear units such as ZrIV-O-CoII [100,101,102,103], ZrIV-O-CuI [104], TiIV-O-CoII [100], and TiIV-O-MnII [105] supported on silica material that display MMCT transitions can extend the optical absorption from UV to visible regions. Some of these materials have been applied as catalyst or cocatalyst for CO2 photoreduction [101,102,103,104]. Several reported POM-based systems, such as the anchored polynuclear charge-transfer complexes consisting of CeIII ions and CuII-substituted Keggin-type CuIIPW11O39 [106], the metal-oxide nanoclusters consisting of CeIII or CoII ions and Keggin-type PW12O403− [107], [CoIIW12O40]6− [108], and [CoII(MxOHy)W11O39](12-x-y)− (MxOHy = VIVO, CrIII(OH2), MnII(OH2), FeIII(OH2), CoII(OH2), NiII(OH2), CuII(OH2), ZnII(OH2)) [109] have sufficiently demonstrated the feasibility that such molecular inorganic MMCT (or MPCT, metal-to-polyoxometalate charge transfer) [108,109] transition enable POM-based catalyst to function as efficient visible-light-driven multielectron-transfer catalysts. Regarding the metal-oxide nanoclusters consisting of CeIII or CoII ions and Keggin-type PW12O403−, even the straightforward video guide of preparing catalytic material has been given [110]. These will further encourage to utilize MMCT of POM-based catalytic materials in CO2 reduction. It is noteworthy that prolonging the picosecond-scale lifetimes of photogenerated states during MMCT transition is still an important issue to be tackled [111]. The works from Lan and Hill et al. has validated the effects of heterometal location [108] and different addenda substitution [109] on the lifetime of photo-excited state.
On the other hand, the tools of advanced operando spectral characterization techniques [112], electrochemical measuring methods [113] and theoretical calculations [114], imbibing more understandings and attaining insights on catalytic mechanisms and structure–reactivity relationships of those reported POM-based catalysts will be helpful to design better POM catalysts for CO2 conversion with explicit destination. On CO2 photo/electroreduction, the hybridation of POM with various novel materials can provide chances to fabricate catalysts with broader visible-light absorption spectrum region or better selectivity but lower overpotential. In those systems with hybrid electro- or photocaytalytic CO2 catalysts, once the light-harvesting/electron-storage centers and catalytically active sites are designated in an integrated system, the key issue is the bridging of the two components—charge kinetics. Exploiting time-resolved spectroscopic techniques to characterize charge kinetics is useful to understand these hybrid catalysts and thus helps to improve their catalytic efficiencies [99].
In catalytic oxidation, POMs were looked upon as inorganic analogs of porphyrin [115,116,117]. The thriving development of transition metal porphyrin or phthalocyanine derivatives as heterogeneous molecular catalysts for electrochemical CO2 reduction [118] makes us have such confidence that this analogy can be still established in electrochemical CO2 reduction. Therefore, the accumulated understandings about electrochemical CO2 reduction by transition metal porphyrin or phthalocyanine may help the development of POM-based catalysts for the electroreductive CO2 conversion. By sunlight or solar electricity, producing fuels and chemicals via CO2 conversion is a promising and reliable option for largely reducing fossil fuel consumption in the future [119]. Thus POM-catalyzed transformation of CO2 with photo- or electrochemical methods will retain long-lasting interest of both academic and industrial research.
To the non-reductive CO2 conversion, searching for POMs with stronger Lewis basicity will benefit the elevation of turnover efficiency and the mitigation of harsh conditions under halogen-free condition. To enhance Lewis basicity, high negative charge density on the terminal oxygen atoms on POMs is required. Calculating and comparing natural bond orbital (NBO) charges of oxygen atoms in POMs has been demonstrated as a useful access to gain more insights into the Lewis basicity on POMs [84,120]. Making full use of both structural traits and diversity of POMs would help to break through some ceilings of practical CO2 chemical conversion.
Author Contributions
Writing—original draft preparation, Y.C., Q.C., C.S.; writing—review and editing, Y.C., C.S., L.H.; conceptualization and project administration, L.H. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding
This work was supported by NSFC (91645118) and NSF of Jiangsu Province (BK20180249).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Le Quéré, C.; Andrew, R.M.; Friedlingstein, P.; Sitch, S.; Hauck, J.; Pongratz, J.; Pickers, P.A.; Korsbakken, J.I.; Peters, G.P.; Canadell, J.G.; et al. Global carbon budget 2018. Earth. Syst. Sci. Data 2018, 10, 2141–2194. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Berzelius, J. The preparation of the phosphomolybdate ion [PMo12O40]3−. Pogg. Ann. 1826, 6, 369–371. [Google Scholar]
- Song, Y.-F. Polyoxometalate-Based Assemblies and Functional Materials; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Van Eldik, R.; Cronin, L. Polyoxometalate Chemistry; Academic Press: Burlington, VT, USA, 2017. [Google Scholar]
- Liu, S.; Tang, Z. Polyoxometalate-based functional nanostructured films: Current progress and future prospects. Nano Today 2010, 5, 267–281. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Y.; Li, G.; Wei, Y. Recent advances in alkoxylation chemistry of polyoxometalates: From synthetic strategies, structural overviews to functional applications. Coord. Chem. Rev. 2019, 378, 395–414. [Google Scholar] [CrossRef]
- Chen, W.; Wang, E. Polyoxometalate Chemistry; Science press: Beijing, China, 2013; (In Chinese edition). [Google Scholar]
- Sartzi, H.; Miras, H.N.; Vila-Nadal, L.; Long, D.L.; Cronin, L. Trapping the delta isomer of the polyoxometalate-based Keggin cluster with a tripodal ligand. Angew. Chem. Int. Ed. 2015, 54, 15488–15492. [Google Scholar] [CrossRef]
- Bassil, B.S.; Kortz, U. Recent advances in lanthanide-containing polyoxotungstates. Z. Anorg. Allg. Chem. 2010, 636, 2222–2231. [Google Scholar] [CrossRef]
- Guo, Y.-H.; Hu, C.-W. Porous hybrid photocatalysts based on polyoxometalates. J. Clust. Sci. 2003, 14, 505–526. [Google Scholar] [CrossRef]
- Wang, S.-S.; Yang, G.-Y. Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 2015, 115, 4893–4962. [Google Scholar] [CrossRef]
- Müller, C.E.; Iqbal, J.; Baqi, Y.; Zimmermann, H.; Röllich, A.; Stephan, H. Polyoxometalates—a new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioor. Med. Chem. Lett. 2006, 16, 5943–5947. [Google Scholar] [CrossRef]
- Proust, A.; Thouvenot, R.; Gouzerh, P. Functionalization of polyoxometalates: towards advanced applications in catalysis and materials science. Chem. Commun. 2008, 16, 1837–1852. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shen, C.; He, L. Recent advances of polyoxometalate-catalyzed selective oxidation based on structural classification. Acta Crystallogr. Sect. C Struct. Chem. 2018, 74, 1182–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepankiewicz, S.H.; Ippolito, C.M.; Santora, B.P.; Van De Ven, T.J.; Ippolito, G.A.; Fronckowiak, L.; Wiatrowski, F.; Power, T.; Kozik, M. Interaction of carbon dioxide with transition-metal-substituted heteropolyanions in nonpolar solvents. spectroscopic evidence for complex formation. Inorg. Chem. 1998, 37, 4344–4352. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, F.; Xu, L.; Liu, X.; Yang, Y. CO2 coordination by inorganic polyoxoanion in water. J. Am. Chem. Soc. 2008, 130, 10838–10839. [Google Scholar] [CrossRef] [PubMed]
- He, L.-N.; Wang, M.-Y.; Song, Q.-W.; Ma, R.; Xie, J.-N. Efficient conversion of carbon dioxide at atmospheric pressure to 2-oxazolidinones promoted by bifunctional Cu(II)-substituted polyoxometalate-based ionic liquids. Green Chem. 2016, 18, 282–287. [Google Scholar]
- Cheng, W.; Xue, Y.-S.; Luo, X.-M.; Xu, Y.; Xue, Y. A rare three-dimensional POM-based inorganic metal polymer bonded by CO2 with high catalytic performance for CO2 cycloaddition. Chem. Commun. 2018, 54, 12808–12811. [Google Scholar] [CrossRef]
- Fang, X.; Anderson, T.M.; Neiwert, W.A.; Hill, C.L. Yttrium polyoxometalates. Synthesis and characterization of a carbonate-encapsulated sandwich-type complex. Inorg. Chem. 1998, 37, 4344–4352. [Google Scholar]
- Chen, B.; Neumann, R. Coordination of carbon dioxide to the Lewis acid site of a zinc-substituted polyoxometalate and formation of an adduct using a polyoxometalate-2,4,6-trimethylpyridine frustrated Lewis pair. Eur. J. Inorg. Chem. 2018, 2018, 791–794. [Google Scholar] [CrossRef]
- Garai, S.; Haupt, E.T.K.; Bögge, H.; Merca, A.; Müller, A. Picking up 30 CO2 molecules by a porous metal oxide capsule based on the same number of receptors. Angew. Chem. Int. Ed. 2012, 51, 10528–10531. [Google Scholar] [CrossRef]
- Selimkhanov, J.; Taylor, B.; Yao, J.; Pilko, A.; Albeck, J.; Hoffmann, A.; Tsimring, L.; Wollman, R. Accurate information transmission through dynamic biochemical signaling networks. Science 2014, 346, 1370–1373. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Peng, B.; Peng, T. Recent advances in heterogeneous photocatalytic CO2 conversion to solar fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Sohn, Y.; Huang, W.; Taghipour, F. Recent progress and perspectives in the photocatalytic CO2 reduction of Ti-oxide-based nanomaterials. Appl. Surf. Sci. 2017, 396, 1696–1711. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Zeng, S.; Kar, P.; Thakur, U.K.; Shankar, K. A review on photocatalytic CO2 reduction using perovskite oxide nanomaterials. Nanotechnology 2018, 29, 052001. [Google Scholar] [CrossRef]
- Shi, R.; Waterhouse, G.I.; Zhang, T. Recent Progress in Photocatalytic CO2 reduction over perovskite oxides. Sol. RRL 2017, 1, 1700126. [Google Scholar] [CrossRef]
- Ye, S.; Wang, R.; Wu, M.-Z.; Yuan, Y.-P. A review on g-C3N4 for photocatalytic water splitting and CO2 reduction. Appl. Surf. Sci. 2015, 358, 15–27. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Zhou, K. Metal-organic-framework-based catalysts for photoreduction of CO2. Adv. Mater. 2018, 30, e1705512. [Google Scholar] [CrossRef]
