Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years


1
New Drug Development Center (NDDC), Daegu-Gyeongbuk Medical Innovation Foundation (DGMIF), 80 Cheombok-ro, Dong-gu, Daegu 701-310, Korea
2
Department of New Drug Discovery and Development, Chungnam National University, Daejon 305-764, Korea
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(14), 2634; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24142634
Submission received: 25 June 2019
/
Revised: 13 July 2019
/
Accepted: 18 July 2019
/
Published: 19 July 2019
(This article belongs to the Special Issue Advances in the Chemistry of Hypervalent Iodine Compounds)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
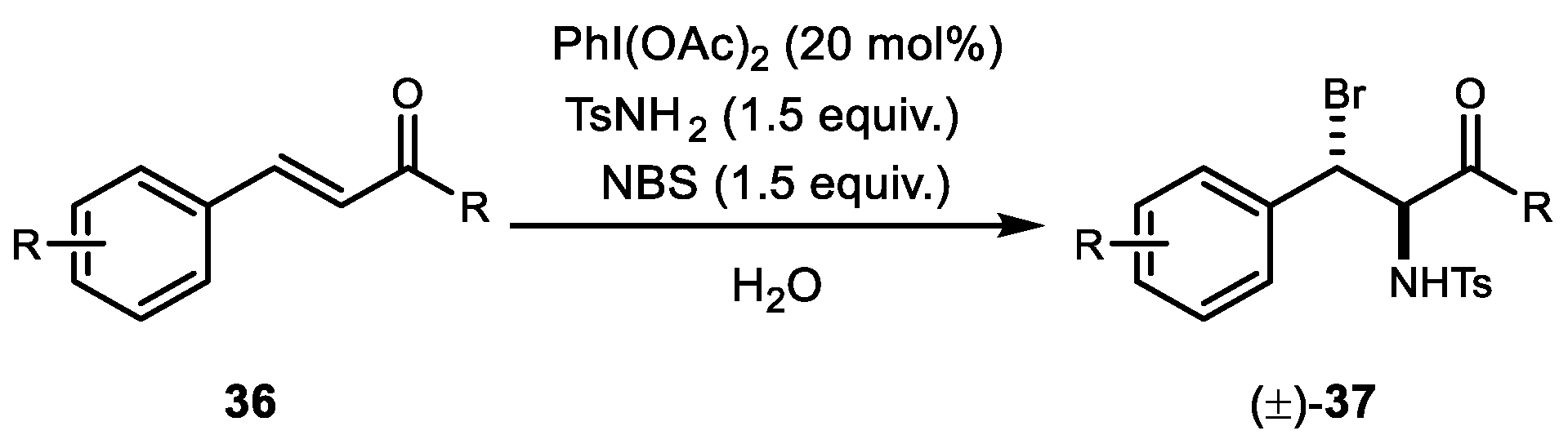{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
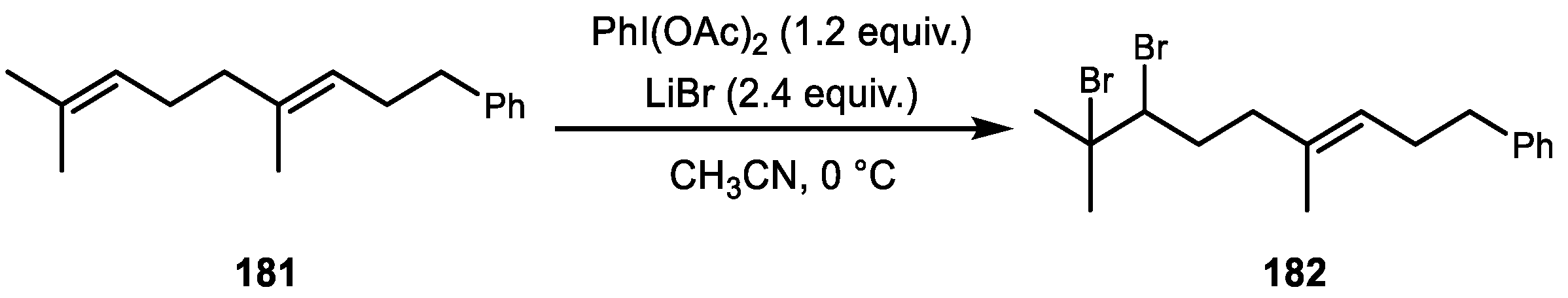{kind=link}
Abstract
:Hypervalent iodine reagents are of considerable relevance in organic chemistry as they can provide a complementary reaction strategy to the use of traditional transition metal chemistry. Over the past two decades, there have been an increasing number of applications including stoichiometric oxidation and catalytic asymmetric variations. This review outlines the main advances in the past 10 years in regard to alkene heterofunctionalization chemistry using achiral and chiral hypervalent iodine reagents and catalysts.
1. Introduction
Starting in the early 2000s, hypervalent iodine chemistry has undergone extensive development and a wide range of organic synthesis applications have been devised owing to their stability in air and to moisture, commercial availability, low toxicity, mild reaction conditions, and ease of handling. Most importantly, however, their complementary oxidizing ability compared to transition metals has been the main reason why they are increasingly being studied and used in synthetic organic chemistry. As an efficient oxidant, hypervalent iodine allows for a wide range of synthetic transformations, namely oxidation of alcohols, α-functionalization of carbonyl compounds, spirocyclization, and difunctionalization of alkenes. Both stoichiometric iodine chemistry and catalytic enantioselective variations were initially investigated in regard to α-functionalization and spirocyclization chemistry, although recent progress has resulted in a more diverse range of applications. For example, due to their oxidizing capacity, the combination with transition metals constitutes a new avenue for prominent carbon-heteroatom bond formation. Hypervalent iodine is a way to gain access to high oxidation state complexes with transition metals. Although they do have some limitations such as the generation of stoichiometric byproducts as well as their limited scope, hypervalent-mediated high-valent transition metal catalysis is nonetheless clearly an emerging area that holds ample promise. Aside from transition metal chemistry, hypervalent iodine itself has the aforementioned properties suitable for application in a diverse range of organic transformations. Consequently, over the past two decades, there have been several general reviews [1,2,3,4,5,6,7,8,9,10], books and book chapters [11,12,13], and specialized reviews [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29] in this regard. In this review, we hence focus on progress and developments that have taken place in the past 10 years in the field of alkene difunctionalization such as diamination, aminofunctionalization, diacetoxylation, oxyfunctionalization, and dihalogenation reactions.