- Sprick, R.S.; Jiang, J.-X.; Bonillo, B.; Ren, S.; Ratvijitvech, T.; Guiglion, P.; Zwijnenburg, M.A.; Adams, D.J.; Cooper, A.I. Tunable organic photocatalysts for visible-light-driven hydrogen evolution. J. Am. Chem. Soc. 2015, 137, 3265–3270. [Google Scholar] [CrossRef]
- Ghosh, S.; Kouamé, N.A.; Ramos, L.; Rémita, S.; Dazzi, A.; Deniset-Besseau, A.; Beaunier, P.; Goubard, F.; Aubert, P.-H.; Remita, H. Conducting polymer nanostructures for photocatalysis under visible light. Nat. Mater. 2015, 14, 505–511. [Google Scholar] [CrossRef]
- Papaconstantinou, E. Photochemistry of polyoxometallates of molybdenum and tungsten and/or vanadium. Chem. Soc. Rev. 1989, 18, 1. [Google Scholar] [CrossRef]
- Hill, C.L.; Prosser-McCartha, C.M. Photocatalytic and Photoredox Properties of Polyoxometalate Systems; Springer Nature: Dordrecht, Netherlands, 1993. [Google Scholar]
- Nemodruk, A.A.; Bezrogova, E.V. The use of photochemical reduction for the determination of silicon and phosphorus as their blue heteropoly acids. Zh. Anal. Khim. 1969, 24, 292. [Google Scholar]
- Morosanova, S.A.; Kolli, N.Y.; Kushnirenko, T.G. Photochemical reduction of 12-molybdoarsenic acid in aqueous-organic media. Zh. Anal. Khim. 1977, 32, 96. [Google Scholar]
- Streb, C. New trends in polyoxometalate photoredox chemistry: From photosensitisation to water oxidation catalysis. Dalton Trans. 2012, 41, 1651–1659. [Google Scholar] [CrossRef]
- Tucher, J.; Wu, Y.; Nye, L.C.; Ivanović-Burmazović, I.; Khusniyarov, M.M.; Streb, C. Metal substitution in a Lindqvist polyoxometalate leads to improved photocatalytic performance. Dalton Trans. 2012, 41, 9938. [Google Scholar] [CrossRef]
- Walsh, J.J.; Bond, A.M.; Forster, R.J.; Keyes, T.E. Hybrid polyoxometalate materials for photo(electro-) chemical applications. Coord. Chem. Rev. 2016, 306, 217–234. [Google Scholar] [CrossRef]
- Khenkin, A.M.; Efremenko, I.; Weiner, L.; Martin, J.M.L.; Neumann, R. Photochemical reduction of carbon dioxide catalyzed by a ruthenium-substituted polyoxometalate. Chem. Eur. J. 2010, 16, 1356–1364. [Google Scholar] [CrossRef]
- Ettedgui, J.; Diskin-Posner, Y.; Weiner, L.; Neumann, R. Photoreduction of carbon dioxide to carbon monoxide with hydrogen catalyzed by a rhenium(I) phenanthroline−polyoxometalate hybrid complex. J. Am. Chem. Soc. 2011, 133, 188–190. [Google Scholar] [CrossRef]
- Poblet, J.M. The Photoreduction mechanism of CO2 to CO catalyzed by a rhenium(I)-polyoxometalate hybrid compound. ACS Catal. 2016, 6, 6422–6428. [Google Scholar]
- Haviv, E.; Shimon, L.J.W.; Neumann, R. Photochemical reduction of CO2 with visible light using a polyoxometalate as photoreductant. Chem. Eur. J. 2017, 23, 92–95. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, W.; Sun, C.; Han, L.; Qin, C.; Chen, M.; Wang, X.; Wang, E.; Su, Z. Oxidative polyoxometalates modified graphitic carbon nitride for visible-light CO2 reduction. ACS Appl. Mater. Interfaces 2017, 9, 11689–11695. [Google Scholar] [CrossRef]
- Liu, S.-M.; Zhang, Z.; Li, X.; Jia, H.; Ren, M.; Liu, S. Ti-substituted Keggin-type polyoxotungstate as proton and electron reservoir encaged into metal-organic framework for carbon dioxide photoreduction. Adv. Mater. Interfaces 2018, 5, 1801062. [Google Scholar] [CrossRef]
- Das, S.; Biswas, S.; Balaraju, T.; Barman, S.; Pochamoni, R.; Roy, S. Photochemical reduction of carbon dioxide coupled with water oxidation using various soft-oxometalate (SOM) based catalytic systems. J. Mater. Chem. A 2016, 4, 8875–8887. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Kumar, S.; Garai, S.; Pochamoni, R.; Paul, S.; Roy, S. Softoxometalate [{K6.5Cu(OH)(8.5)(H2O)(7.5)}(0.5)@{K3PW12O40}](n) (n=1348-2024) as an efficient inorganic material for CO2 reduction with concomitant water oxidation. ACS Appl. Mater. Interfaces 2017, 9, 35086–35094. [Google Scholar] [CrossRef]
- Das, S.; Balaraju, T.; Barman, S.; Sreejith, S.S.; Pochamoni, R.; Roy, S. A Molecular CO2 reduction catalyst based on giant polyoxometalate {Mo-368}. Front. Chem. 2018, 6, 514. [Google Scholar] [CrossRef] [PubMed]
- Yamase, T.; Sugeta, M. Photoreduction of CO2 to CH4 in water using dititanodecatungstophosphate as multielectron transfer catalyst. Inorg. Chim. Acta. 1990, 172, 131–134. [Google Scholar] [CrossRef]
- Xie, S.-L.; Liu, J.; Dong, L.-Z.; Li, S.-L.; Lan, Y.-Q.; Su, Z.-M. Hetero-metallic active sites coupled with strongly reductive polyoxometalate for selective photocatalytic CO2-to-CH4 conversion in water. Chem. Sci. 2019, 10, 185–190. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Paul, J.; Page, P.; Sauers, P.; Ertel, K.; Pasternak, C.; Lin, W.; Kozik, M. Transition-Metal-Substituted Heteropoly Anions in Nonpolar Solvents-Structures and Interaction with Carbon Dioxide; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Girardi, M.; Blanchard, S.; Griveau, S.; Simon, P.; Fontecave, M.; Bedioui, F.; Proust, A. Electro-assisted reduction of CO2 to CO and formaldehyde by (TOA)6[α-SiW11O39Co(_)] polyoxometalate. Eur. J. Inorg. Chem. 2015, 2015, 3642–3648. [Google Scholar] [CrossRef]
- Girardi, M.; Platzer, D.; Griveau, S.; Bedioui, F.; Alves, S.; Proust, A.; Blanchard, S. Assessing the electrocatalytic properties of the {Cp*RhIII}2+-polyoxometalate derivative [H2PW11O39{(RhIIICp*(OH2)}]3− towards CO2 reduction. Eur. J. Inorg. Chem. 2019, 3–4, 387–393. [Google Scholar] [CrossRef]
- Szczęśniak, B.; Choma, J.; Jaroniec, M. Gas adsorption properties of hybrid graphene-MOF materials. J. Colloid Interface Sci. 2018, 514, 801–813. [Google Scholar] [CrossRef]
- Genovese, M.; Lian, K. Polyoxometalate modified inorganic–organic nanocomposite materials for energy storage applications: A review. Curr. Opin. Solid State Mater. Sci. 2015, 19, 126–137. [Google Scholar] [CrossRef]
- Fan, D.; Hao, J.; Wei, Q. Assembly of polyoxometalate-based composite materials. J. Inorg. Organomet. Polym. Mater. 2012, 22, 301–306. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Huang, Q.; He, C.-T.; Chen, Y.; Liu, J.; Shen, F.-C.; Lan, Y.-Q. Oriented electron transmission in polyoxometalate-metalloporphyrin organic framework for highly selective electroreduction of CO2. Nat. Commun. 2018, 9, 4466. [Google Scholar] [CrossRef]
- Guo, S.-X.; MacFarlane, D.R.; Zhang, J. Bioinspired electrocatalytic CO2 reduction by bovine serum albumin-capped silver nanoclusters mediated by [α-SiW12O40]4−. Chemsuschem 2016, 9, 80–87. [Google Scholar] [CrossRef]
- Wang, Y.; Weinstock, I.A. Polyoxometalate-decorated nanoparticles. Chem. Soc. Rev. 2012, 41, 7479. [Google Scholar] [CrossRef] [PubMed]
- Jameel, U.; Zhu, M.; Chen, X.; Tong, Z. Recent progress of synthesis and applications in polyoxometalate and nanogold hybrid materials. J. Mater. Sci. 2016, 51, 2181–2198. [Google Scholar] [CrossRef]
- Guo, S.-X.; Li, F.; Chen, L.; Macfarlane, D.R.; Zhang, J. Polyoxometalate-promoted electrocatalytic CO2 reduction at nanostructured silver in dimethylformamide. ACS Appl. Mater. Interfaces 2018, 10, 12690–12697. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Opgenorth, P.H.; Wernick, D.G.; Rogers, S.; Wu, T.-Y.; Higashide, W.; Malati, P.; Huo, Y.-X.; Cho, K.M.; Liao, J.C. Integrated electromicrobial conversion of CO2 to higher alcohols. Science 2012, 335, 1596. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, A.S.; McTernan, P.M.; Lian, H.; Kelly, R.M.; Adams, M.W. Biological conversion of carbon dioxide and hydrogen into liquid fuels and industrial chemicals. Curr. Opin. Biotechnol. 2013, 24, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, A.; Fricke, W.F.; Reinecke, F.; Kusian, B.; Liesegang, H.; Cramm, R.; Eitinger, T.; Ewering, C.; Pötter, M.; Schwartz, E.; et al. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 2006, 24, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.; Nunes, M.; Fernandes, D.M.; Abdelkader, V.K. POM&MOF-based electrocatalysts for energy-related reactions. ChemCatChem 2018, 10, 1703–1730. [Google Scholar]
- Wang, M.; Zhong, W.; Zhang, S.; Liu, R.; Xing, J.; Zhang, G. An overall water-splitting polyoxometalate catalyst for the electromicrobial conversion of CO2 in neutral water. J. Mater. Chem. A 2018, 6, 9915–9921. [Google Scholar] [CrossRef]
- Yuan, G.; Qi, C.; Wu, W.; Jiang, H. Recent advances in organic synthesis with CO2 as C1 synthon. Green Sustain. Chem. 2017, 3, 22–27. [Google Scholar]
- Yu, B.; Zou, B.; Hu, C.-W. Recent applications of polyoxometalates in CO2 capture and transformation. J. CO2 Util. 2018, 26, 314–322. [Google Scholar] [CrossRef]
- La, K.W.; Youn, M.H.; Chung, J.S.; Baeck, S.H.; Song, I.K. Synthesis of dimethyl carbonate from methanol and carbon dioxide by heteropolyacid/metal oxide catalysts. Solid State Phenom. 2007, 119, 287–290. [Google Scholar] [CrossRef]
- Aouissi, A.; Al-Othman, Z.A.; Al-Amro, A. Gas-phase synthesis of dimethyl carbonate from methanol and carbon dioxide over Co1.5PW12O40 Keggin-type heteropolyanion. Int. J. Mol. Sci. 2010, 11, 1343–1351. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, S.; Jung, J.C.; Song, I.K. Direct synthesis of dimethyl carbonate from methanol and carbon dioxide over H3PW12O40/CeXZr1-XO2 catalysts: Effect of acidity of the catalysts. Korean J. Chem. Eng. 2011, 28, 1518–1522. [Google Scholar] [CrossRef]
- La, K.W.; Jung, J.C.; Kim, H.; Baeck, S.-H.; Song, I.K. Effect of acid–base properties of H3PW12O40/CexTi1−xO2 catalysts on the direct synthesis of dimethyl carbonate from methanol and carbon dioxide: A TPD study of H3PW12O40/CexTi1−xO2 catalysts. J. Mol. Catal. A: Chem. 2007, 269, 41–45. [Google Scholar] [CrossRef]
- Kimura, T.; Kamata, K.; Mizuno, N. A bifunctional tungstate catalyst for chemical fixation of CO2 at atmospheric pressure. Angew. Chem. Int. Ed. 2012, 51, 6700–6703. [Google Scholar] [CrossRef] [PubMed]
- Sunaba, H.; Kimura, T.; Kamata, K.; Mizuno, N. Efficient [WO4]2− -catalyzed chemical fixation of carbon dioxide with 2-aminobenzonitriles to quinazoline-2,4(1H,3H)-diones. Inorg. Chem. 2012, 51, 13001–13008. [Google Scholar]
- Wang, D.; Zhong, S. Study on CuPMo/TiO2 catalyst for direct synthesis of methacrylic acid from propylene and carbon dioxide. Chin. J. Catal. 2003, 24, 705–710. [Google Scholar]
- Wang, D.-W.; Zhong, S.-H. Study on CuPW/TiO2 catalyst for direct synthesis of MAA from propylene and carbon dioxide. J. Fuel Chem. Technol. 2004, 32, 219–224, (In Chinese with English abstract). [Google Scholar]
- Wang, M.-Y.; Ma, R.; He, L.-N. Polyoxometalate-based ionic liquids-promoted CO2 conversion. Sci. China Chem. 2016, 59, 507–516. [Google Scholar] [CrossRef]
- Kamata, K.; Sugahara, K. Base catalysis by mono- and polyoxometalates. Catalysts 2017, 7, 345. [Google Scholar] [CrossRef]
- Guo, L.; Jin, X.; Wang, X.; Yin, L.; Wang, Y.; Yang, Y.-W. Immobilizing polyether imidazole ionic liquids on zsm-5 zeolite for the catalytic synthesis of propylene carbonate from carbon dioxide. Molecules 2018, 23, 2710. [Google Scholar] [CrossRef]
- Han, Q.; Qi, B.; Ren, W.; He, C.; Niu, J.; Duan, C. Polyoxometalate-based homochiral metal-organic frameworks for tandem asymmetric transformation of cyclic carbonates from olefins. Nat. Commun. 2015, 6, 10007. [Google Scholar] [CrossRef]
- Jia, J.; Niu, Y.; Zhang, P.; Zhang, D.; Ma, P.; Zhang, C.; Niu, J.; Wang, J. A Monomeric tricobalt(II)-substituted dawson-type polyoxometalate decorated by a metal carbonyl group: [P2W15O56Co3(H2O)3(OH)3Mn(CO)3]8−. Inorg. Chem. 2017, 56, 10131–10134. [Google Scholar] [CrossRef]
- Lu, J.; Ma, X.; Singh, V.; Zhang, Y.; Wang, P.; Feng, J.; Ma, P.; Niu, J.; Wang, J. Facile CO2 cycloaddition to epoxides by using a tetracarbonyl metal selenotungstate derivate [{Mn(CO)3}4(Se2W11O43)]8−. Inorg. Chem. 2018, 57, 14632–14643. [Google Scholar] [CrossRef]
- Ge, W.; Wang, X.; Zhang, L.; Du, L.; Zhou, Y.; Wang, J. Fully-occupied Keggin type polyoxometalate as solid base for catalyzing CO2 cycloaddition and Knoevenagel condensation. Catal. Sci. Technol. 2016, 6, 460–467. [Google Scholar] [CrossRef]
- Chen, A.; Chen, C.; Xiu, Y.; Liu, X.; Chen, J.; Guo, L.; Zhang, R.; Hou, Z. Niobate salts of organic base catalyzed chemical fixation of carbon dioxide with epoxides to form cyclic carbonates. Green Chem. 2015, 17, 1842–1852. [Google Scholar] [CrossRef]
- Hayashi, S.; Yamazoe, S.; Koyasu, K.; Tsukuda, T. Lewis base catalytic properties of [Nb10O28]6− for CO2 fixation to epoxide: kinetic and theoretical studies. Chem. – Asian J. 2017, 12, 1635–1640. [Google Scholar] [CrossRef]
- Al-Garni, T.; Al-Jallal, N.; Aouissi, A. Synthesis of propylene carbonate from epoxide and CO2 catalyzed by carbon nanotubes supported Fe1.5PMo12O40. J. Chem. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Gómez-Romer, P. Polyoxometalates as photoelectrochemical models for quantum-sized colloidal semiconducting oxides. Solid State Ionics 1997, 101, 243–248. [Google Scholar]
- Vickers, J.W.; Lv, H.; Sumliner, J.M.; Zhu, G.; Luo, Z.; Musaev, D.G.; Geletii, Y.V.; Hill, C.L. Differentiating homogeneous and heterogeneous water oxidation catalysis: confirmation that [Co4(H2O)2(α-PW9O34)2]10− is a molecular water oxidation catalyst. J. Am. Chem. Soc. 2013, 135, 14110–14118. [Google Scholar] [CrossRef]
- Rozes, L.; Sanchez, C. Titanium oxo-clusters: Precursors for a Lego-like construction of nanostructured hybrid materials. Chem. Soc. Rev. 2011, 40, 1006–1030. [Google Scholar] [CrossRef]
- Izarova, N.V.; Pope, M.T.; Kortz, U. Noble metals in polyoxometalates. Angew. Chem., Int. Ed. 2012, 51, 9492–9510. [Google Scholar] [CrossRef]
- Dan-Hardi, M.; Serre, C.; Frot, T.; Rozes, L.; Maurin, G.; Sanchez, C.; Férey, G. A new photoactive crystalline highly porous titanium(IV) dicarboxylate. J. Am. Chem. Soc. 2009, 131, 10857–10859. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K.; Schmidt-Mende, L.; Schmidt-Mende, L. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef]
- Abdullah, H.; Khan, M.M.R.; Ong, H.R.; Yaakob, Z. Modified TiO2 photocatalyst for CO2 photocatalytic reduction: an overview. J. CO2 Util. 2017, 22, 15–32. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Jaroniec, M.; Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 2019, 119, 3962–4179. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ayass, W.W.; Taffa, D.H.; Schneemann, A.; Semrau, A.L.; Wannapaiboon, S.; Altmann, P.J.; Pöthig, A.; Nisar, T.; Balster, T.; et al. Discovery of polyoxo-noble-metalate-based metal–organic frameworks. J. Am. Chem. Soc. 2019, 141, 3385–3389. [Google Scholar] [CrossRef] [PubMed]
- Pley, M.; Wickleder, M.S. The cluster ion [Pt12O8(SO4)12]4−. Angew. Chem. Int. Ed. 2004, 43, 4168–4170. [Google Scholar] [CrossRef]
- Goloboy, J.C.; Klemperer, W.G. Are particulate noble-metal catalysts metals, metal oxides, or something in-between? Angew. Chem. Int. Ed. 2009, 48, 3562–3564. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, J.; Xu, H.; Xiong, Y. Coordination chemistry in the design of heterogeneous photocatalysts. Chem. Soc. Rev. 2017, 46, 2799–2823. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.D.; Katsoukis, G.; Frei, H. Photoinduced electron transfer from ZrOCo binuclear light absorber to pyridine elucidated by transient optical and infrared spectroscopy. J. Phys. Chem. C 2018, 122, 20176–20185. [Google Scholar] [CrossRef]
- Kim, W.; Frei, H. Directed assembly of cuprous oxide nanocatalyst for CO2 reduction coupled to heterobinuclear ZrOCoII light absorber in mesoporous silica. ACS Catal. 2015, 5, 5627–5635. [Google Scholar] [CrossRef]
- Kim, W.; Yuan, G.; McClure, B.A.; Frei, H. Light induced carbon dioxide reduction by water at binuclear ZrOCoII unit coupled to Ir oxide nanocluster catalyst. J. Am. Chem. Soc. 2014, 136, 11034–11042. [Google Scholar] [CrossRef]
- Macnaughtan, M.L.; Soo, H.S.; Frei, H. Binuclear ZrOCo metal-to-metal charge-transfer unit in mesoporous silica for light-driven CO2 reduction to CO and formate. J. Phys. Chem. C 2014, 118, 7874–7885. [Google Scholar] [CrossRef]
- Lin, W.; Frei, H. Photochemical CO2 splitting by metal-to-metal charge-transfer excitation in mesoporous ZrCu(I)-MCM-41 silicate sieve. J. Am. Chem. Soc. 2005, 127, 1610–1611. [Google Scholar] [CrossRef]
- McClure, B.A.; Frei, H. Excited state electron transfer of all-inorganic heterobinuclear TiOMn2+ chromophore anchored on silica nanoparticle surface. J. Phys. Chem. C 2014, 118, 11601–11611. [Google Scholar] [CrossRef]
- Takashima, T.; Yamaguchi, A.; Hashimoto, K.; Nakamura, R. Multielectron-transfer reactions at single Cu(II) centers embedded in polyoxotungstates driven by photo-induced metal-to-metal charge transfer from anchored Ce(III) to framework W(VI). Chem. Commun. 2012, 48, 2964. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Design of metal-to-metal charge-transfer chromophores for visible-light activation of oxygen-evolving Mn oxide catalysts in a polymer film. Chem. Mater. 2017, 29, 7234–7242. [Google Scholar] [CrossRef]
- Glass, E.N.; Fielden, J.; Kaledin, A.L.; Musaev, D.G.; Lian, T.; Hill, C.L. Extending metal-to-polyoxometalate charge transfer lifetimes: the effect of heterometal location. Chem. Eur. J. 2014, 20, 4297–4307. [Google Scholar] [CrossRef]
- Glass, E.N.; Musaev, D.G.; Lian, T.; Hill, C.L.; Fielden, J.; Huang, Z.; Xiang, X. Transition metal substitution effects on metal-to-polyoxometalate charge transfer. Inorg. Chem. 2016, 55, 4308–4319. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Preparation of polyoxometalate-based photo-responsive membranes for the photo-activation of manganese oxide catalysts. J. Vis. Exp. 2018, 138, e58072. [Google Scholar] [CrossRef]
- Meng, Y.-S.; Sato, O.; Liu, T. Manipulating metal-to-metal charge transfer for materials with switchable functionality. Angew. Chem. Int. Ed. 2018, 57, 12216–12226. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, D.; Xu, X.; Sheng, Y.; Xu, J.; Han, Y.-F. Application of operando spectroscopy on catalytic reactions. Curr. Opin. Chem. Eng. 2016, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Freund, T.; Gomes, W.P. Electrochemical methods for investigating catalysis by semiconductors. Catal. Rev. 1970, 3, 1–36. [Google Scholar] [CrossRef]
- Lopez, X.; Carbó, J.J.; Bo, C.; Poblet, J.M. Structure, properties and reactivity of polyoxometalates: A theoretical perspective. Chem. Soc. Rev. 2012, 41, 7537. [Google Scholar] [CrossRef]
- Lyon, D.K.; Miller, W.K.; Novet, T.; Domaille, P.J.; Evitt, E.; Johnson, D.C.; Finke, R.G. Highly oxidation resistant inorganic-porphyrin analog polyoxometalate oxidation catalysts. 1. The synthesis and characterization of aqueous-soluble potassium salts of α2-P2W17O61(Mn+·OH2)(n-10) and organic solvent soluble tetra-n-butylammonium salts of α2-P2W17O61(Mn+·Br)(n-11) (M = Mn3+, Fe3+, Co2+, Ni2+, Cu2+). J. Am. Chem. Soc. 1991, 113, 7209–7221. [Google Scholar]
- Randell, W.J.; Weakley, T.J.R.; Finke, R.G. Oxidation resistant inorganic porphyrin analog polyoxometalates. 3. The synthesis and X-ray crystallographic characterization of a new heteropolyoxoanion structural type, the diruthenium-oxo-bridged “bimetallic unorganic-polyphyrin analog KLi15[O{RuIVCl(α2-P2W17O61)}2]·2KCl·60H2O”. Inorg. Chem. 1993, 32, 1068–1071. [Google Scholar]
- Neumann, R.; Dahan, M. A ruthenium-substituted polyoxometalate as an inorganic dioxygenase for activation of molecular oxygen. Nature 1997, 388, 353–355. [Google Scholar] [CrossRef]
- Corbin, N.; Zeng, J.; Williams, K.; Manthiram, K. Heterogeneous molecular catalysts for electrocatalytic CO2 reduction. Nano Res. 2019, 1–33. [Google Scholar] [CrossRef]
- Shih, C.F.; Zhang, T.; Li, J.; Bai, C. Powering the future with liquid sunshine. Joule 2018, 2, 1925–1949. [Google Scholar] [CrossRef]
- Xu, Q.; Niu, Y.; Wang, G.; Li, Y.; Zhao, Y.; Singh, V.; Niu, J.; Wang, J. Polyoxoniobates as a superior Lewis base efficiently catalyzed Knoevenagel condensation. Mol. Catal. 2018, 453, 93–99. [Google Scholar] [CrossRef]
Sample Availability: No available |
Figure 1.
Isomers and mono- to trilacunary derivatives of the Keggin structure; color code: MO6 octahedron, light blue; XO4 tetrahedron, red (X = central atom).
Figure 1.
Isomers and mono- to trilacunary derivatives of the Keggin structure; color code: MO6 octahedron, light blue; XO4 tetrahedron, red (X = central atom).