2. Alkene Diamination
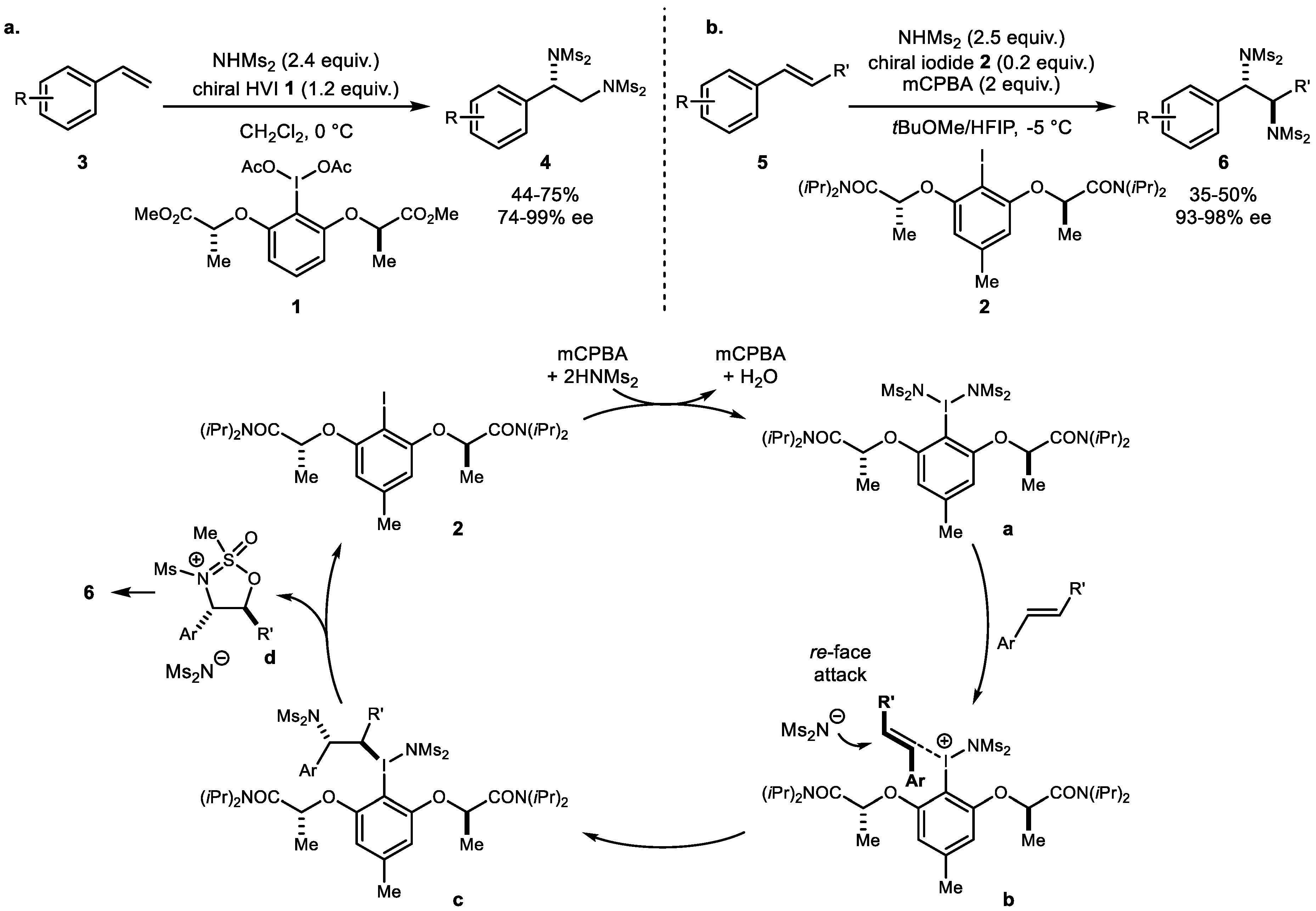
The diamination reaction of alkenes has recently become a useful synthetic approach due to the potential to economically generate a diverse range of vicinal diamines of pharmaceuticals and natural products. In 2011, Muñiz and colleagues described the first enantioselective transfer of two nitrogen atoms using bismesylimide onto the prochiral face of styrene using a chiral iodine (III) reagent 1 (Scheme 1a) [30]. Direct intermolecular diamination of an unactivated alkene is uncommon, especially in the absence of a metal catalyst. In this context, stoichiometric chiral hypervalent iodine exclusively promoted overall oxidation of alkene up to 95% ee (enantiomeric excess). Subsequently, in 2017, Muñiz and colleagues developed the first catalytic asymmetric diamination using 4-methylaryl iodide 2 with two modified chiral lactamide side chains and 3-chloroperbenzoic acid as a terminal oxidant in an MTBE/HFIP (methyl tert-butyl ether/hexafluoroisopropanol) solvent combination (Scheme 1b) [31]. This protocol yielded a wide range of products with excellent enantioselectivity of up to 98% ee. This pioneering work demonstrated catalytic intermolecular enantioselective diamination for the first time and provided insights into the rational design of catalysts within the context of hypervalent catalyzed alkene functionalization.
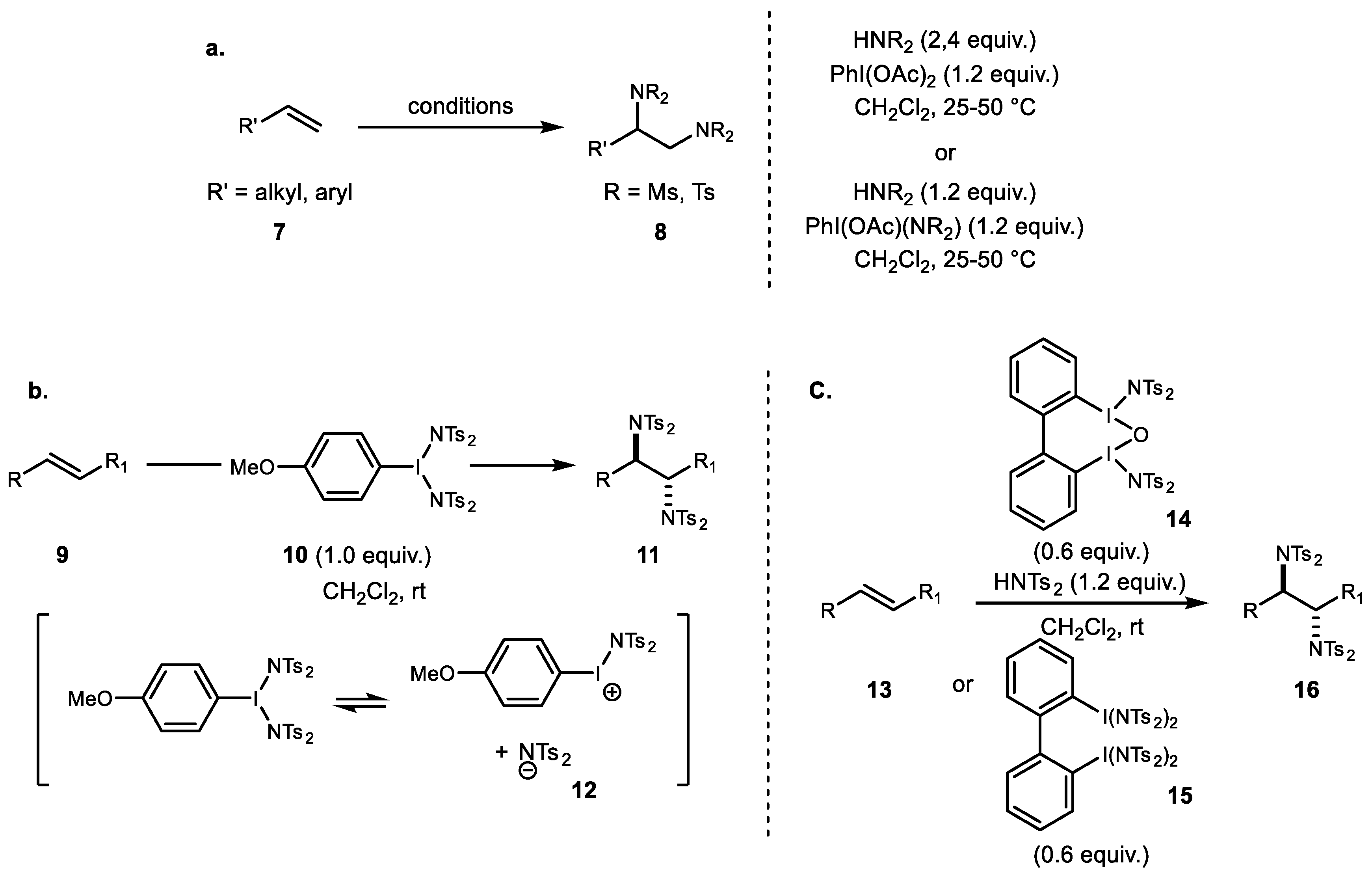
Between 2012 and 2016, more general use of the achiral iodine reagent and bissulfonimide for diamination of alkene was also reported by Muñiz et al. (Scheme 2a) [32]. Over 60 examples comprising a diverse range of terminal and internal alkenes were converted to vicinal diamine under metal-free conditions. The synthesis and the isolation of these new hypervalent iodine PhI[N(SO2R)2]2 entities, as well as their reactivity, were also reported by Muñiz as was the X-ray structure of chiral bisimidoiodine [33]. Additionally, an investigation of electronic effects on the aryl substituent of the hypervalent iodine reagent revealed that the reaction was accelerated by electron donating groups due to stabilization of the cationic iodonium 12 (Scheme 2b) [34]. Another example of the new dinuclear iodine 15 involves intermolecular alkene diamination (Scheme 2c) [35]. This protocol provides a comparable reactivity and a new addition for amination reactions. Although initial attempts using the chiral binaphthyl reagent for enantioselective variation yielded low ee values, this newly designed hypervalent iodine motif nonetheless has significant potential for future development.
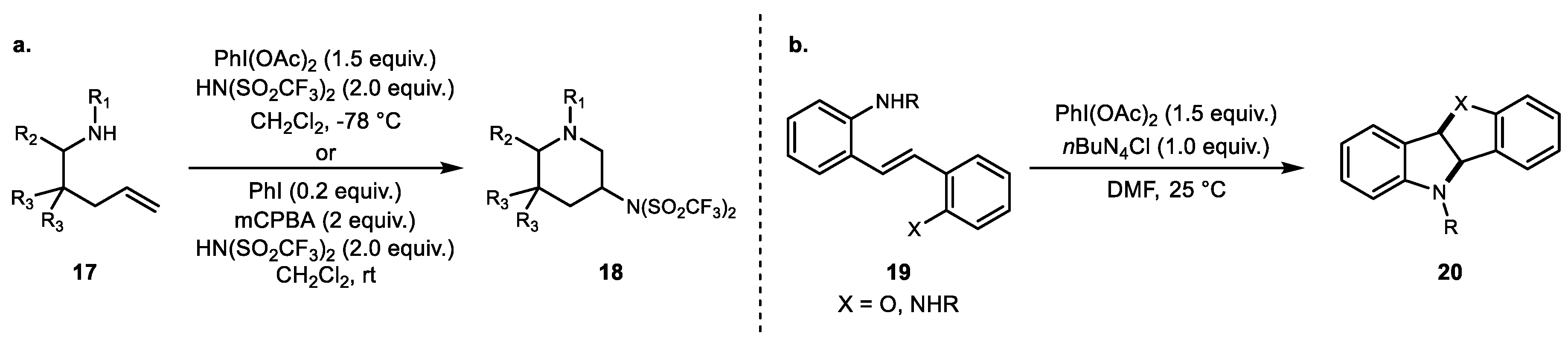
In 2012, Blakey and coworkers developed both stoichiometric and catalytic versions of intramolecular alkene diamination for stereoselective 6-endo-oxidative cyclization (Scheme 3a) [36]. The same reaction condition reported by Muñiz and colleagues yielded stereoselective 6-endo 3-aminopiperidine derivatives when the substrate had a geminal alkyl substitution at the 4-position. Additionally, the homoallylic substituents in the pentene side chain improved the regioselectivity and resulted in good diastereoselectivity. The same year, Chang and colleagues demonstrated not only an elegant diamination reaction but also inorganic additive effects and functional group effects on the nitrogen atom [37]. Aside from Lewis acids and other promoters, halide additives also enhanced the overall efficiency and reactivity of the hypervalent iodine chemistry. These examples demonstrate the potential to achieve alkene heterofunctionalization using only hypervalent iodine compared to previously reported Pd(II)/Pd(IV)-based chemistry using the same substrates [38].