Figure 2.
Suggested modes of coordination of CO2. Color code: W, black ball; P, green ball; O, red ball; Zn, purple ball. Reprinted with permission from [21]. Copyright © 2018 Wiley-VCH Verlag GmbH and Co. KGaA.
Figure 2.
Suggested modes of coordination of CO2. Color code: W, black ball; P, green ball; O, red ball; Zn, purple ball. Reprinted with permission from [21]. Copyright © 2018 Wiley-VCH Verlag GmbH and Co. KGaA.

Figure 3.
The substitution of H2O with CO2 in [RuIII(H2O)SiW11O39]5−. Reprinted with permission from [40]. Copyright © 2010 Wiley-VCH Verlag GmbH and Co. KGaA.
Figure 3.
The substitution of H2O with CO2 in [RuIII(H2O)SiW11O39]5−. Reprinted with permission from [40]. Copyright © 2010 Wiley-VCH Verlag GmbH and Co. KGaA.

Scheme 1.
Proposed photoreduction of CO2 with H2.

Figure 4.
ReI(L)(CO)3-NaHPW12O40 presenting the complexation of two [PW12O40]3− units to Na. H atoms and solvent molecules are omitted. C, black ball; N, blue ball; O, red ball; P, purple ball; Na, yellow ball; Re, green ball; W, gray ball. Reprinted with permission from [41]. Copyright© 2011 American Chemical Society.
Figure 4.
ReI(L)(CO)3-NaHPW12O40 presenting the complexation of two [PW12O40]3− units to Na. H atoms and solvent molecules are omitted. C, black ball; N, blue ball; O, red ball; P, purple ball; Na, yellow ball; Re, green ball; W, gray ball. Reprinted with permission from [41]. Copyright© 2011 American Chemical Society.

Figure 5.
Potential energy (units in kcal mol−1) surface for the reduction of CO2 to CO catalyzed by POM-ReI complex. Reprinted with permission from [42]. Copyright © 2016 American Chemical Society.
Figure 5.
Potential energy (units in kcal mol−1) surface for the reduction of CO2 to CO catalyzed by POM-ReI complex. Reprinted with permission from [42]. Copyright © 2016 American Chemical Society.

Figure 6.
The photoreduction of CO2 by the Co4@g-C3N4 hybrid material. The structure of Na10[Co4(H2O)2(PW9O34)2]; color code: WO6 octahedron, light blue; PO4 tetrahedron, red; Co, purple ball; O, red ball. (a). The photocatalytic activity of different photocatalysts (b). Time course of the CO and H2 (c). Recycling experiments (d). Reprinted with permission from [44]. Copyright © 2017 American Chemical Society.
Figure 6.
The photoreduction of CO2 by the Co4@g-C3N4 hybrid material. The structure of Na10[Co4(H2O)2(PW9O34)2]; color code: WO6 octahedron, light blue; PO4 tetrahedron, red; Co, purple ball; O, red ball. (a). The photocatalytic activity of different photocatalysts (b). Time course of the CO and H2 (c). Recycling experiments (d). Reprinted with permission from [44]. Copyright © 2017 American Chemical Society.

Figure 7.
Proposed crystal growth mechanism of Au@NENU-3 and Au@NENU-10. The intermediate of Cu2+ around polyoxometalate (POM) (a). The proposed the basic growing unit consisting of 1 POM, 24 Cu2+, and 8 BTC ligands in which each Cu2 unit coordinates with 2 BTC ligands (b). Eight BTC ligands are needed to coordinate with four Cu2 units when crystal grows along the 〈100〉 direction (c). While only six BTC ligands are needed for three Cu2 units along the 〈111〉 direction (d). The enlarged diagram of the dashed red circle (e). The exposed surface of the octahedron shape for Au@NENU-3 ({111} plane where [PW12O40]3− is sheltered by BTC (f). The exposed surface of the cube shape for NENU-10 ({100} plane where [PTi2W10O40]7− is exposed wholly) (g). The SEM images for Au@NENU-3 and Au@NENU-10, respectively (h, i). Reprinted with permission from [45]. Copyright © 2018 Wiley-VCH Verlag GmbH and Co. KGaA.
Figure 7.
Proposed crystal growth mechanism of Au@NENU-3 and Au@NENU-10. The intermediate of Cu2+ around polyoxometalate (POM) (a). The proposed the basic growing unit consisting of 1 POM, 24 Cu2+, and 8 BTC ligands in which each Cu2 unit coordinates with 2 BTC ligands (b). Eight BTC ligands are needed to coordinate with four Cu2 units when crystal grows along the 〈100〉 direction (c). While only six BTC ligands are needed for three Cu2 units along the 〈111〉 direction (d). The enlarged diagram of the dashed red circle (e). The exposed surface of the octahedron shape for Au@NENU-3 ({111} plane where [PW12O40]3− is sheltered by BTC (f). The exposed surface of the cube shape for NENU-10 ({100} plane where [PTi2W10O40]7− is exposed wholly) (g). The SEM images for Au@NENU-3 and Au@NENU-10, respectively (h, i). Reprinted with permission from [45]. Copyright © 2018 Wiley-VCH Verlag GmbH and Co. KGaA.

Figure 8.
The structures of {Mo154}, {Mn6P3W24}, and {Mo132} POM macroions catalysts. Color code: Mo, red ball; O, blue ball; Mn, yellow ball; P, pink ball; WO6 octahedron, grey. Reprinted with permission from [46]. © 2016 Attribution-Non Commercial 3.0 Unported Licence (CC BY-NC 3.0).
Figure 8.
The structures of {Mo154}, {Mn6P3W24}, and {Mo132} POM macroions catalysts. Color code: Mo, red ball; O, blue ball; Mn, yellow ball; P, pink ball; WO6 octahedron, grey. Reprinted with permission from [46]. © 2016 Attribution-Non Commercial 3.0 Unported Licence (CC BY-NC 3.0).

Figure 9.
The asymmetric units of NENU-605 (a), NENU-606 (b) and NENU-607 (c). Reprinted with permission from [50]. © 2019 Attribution-Non Commercial 3.0 Unported Licence (CC BY-NC 3.0).
Figure 9.
The asymmetric units of NENU-605 (a), NENU-606 (b) and NENU-607 (c). Reprinted with permission from [50]. © 2019 Attribution-Non Commercial 3.0 Unported Licence (CC BY-NC 3.0).

Figure 10.
The proposed mechanism for photocatalytic reduction of CO2 to CH4 using NENU-605 or NENU-606. Color code: P, yellow ball; Mo, blue ball; O, red ball. Reprinted with permission from [50]. © 2019 Attribution-Non Commercial 3.0 Unported Licence (CC BY-NC 3.0).
Figure 10.
The proposed mechanism for photocatalytic reduction of CO2 to CH4 using NENU-605 or NENU-606. Color code: P, yellow ball; Mo, blue ball; O, red ball. Reprinted with permission from [50]. © 2019 Attribution-Non Commercial 3.0 Unported Licence (CC BY-NC 3.0).

Figure 11.
Electroreduction of CO2 by [SiW11O39Co]6− catalyst. Color code: WO6 octahedron, light blue; SiO4 tetrahedron, red; Co, purple ball; O, red ball. The notation of (_) in the above figure represents a vacant coordination site.
Figure 11.
Electroreduction of CO2 by [SiW11O39Co]6− catalyst. Color code: WO6 octahedron, light blue; SiO4 tetrahedron, red; Co, purple ball; O, red ball. The notation of (_) in the above figure represents a vacant coordination site.

Figure 12.
Synthesis of the organometallic derivative [α-H2PW11O39(RhIIICp*(OH2))]3−. Color code: WO6 octahedron, navy blue; PO4 tetrahedron, green; O, red ball. Reprinted with permission from [54]. Copyright © 2019 Wiley-VCH Verlag GmbH and Co. KGaA.
Figure 12.
Synthesis of the organometallic derivative [α-H2PW11O39(RhIIICp*(OH2))]3−. Color code: WO6 octahedron, navy blue; PO4 tetrahedron, green; O, red ball. Reprinted with permission from [54]. Copyright © 2019 Wiley-VCH Verlag GmbH and Co. KGaA.

Figure 13.
Schematic illustration of the structures of M-PMOFs (Polyoxometalate-Metalloporphyrin Organic Frameworks). Color code: MoO6 octahedra, bottle green; Zn, light blue ball; Ni, green ball; Fe, orange ball; Co, magenta ball, N, blue ball; C, grey ball; O, red ball; H was omitted. Reprinted with permission from [58]. Copyright © 2015 Attribution 4.0 International (CC BY 4.0).
Figure 13.
Schematic illustration of the structures of M-PMOFs (Polyoxometalate-Metalloporphyrin Organic Frameworks). Color code: MoO6 octahedra, bottle green; Zn, light blue ball; Ni, green ball; Fe, orange ball; Co, magenta ball, N, blue ball; C, grey ball; O, red ball; H was omitted. Reprinted with permission from [58]. Copyright © 2015 Attribution 4.0 International (CC BY 4.0).

Figure 14.
The DFT calculation and proposed reaction mechanism. The free energy diagrams CO2 reduction reaction (CO2RR) for the Zn-ε-Keggin POM, the Co-TCPP metalloporphyrin and the Co-PMOF (a); comparison of the free energy of each elementary reaction (ΔG1, ΔG2, and ΔG3 represent the free energy of * COOH formation, * CO formation, and CO desorption process, respectively) in CO2RR for Co-PMOF, Fe-PMOF, Ni-PMOF, and Zn-PMOF (b); proposed mechanistic scheme for the CO2RR on Co-PMOF (c,d); color code: MoO6 octahedra, bottle green; Zn, light blue ball; Co, magenta ball, N, blue ball; C, grey ball; O, red ball. Reprinted with permission from [58]. Copyright © 2015 Attribution 4.0 International (CC BY 4.0).
Figure 14.
The DFT calculation and proposed reaction mechanism. The free energy diagrams CO2 reduction reaction (CO2RR) for the Zn-ε-Keggin POM, the Co-TCPP metalloporphyrin and the Co-PMOF (a); comparison of the free energy of each elementary reaction (ΔG1, ΔG2, and ΔG3 represent the free energy of * COOH formation, * CO formation, and CO desorption process, respectively) in CO2RR for Co-PMOF, Fe-PMOF, Ni-PMOF, and Zn-PMOF (b); proposed mechanistic scheme for the CO2RR on Co-PMOF (c,d); color code: MoO6 octahedra, bottle green; Zn, light blue ball; Co, magenta ball, N, blue ball; C, grey ball; O, red ball. Reprinted with permission from [58]. Copyright © 2015 Attribution 4.0 International (CC BY 4.0).

Figure 15.
Schematic illustrations of the bioelectrochemical system for CO2 fixation. Color code: WO6 octahedra, light blue; Co, pink ball; P, orange ball; Cu spheres, green; C, black ball; N, dark blue ball; O, red ball; H atoms have been omitted for clarity. Reprinted with permission from [67]. Copyright © 2018 The Royal Society of Chemistry.
Figure 15.
Schematic illustrations of the bioelectrochemical system for CO2 fixation. Color code: WO6 octahedra, light blue; Co, pink ball; P, orange ball; Cu spheres, green; C, black ball; N, dark blue ball; O, red ball; H atoms have been omitted for clarity. Reprinted with permission from [67]. Copyright © 2018 The Royal Society of Chemistry.

Scheme 2.
Non-reductive chemical fixation of CO2 by POM catalysts.

Figure 16.
Synthetic procedure of the polyoxometallate-organocatalyst-metal organic frameworks (POMOFs) and the schematic representation of tandem catalysis for the asymmetric cyclic carbonate transformation from olefins and CO2. Color code: α-[ZnW12O40]6−, yellow polyhedral; Zn, cyan ball; N, blue ball; C, orange ball; O, red ball; H atom was omitted. Reprinted with permission from [81]. Copyright © 2015 Attribution 4.0 International (CC BY 4.0).
Figure 16.
Synthetic procedure of the polyoxometallate-organocatalyst-metal organic frameworks (POMOFs) and the schematic representation of tandem catalysis for the asymmetric cyclic carbonate transformation from olefins and CO2. Color code: α-[ZnW12O40]6−, yellow polyhedral; Zn, cyan ball; N, blue ball; C, orange ball; O, red ball; H atom was omitted. Reprinted with permission from [81]. Copyright © 2015 Attribution 4.0 International (CC BY 4.0).

Figure 17.
The basic building blocks (a) and coordination model of CO2 (b) in [PMo12O40Zn4(CO2)]2−. Color code: Zn, yellow ball; O, red ball; C, white ball; MoO6 octahedra, green; PO4 tetrahedron, magenta. Reprinted with permission from [19]. © 2018 The Royal Society of Chemistry.
Figure 17.
The basic building blocks (a) and coordination model of CO2 (b) in [PMo12O40Zn4(CO2)]2−. Color code: Zn, yellow ball; O, red ball; C, white ball; MoO6 octahedra, green; PO4 tetrahedron, magenta. Reprinted with permission from [19]. © 2018 The Royal Society of Chemistry.

Figure 18.
The effect of particle size on the flat band redox potentials (a) and schematic band structure diagram of POMs and WO3 (b). Reprinted with permission from [88]. © 1997 2018 Elsevier BV.
Figure 18.
The effect of particle size on the flat band redox potentials (a) and schematic band structure diagram of POMs and WO3 (b). Reprinted with permission from [88]. © 1997 2018 Elsevier BV.


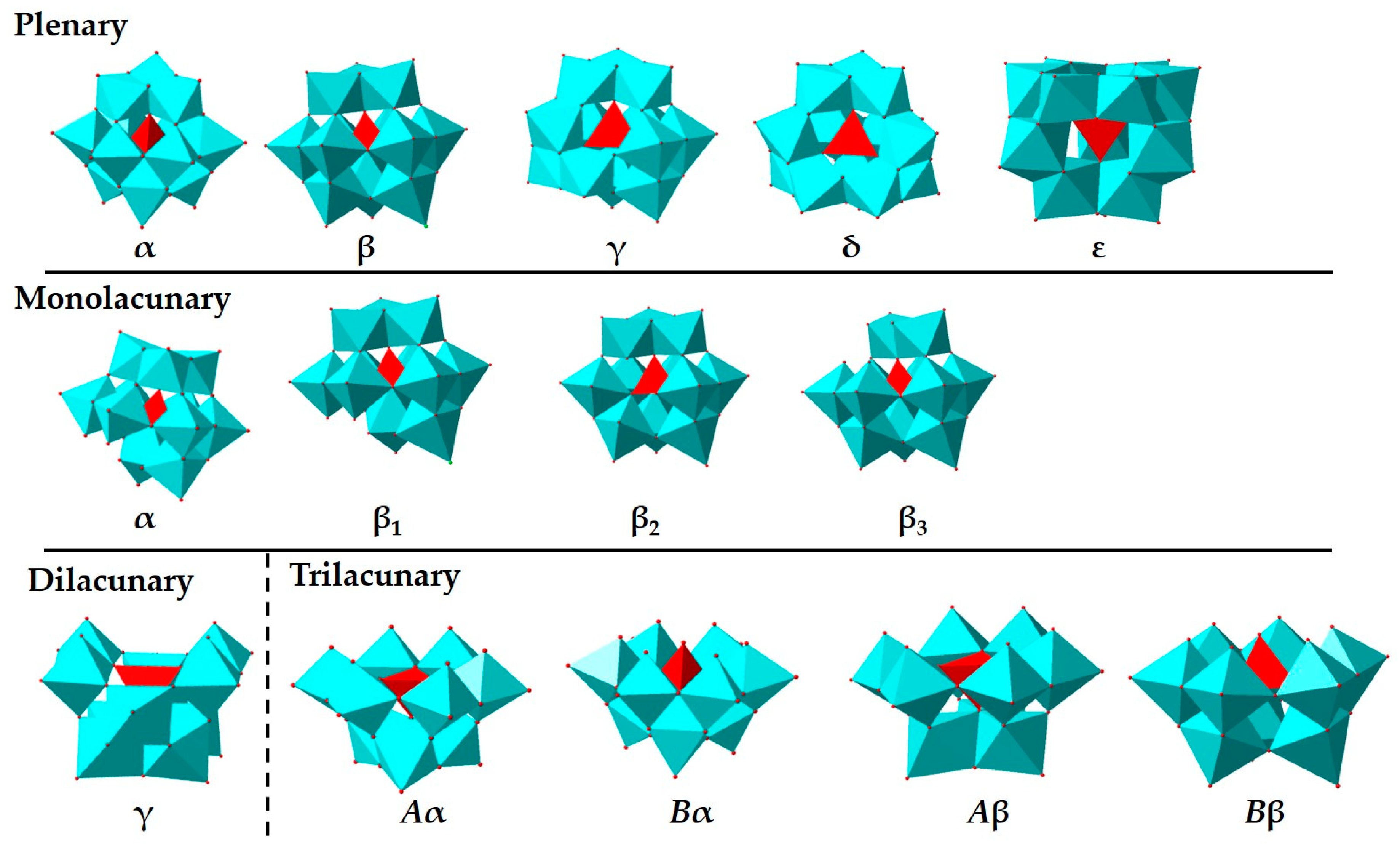{kind=link}
{kind=link}
{kind=link}
{kind=link}
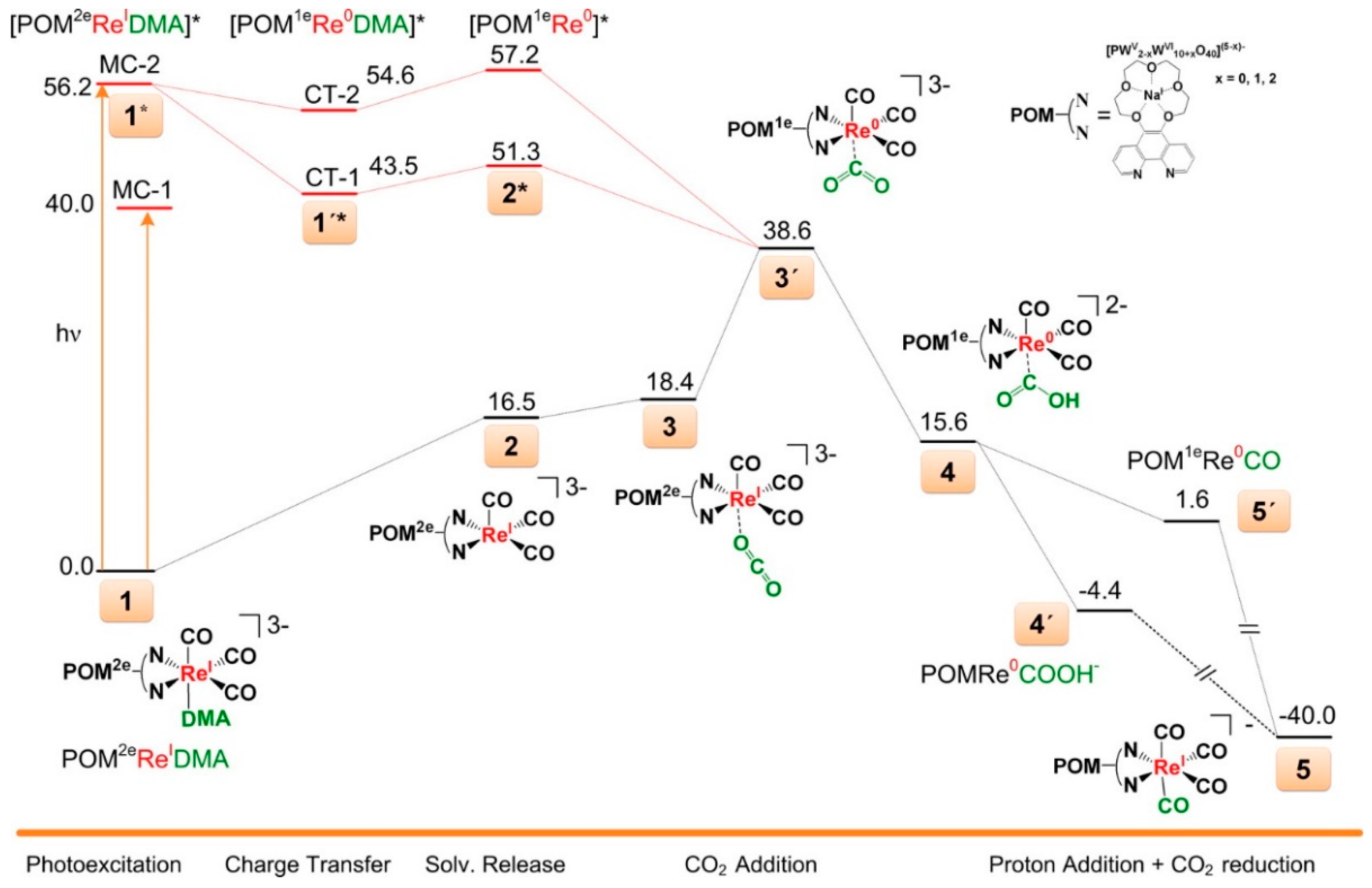{kind=link}
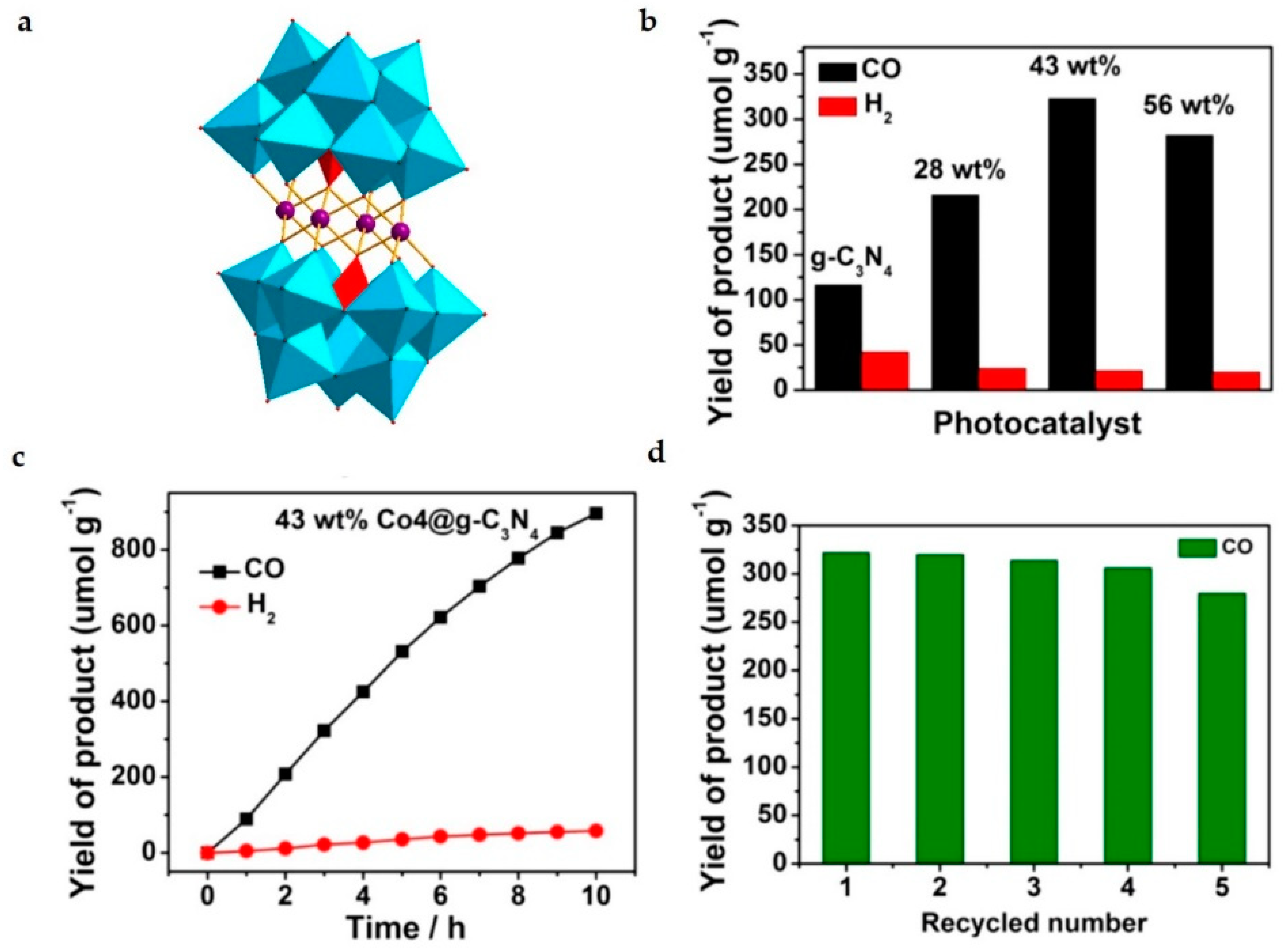{kind=link}
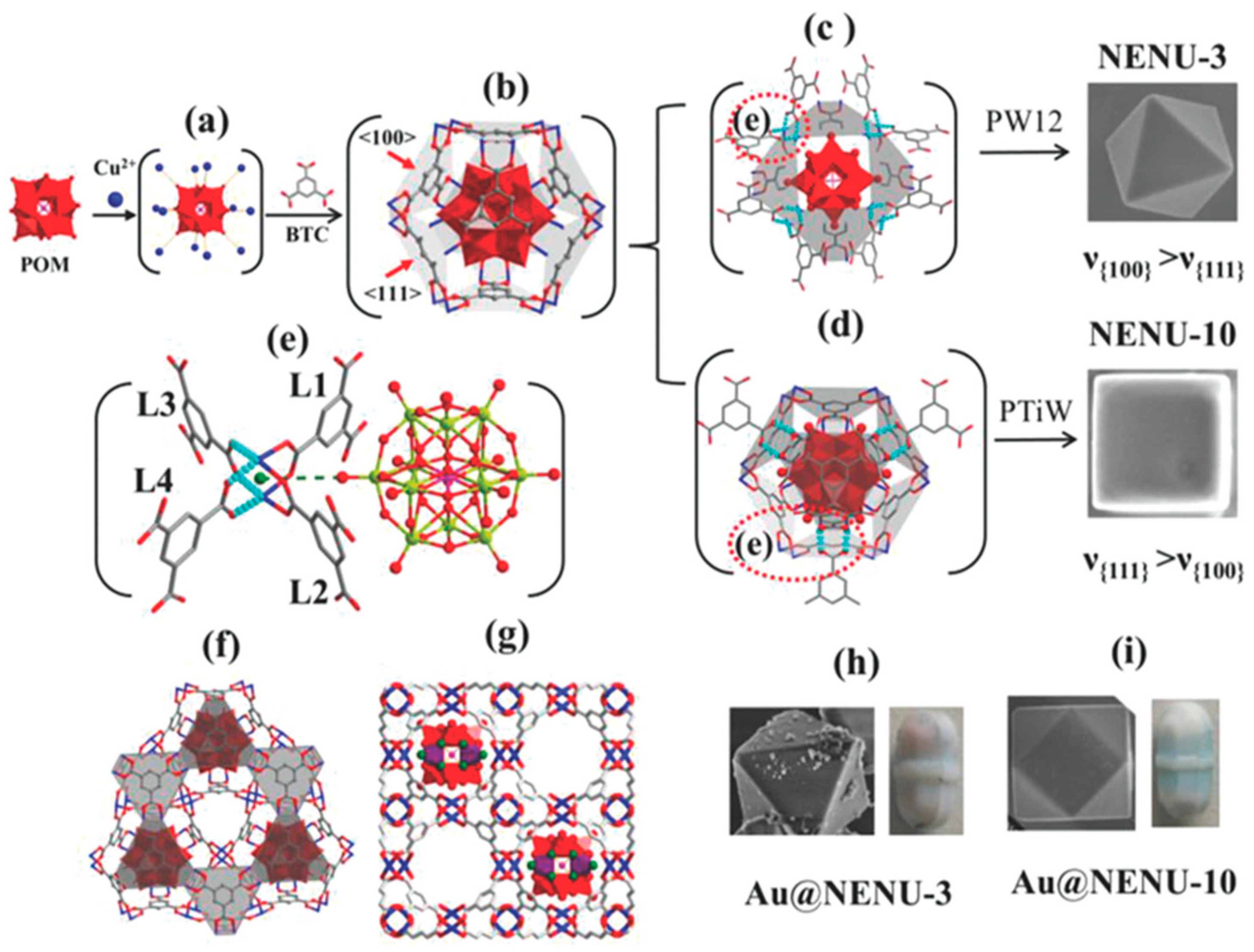{kind=link}
{kind=link}
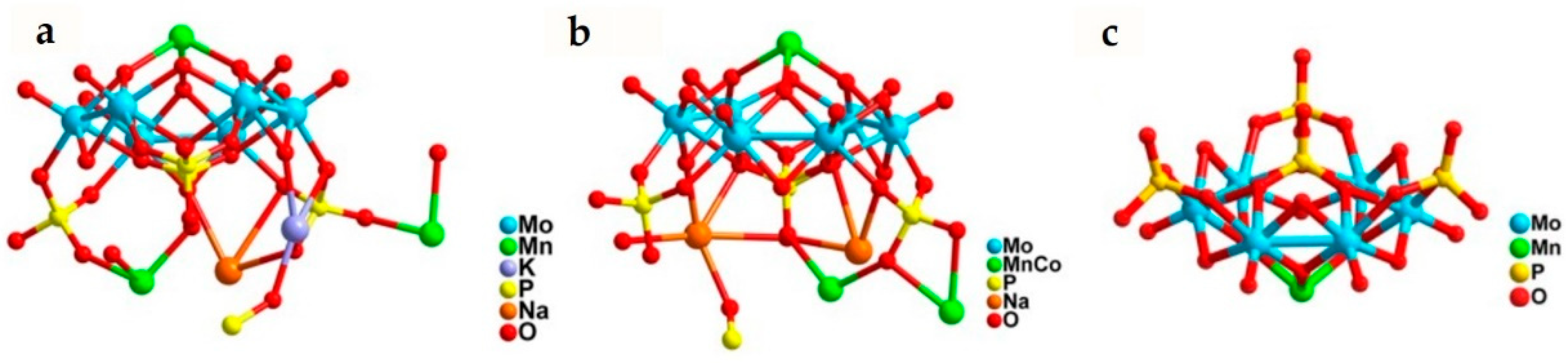{kind=link}
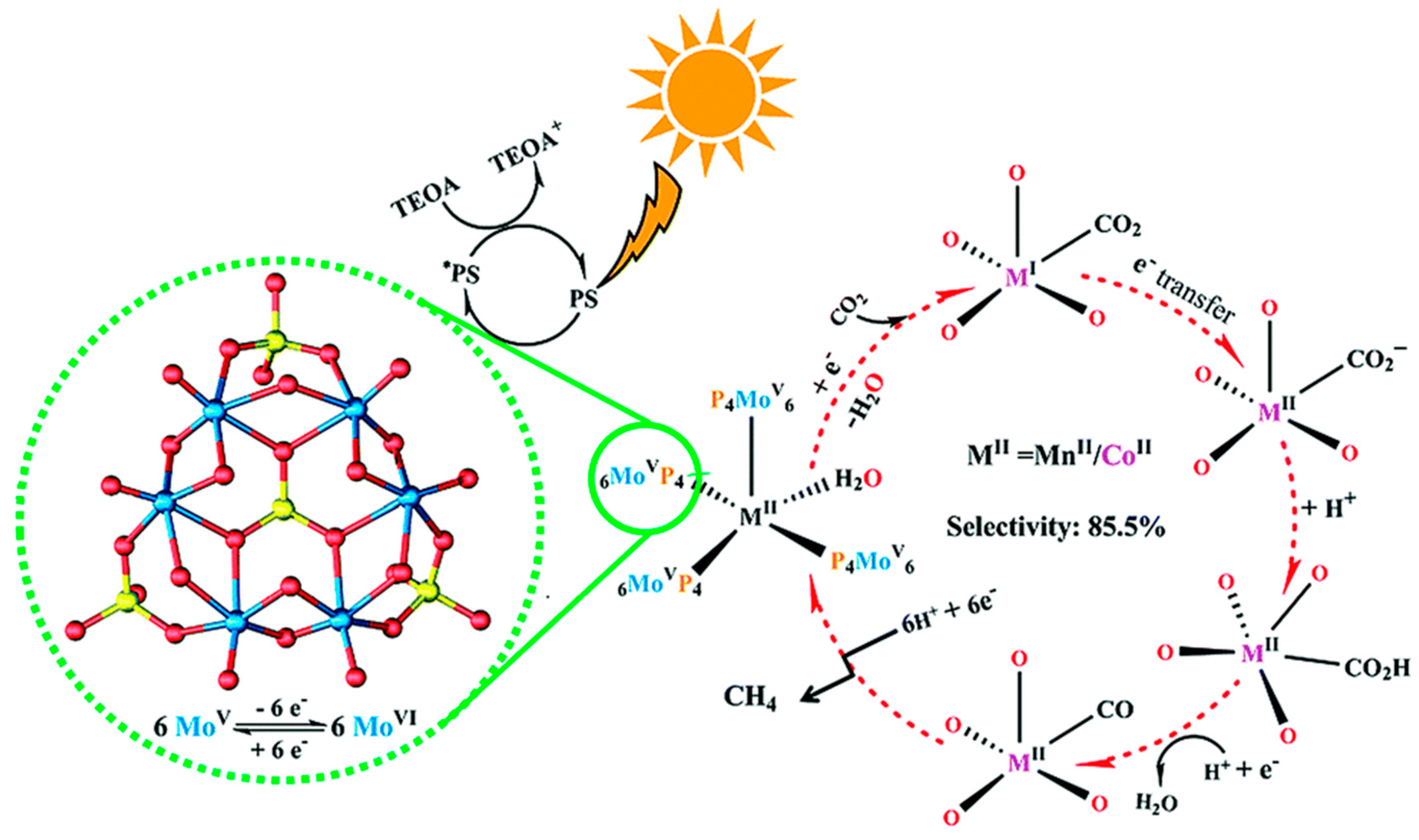{kind=link}
{kind=link}
{kind=link}
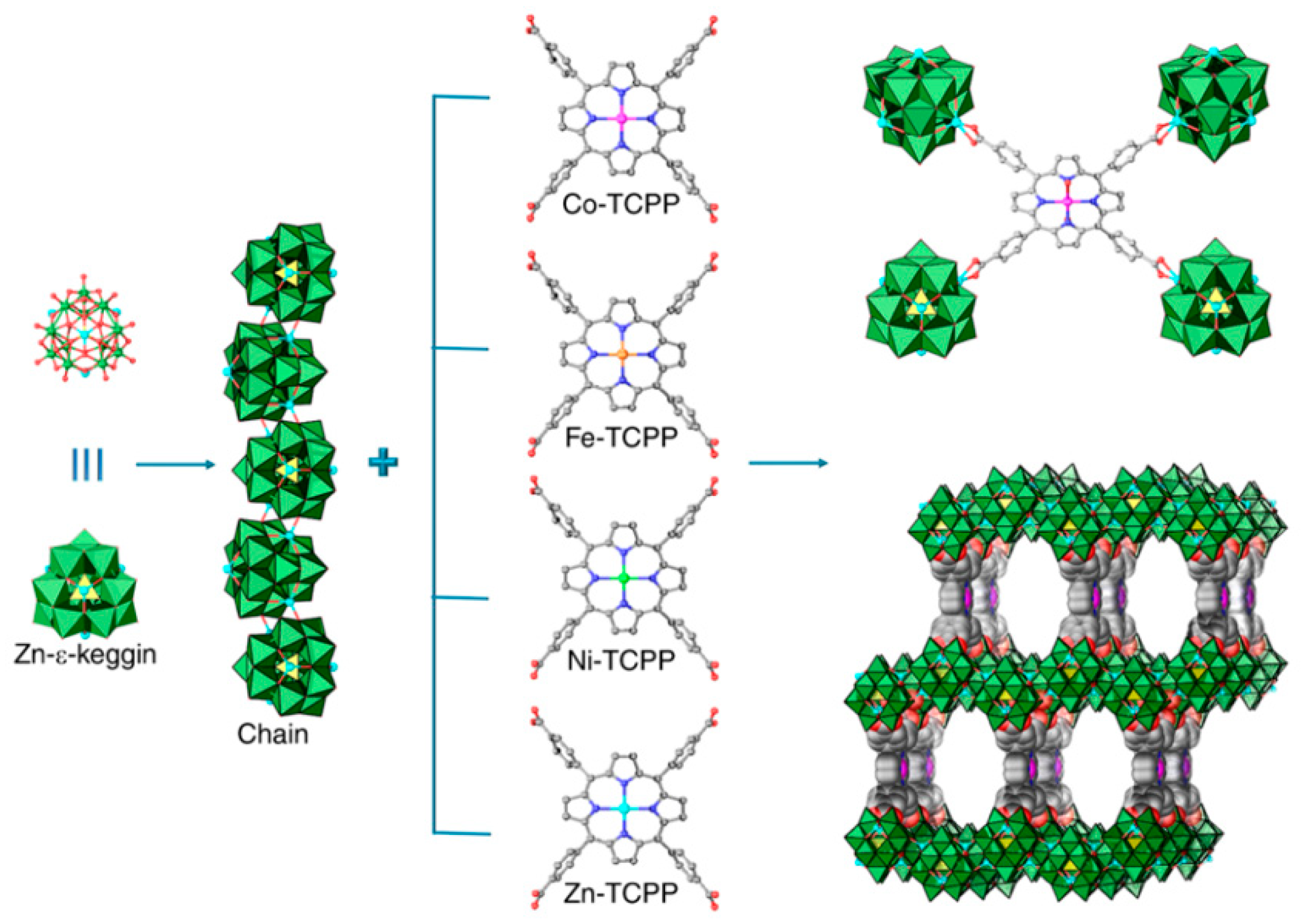{kind=link}
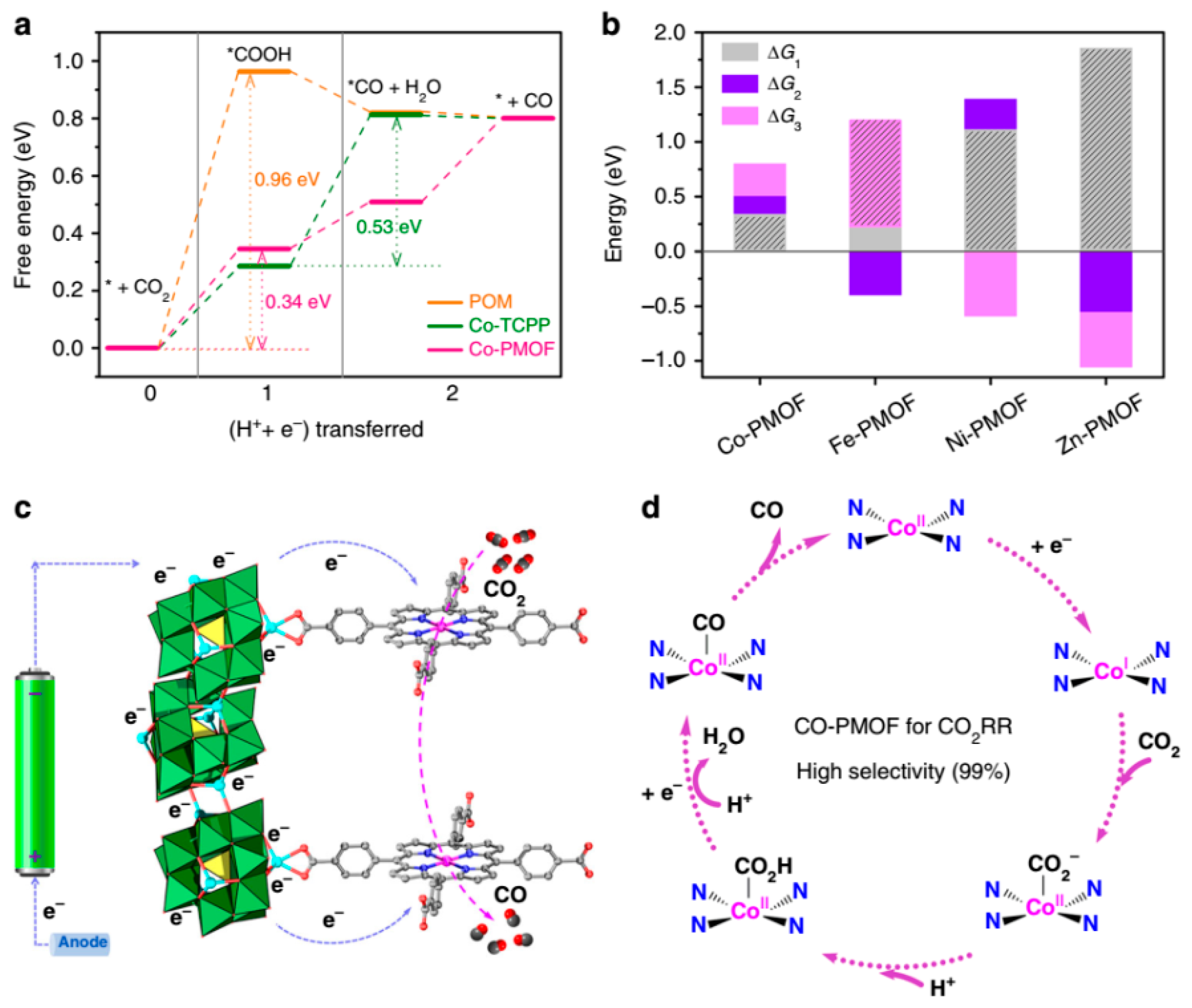{kind=link}
{kind=link}
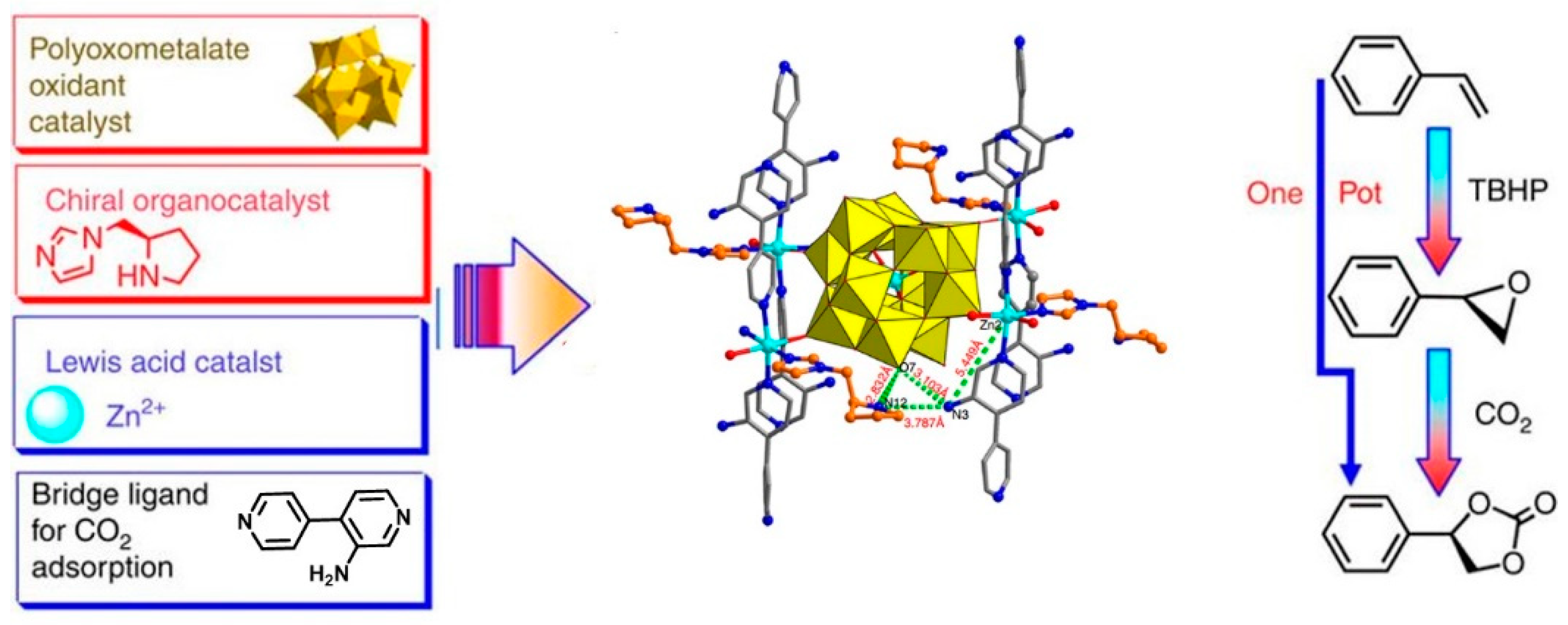{kind=link}
{kind=link}
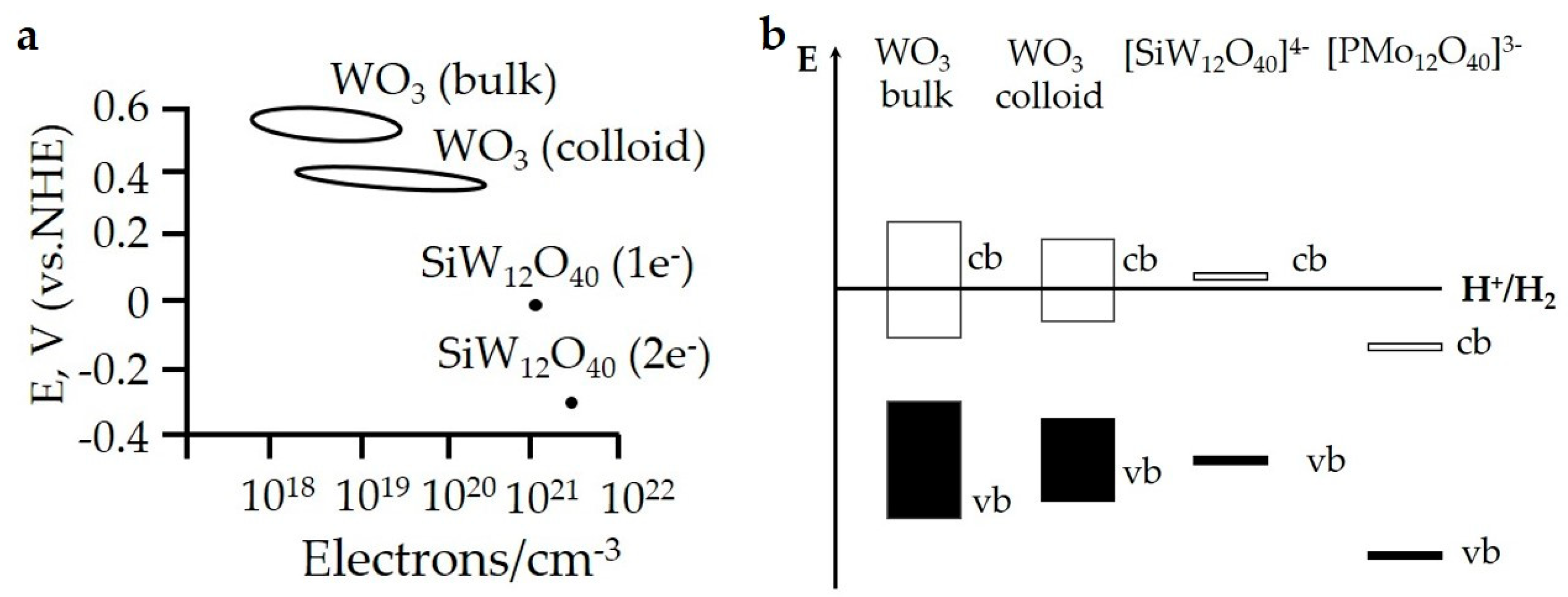{kind=link}
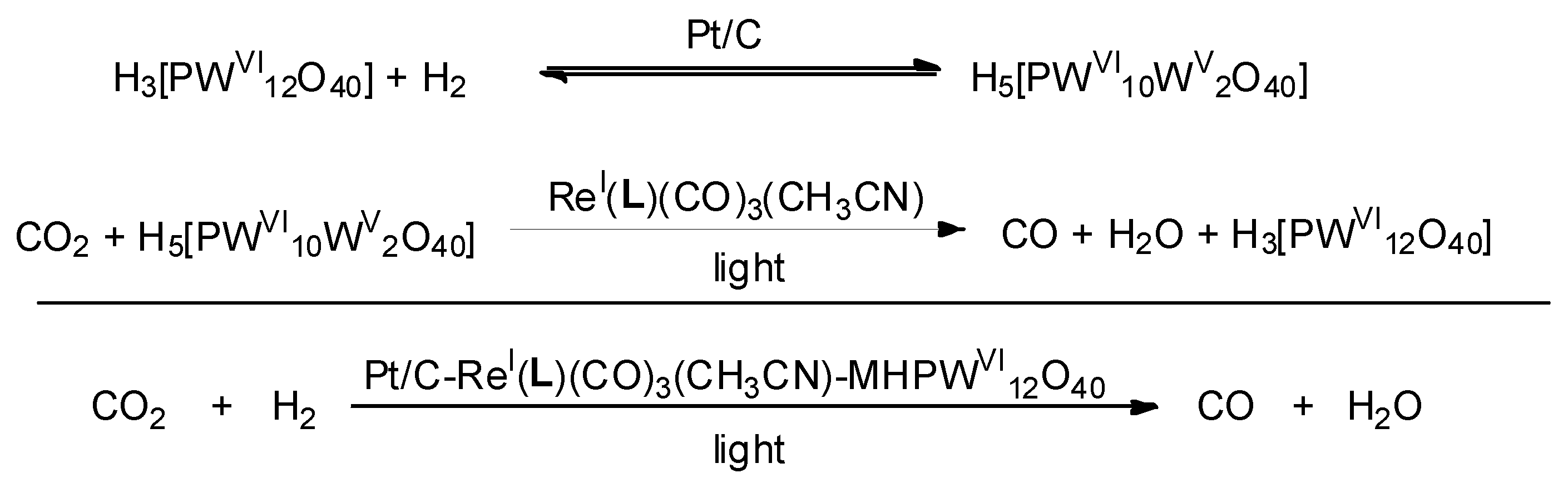{kind=link}
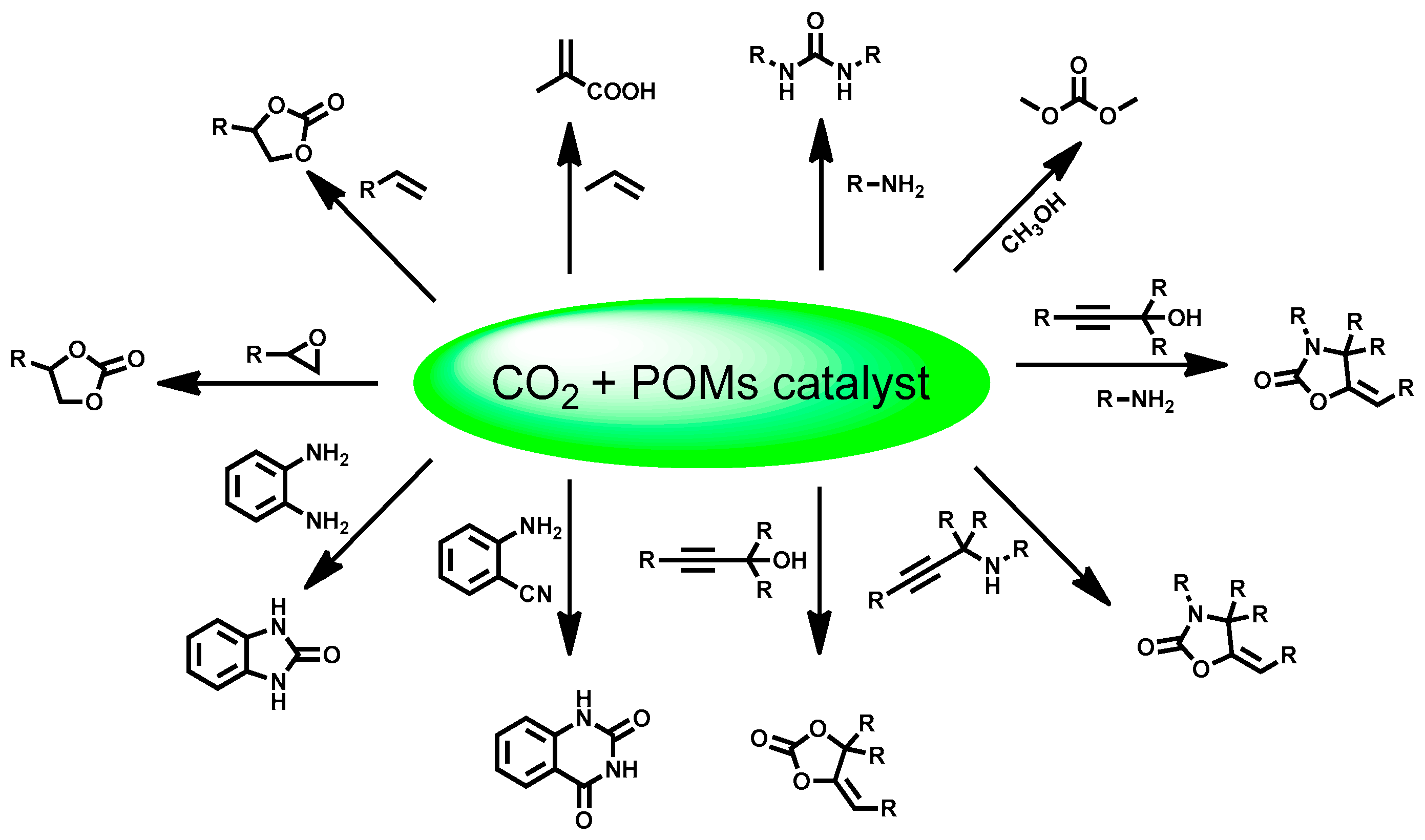{kind=link}
Table 1.
Redox potentials for CO2 reduction.
| Products | Products Reaction E0 (V) | E0 (V) pH = 7 |
|---|---|---|
| - | HO + 2 h+ → 1/2O2 + 2H+ | +0.82 |
| - | CO2 + e− → | −1.9 |
| HCOOH | CO2 + 2 H+ + 2 e− → HCOOH | −0.61 |
| CO | CO2 + 2 H+ + 2 e− → CO + H2O | −0.53 |
| HCHO | CO2 + 2 H+ + 4 e− → HCHO + H2O | −0.48 |
| CH3OH | CO2 + 6 H+ + 6 e− → CH3OH + H2O | −0.38 |
| CH4 | CO2 + 8 H+ + 8 e− → CH4 + 2H2O | −0.24 |
| - | 2 H+ + 2 e− → H2 | –0.41 |
Table 2.
Catalytic performance of CO, H2, and CH4 from CO2 photoreduction a.
| Entry | Catalysts | Products (μmol g−1 h−1) | ||
|---|---|---|---|---|
| CO | CH4 | H2 | ||
| 1 | Au@NENU-10 | 12.8 | 2.1 | 2.6 |
| 2 | Au@NENU-3 | 0.5 | - | 0.15 |
| 3 | Au/Na3PW12 O40) | - | - | 0.45 |