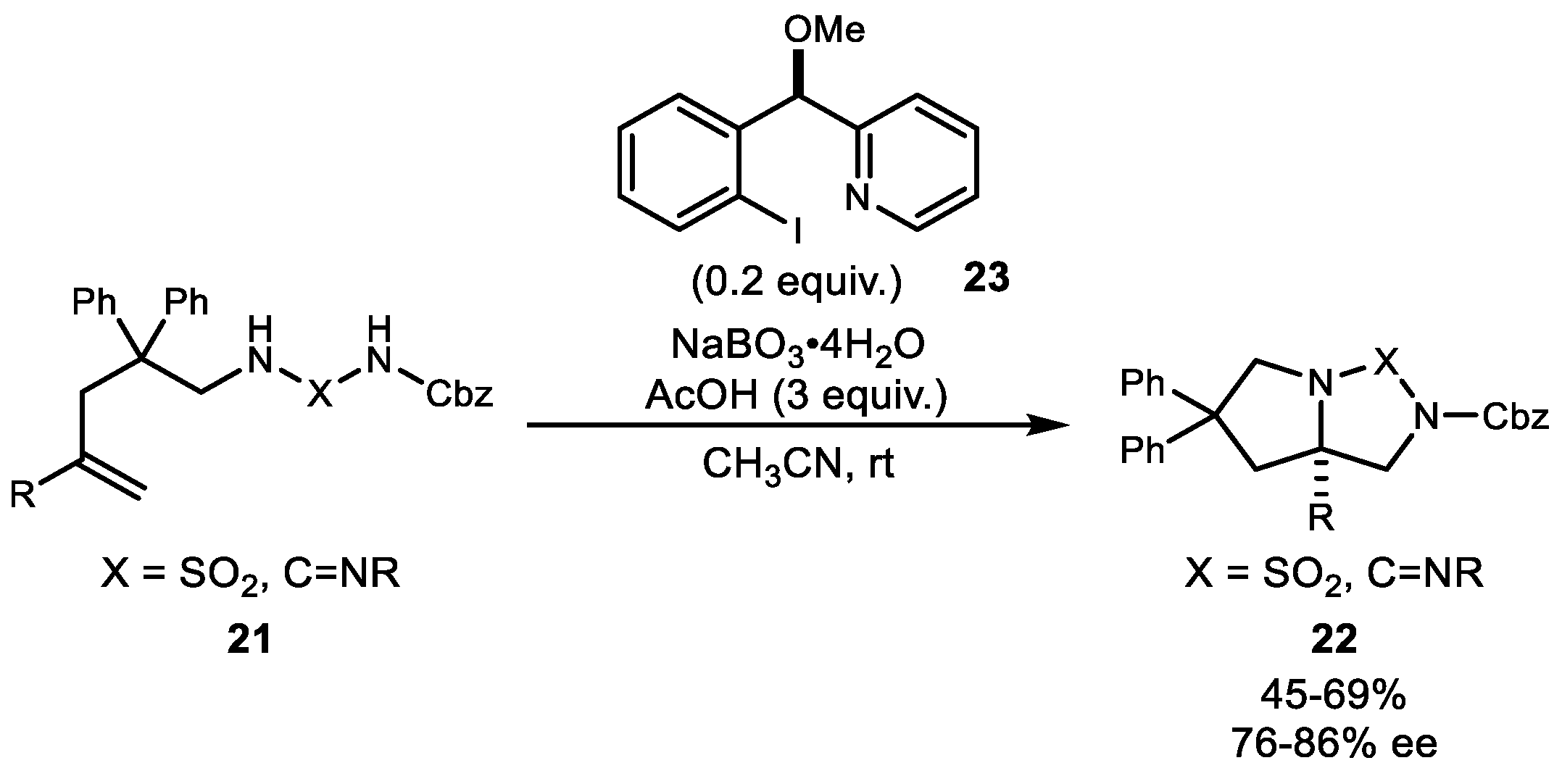
In 2014, Wirth et al. demonstrated the first catalytic intramolecular diamination using various tethered homoallylic guanidine and diaminosulfone derivatives to generate bicyclic molecules (Scheme 4) [39]. This newly designed hypervalent iodine reagent 23 with a pyridine affixed to a chiral benzylic center can allow efficient coordination of the pyridine nitrogen to the iodine center and can yield cyclized adduct up to 94% ee. Subsequent reduction of both the X (SO2 and C=NR) and the Cbz group yielded 1,2-diamine at 86% ee. While only diphenyl-substituted substrates led to the desired products in good yields and with high enantioselectivities, the newly introduced hypervalent iodine ligand 23 can provide new enantioselective variation compared to lactate-based iodine catalysts.