| 4 | Au/K7(PTi2W10O40) | 2.1 | 0.35 | 0.29 |
| 5 | NENU-10 | - | - | - |
| 6 | NENU-3 | - | - | - |
| 7 | HKUST-1 | - | - | - |
a. Reaction conditions: 100 mL quartz reactor, 15 mL H2O, 100 mg catalyst, 40 °C, 5 h, 300 W Xe lamp (λ > 420 nm). Solid catalyst was in the atmosphere of water vapor and CO2 instead of being immersed in water.
Table 3.
The photocatalytic performance of the different catalysts a.
| Catalyst | HCOOH Yield (μmol) | TON | TOF (s−1) |
|---|---|---|---|
| {Mo154}1165 | 116.7 | 778 | 377 |
| {Mn6P3W24}931 | 40.6 | 270 | 56 |
| {Mo132}1064@RGO | 205 | 1366 | 610 |
a. Catalyst containing 0.15 μmol of {Mo154}, {Mn6P3W24} or {Mo132} units. The reaction mixtures were kept in a photo-reactor under UV-light lamp with 373 nm wavelength and 19 mW cm−2 energy density.
Table 4.
The photocatalytic reduction of CO2 to CH4 using different catalysts a.
| Entry | Catalyst | CO (nmol/g) | CH4 (nmol/g) | CH4-TON (10−3) | CH4-TOF (10−3 h−1) | All-TON (10−3) | All-TOF (10−3 h−1) |
|---|---|---|---|---|---|---|---|
| 1 | NENU-605 | 52 | 170 | 104.1 | 5.5 | 135.9 | 7.2 |
| 2 | NENU-606 | 68 | 402 | 241.4 | 10.5 | 282.2 | 12.3 |
| 3 | NENU-607 | 47 | 70 | 15.2 | 0.75 | 25.4 | 1.3 |
| 4 b | NENU-606 | n.d. | n.d. | - | - | - | - |
| 5 c | NENU-606 | n.d. | n.d. | - | - | - | - |
| 6 d | NENU-606 | n.d. | n.d. | - | - | - | - |
| 7 | blank | n.d. | n.d. | - | - | - | - |
a. Reaction conditions: PS = [Ru(bpy)3]Cl2.6H2O (0.01 mmol, ) H2O (28 mL), SD = TEOA (2 mL), CO2 (1 atm), λ ≥420 nm, 20 °C; b. without PS; c. In dark; d. Altering H2O with dry MeCN. n.d. = not detected.
Table 5.
CO2 reduction potentials vs. SHE a.
| Products | Half-Electrochemical Thermodynamic Reactions | V vs. SHE |
|---|---|---|
| C | CO2(g) + 4 H+ + 4 e− = C(s) + 2 H2O(l) | −0.210 |
| C | CO2(g) + 2 H2O(l) + 4 e− = C(s) + 4 OH− | −0.627 |
| HCOOH | CO2(g) + 2 H+ + 2e− = HCOOH(l) | −0.250 |
| HCOO− | CO2(g) + 2 H2O(l) + 2 e− = HCOO− (aq) + OH− | −1.078 |
| CO | CO2(g) + 2 H+ + 2 e− = CO(g) + H2O(l) | −0.106 |
| CO | CO2(g) + 2 H2O(l) + 2 e− = CO(g) + 2 OH− | −0.934 |
| HCHO | CO2(g) + 4 H+ + 4e− = HCHO(l) + H2O(l) | −0.070 |
| HCHO | CO2(g) + 3 H2O(l) + 4 e− = HCHO(l) + 4 OH− | −0.898 |
| CH3OH | CO2(g) + 6 H+ + 6 e− = CH3OH(l) + H2O(l) | 0.016 |
| CH3OH | CO2(g) + 5 H2O(l) + 6 e− = CH3OH(l) + 6 OH− | −0.812 |
| CH4 | CO2(g) + 8 H+ + 8 e− = CH4(g) + 2 H2O (l) | 0.169 |
| CH4 | CO2(g) + 6 H2O(l) + 8 e− = CH4(g) + 8 OH− | −0.659 |
| oxalic acid | 2 CO2(g) + 2 H+ + 2 e− = (COOH)2(aq) | −0.500 |
| oxalate | 2 CO2(g) + 2 e− = C2(aq) | −0.590 |
| ethylene | 2 CO2(g) + 12 H+ + 12 e− = CH2CH2(g) + 4 H2O(l) | 0.064 |
| ethylene | 2 CO2(g) + 8 H2O(l) + 12 e− = CH2CH2(g) + 12 OH− | −0.764 |
| ethanol | 2 CO2(g) + 12 H+ + 12 e− = CH3CH2OH(l) + 3 H2O(l) | 0.084 |
| ethanol | 2 CO2(g) + 9 H2O(l) + 12 e− = CH3CH2OH(l) + 12 OH− | −0.744 |