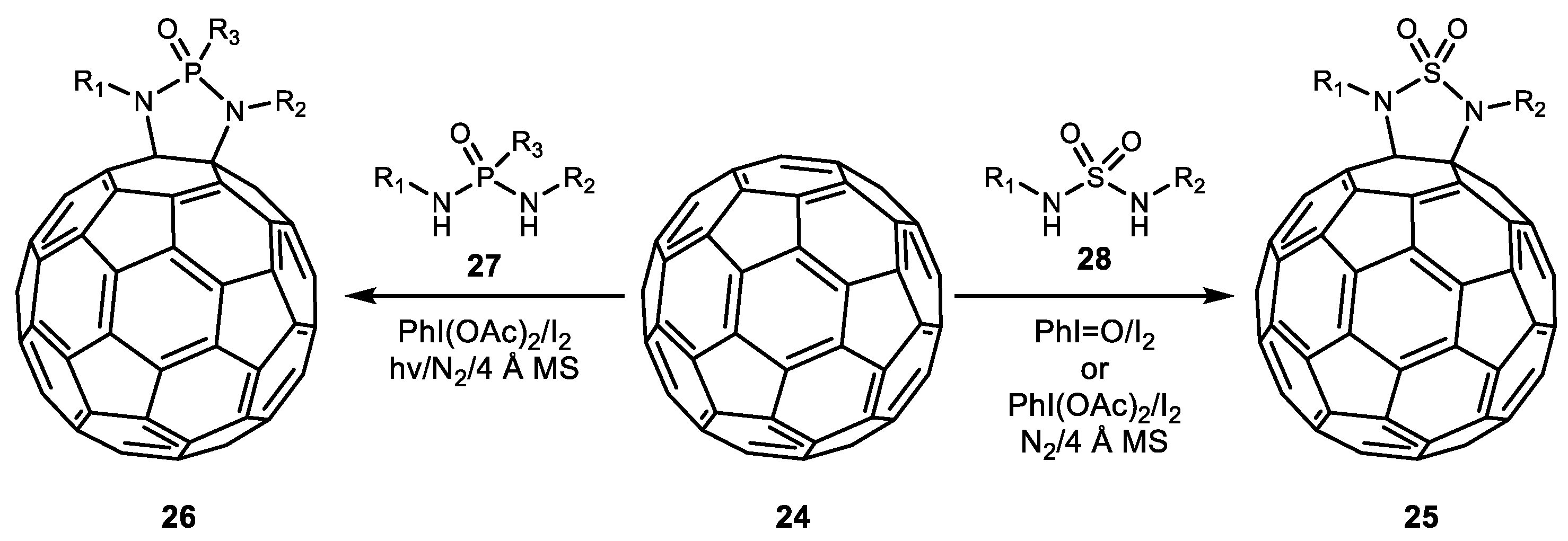
The same year, Miao et al. reported an intermolecular variation of oxidative diamination of olefins with sulfamide 28 and phosphoryl diamide 27 for the synthesis of C60-fused cyclic sulfamide 25 and phosphoryl diamide 26 (Scheme 5) [40]. Depending on the combination of the iodine source, molecular iodine, and the nitrogen source, either a selective diamination or an aziridination reaction was achieved. Notably, phosphoryl diamide was used as a diamine source for the first time.
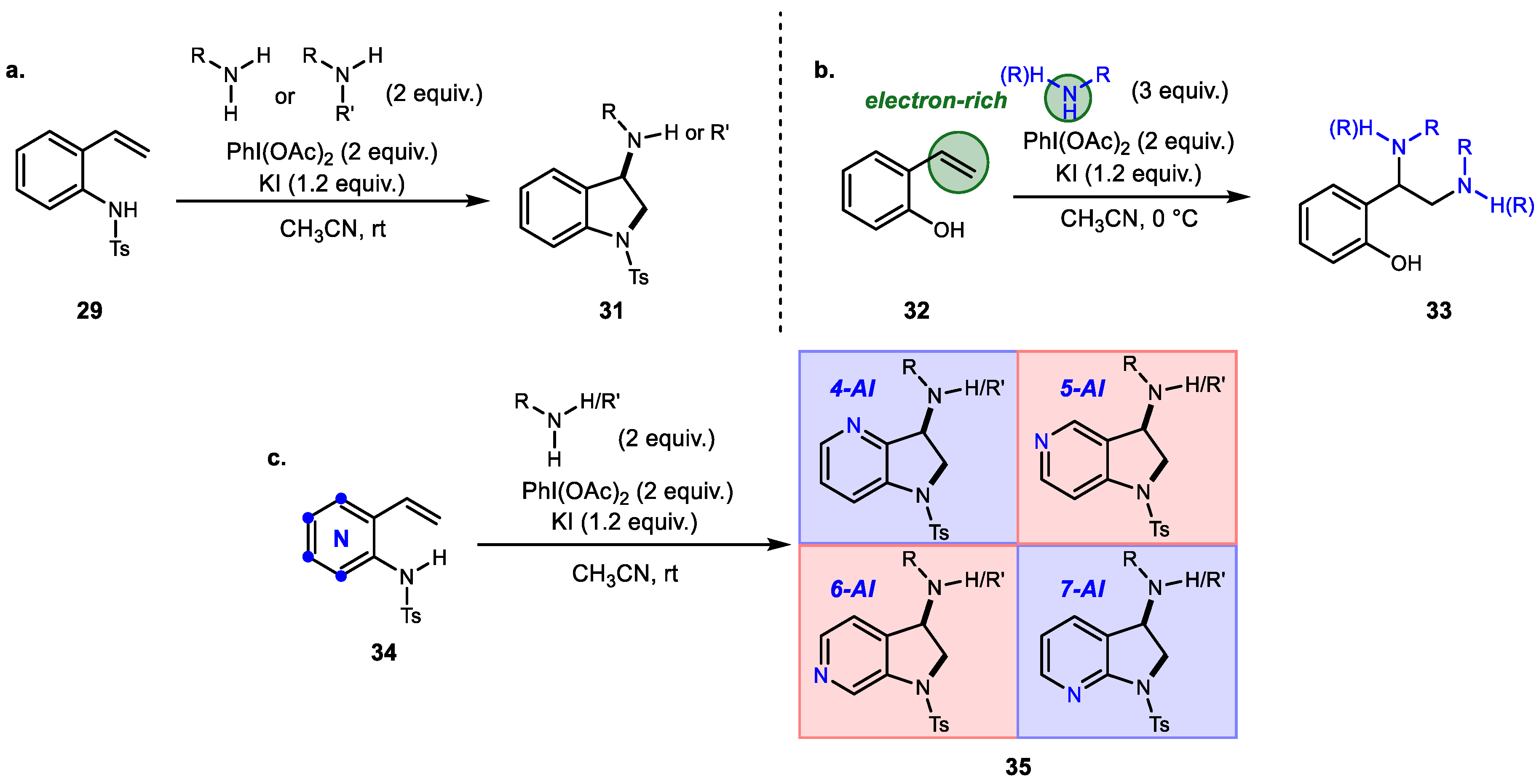
Unlike other diamination protocols that used an electron-deficient nitrogen source, Johnston and Hong developed a series of alkene diaminations using Brønsted-base, electron-rich amines (Scheme 6) [41,42,43]. A combination of stoichiometric hypervalent iodine and inorganic additives can promote inter/intramolecular C-N bond formation without amine preactivation or protection. This protocol can be used to generate 3-aminoindolines and 1,2-diamine derivatives, and it provides a gateway to access all four 3-aminoazaindoline derivatives.
3. Alkene Aminofunctionalization
The first PhI(OAc)2-catalyzed intermolecular amino-bromination of alkene in water with a TsNH2 and NBS combination was reported by Wang et al. in 2009 (Scheme 7) [44]. A diverse range of electron-deficient olefins such as α,β-unsaturated ketones, cinnamates, and cinnamides were tolerated under these reaction conditions. Additionally, this reaction resulted in an unusual rate of acceleration and regioselectivity for styrenes compared to the reaction in organic solvents.
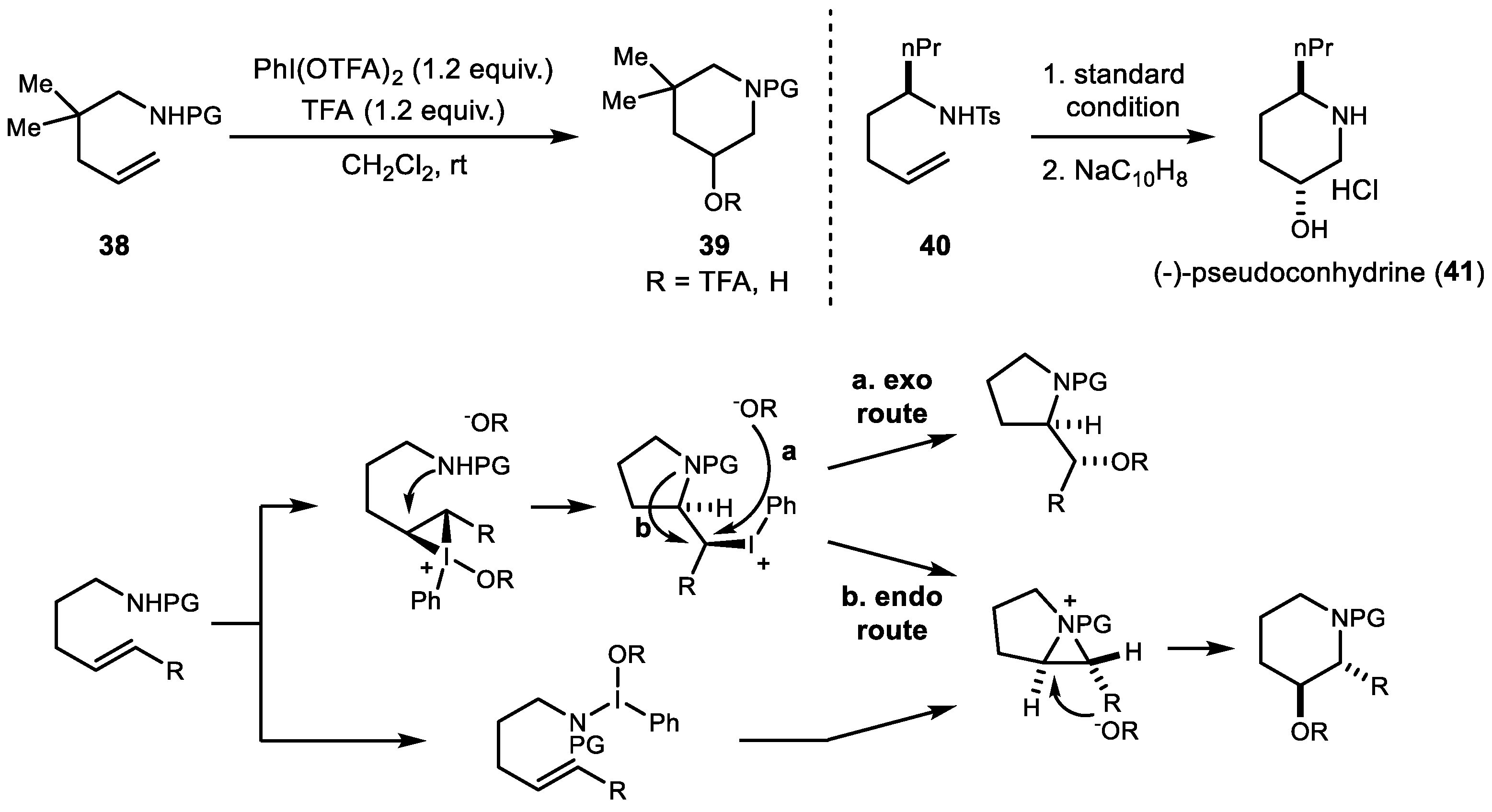
In 2010, Michael et al. demonstrated the first example of a highly endo-selective aminooxygenation (Scheme 8) [45]. Even though the stereochemical outcome depended on the substrates, the piperidine product favored anti-addition in endo-cyclization. This methodology was successfully applied to the synthesis of (−)-pseudoconhydrine 41 in a total of 6 steps.