a. Reprinted with permission from [51]. Copyright © 2014 Royal Society of Chemistry.
Table 6.
CO2 electroreduction performances a.
| Catalysts | E V vs. SHE | Onset Potential V | jCO mA cm−2 | Tafel Slope (mV dec−1) | ECSA mF cm−2 | EIS Ω | FEco % | TOF H−1 |
|---|---|---|---|---|---|---|---|---|
| Co-PMOF | −0.8 | −0.35 | 18.08 | 98 | 12.17 | 9.83 | 98.7 | 1656 |
| Fe-PMOF | −0.7 | −0.53 | 0.47 | 211 | 10.26 | 10.26 | 28.8 | 17.45 |
| Ni-PMOF | −0.8 | −0.58 | 0.27 | 675 | 10.16 | 10.70 | 18.5 | 8.11 |
| Zn-PMOF | −0.9 | −0.60 | 0.02 | 206 | 9.83 | 12.17 | 0.95 | 0.005 |
| Co-TMCP | −0.9 | −0.53 | NA b | 151 | NA | NA | 40 | NA |
| TMCP | −0.6 | −0.67 | NA | 552 | NA | NA | 0.77 | NA |
| NNU-12 | −0.6 | −0.6 | NA | 413 | NA | NA | 1.8 | NA |
a. Electrolyte and pH 0.5 M KHCO3, pH = 7.2; b. NA means not mentioned in the [58].
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cao, Y.; Chen, Q.; Shen, C.; He, L. Polyoxometalate-Based Catalysts for CO2 Conversion. Molecules 2019, 24, 2069. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112069
AMA Style
Cao Y, Chen Q, Shen C, He L. Polyoxometalate-Based Catalysts for CO2 Conversion. Molecules. 2019; 24(11):2069. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112069
Chicago/Turabian StyleCao, Yanwei, Qiongyao Chen, Chaoren Shen, and Lin He. 2019. "Polyoxometalate-Based Catalysts for CO2 Conversion" Molecules 24, no. 11: 2069. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112069