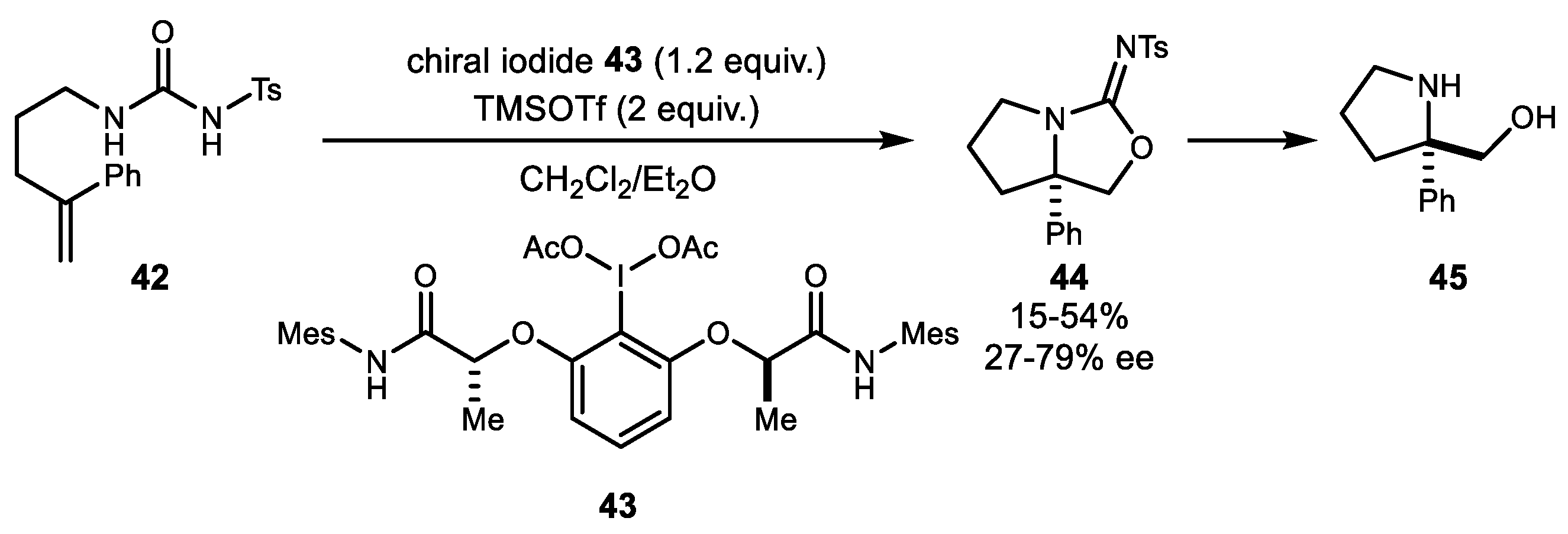
The first example of stereoselective aminooxygenation using a chiral hypervalent iodine reagent was reported by Wirth and colleagues in 2012 (Scheme 9) [46]. A lactate-based amide attached chirally to hypervalent iodine 43 and TMSOTf (Trimethylsilyl trifluoromethanesulfonate) combination at a low temperature provided the highest selectivity for isourea adduct 44 formation. This isourea can readily be converted to the α-substituted proline derivative 45, which has not been synthesized with high enantiopurity.
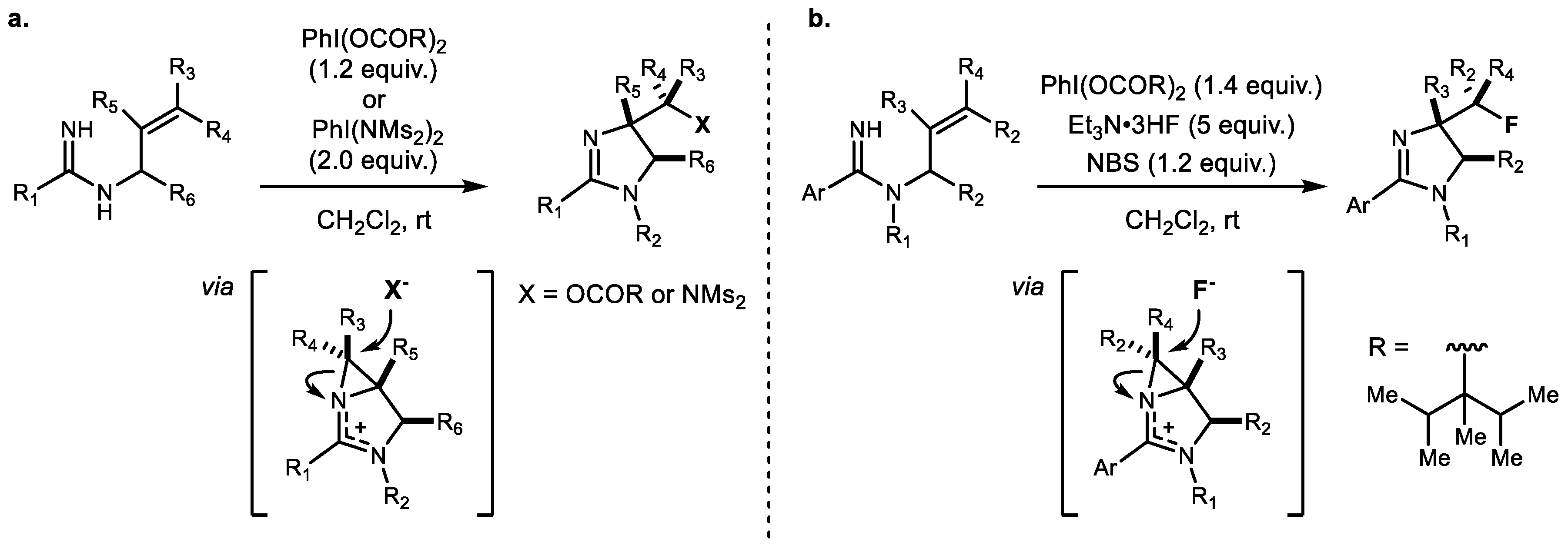
In 2014, Chiba et al. reported a series of diastereoselective aminooxygenations using either PhI(OAc)2 or PhI(OTFA)2 and diamination using defined Muñiz’s bisimido iodine reagent 47 (Scheme 10a) [47]. Compared to previous metal-catalyzed reactions [48,49], this strategy extended the scope to substituted internal alkenes bearing N-allylamidine 46. Subsequently, in 2016, the same group reported anti-selective aminofluorination using bulkier carboxylate (2-isopropyl-2,3-dimethylbutanoate)-attached iodobenzene 50 to minimize oxygenation of the product and excess Et3N·3HF combination (Scheme 10b) [50]. Furthermore, these 4-fluoro-2-alkyl imidazolines 51 could yield functionalized 3-fluoropropane-1,2-diamine derivatives after a reductive ring-opening step. Both procedures involve a stepwise sequence comprising syn-aziridination and nucleophilic ring opening of an aziridinium intermediate, with various nucleophile sources such as oxygen, nitrogen, and fluorine sources.
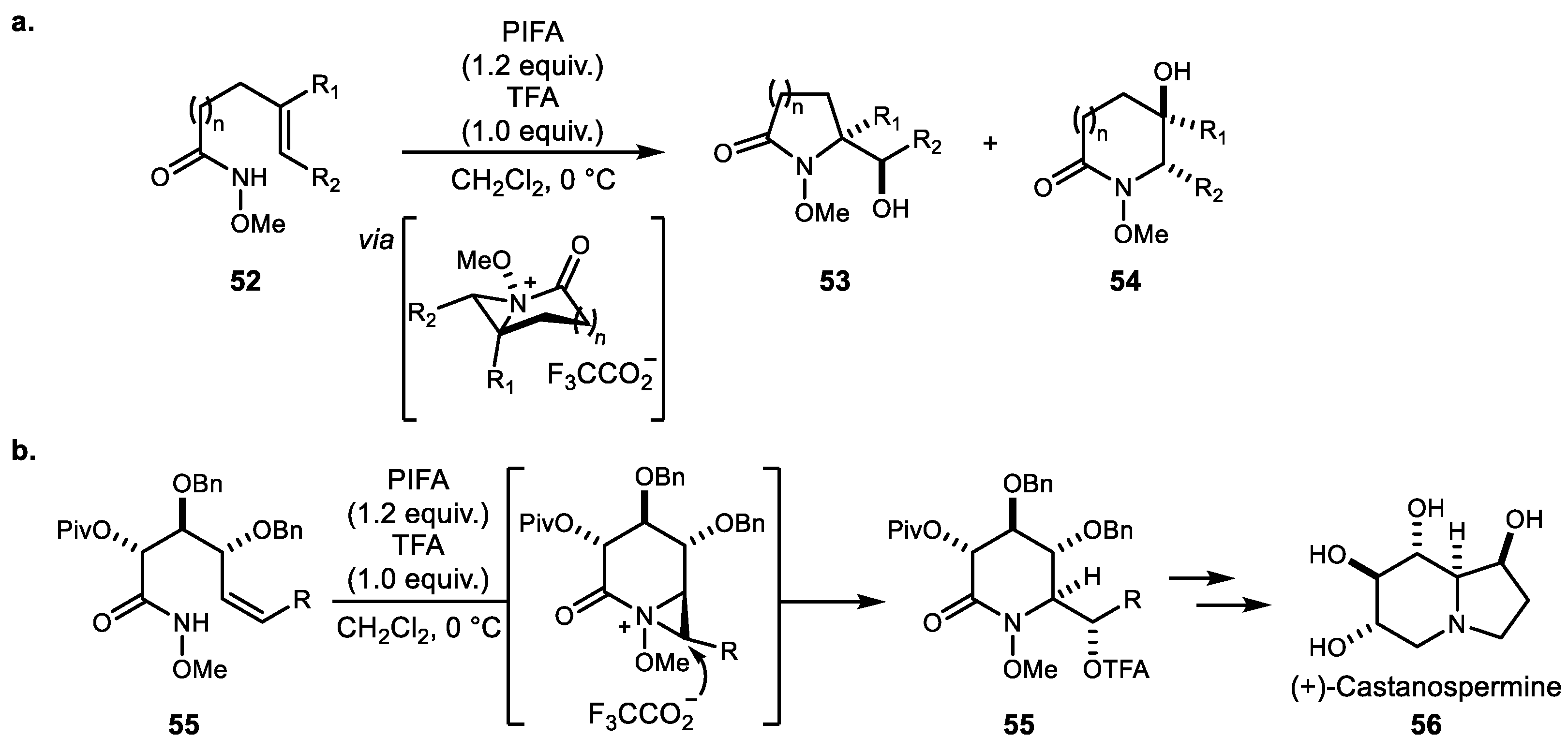
In 2010, Wardrop et al. demonstrated PIFA/TFA-mediated (phenyliodine bis(trifluoroacetate)/trifluoroacetic acid) intramolecular aminooxygenation using nitrenium generated from O-alkyl hydroxamate 52 to generate bicyclic five- to eight-membered lactams (Scheme 11a) [51]. With this protocol, they noted further rate acceleration and overall efficiency in the presence of 1 equivalent of trifluoroacetic acid. Additionally, this nitrenium-mediated aminooxygenation was successfully applied to the synthesis of a series of polyhydroxylated indolizidine alkaloids 56 (Scheme 11b) [52,53]. The same protocol was recently used to synthesize madangamine D and other morphan-based natural products [54].
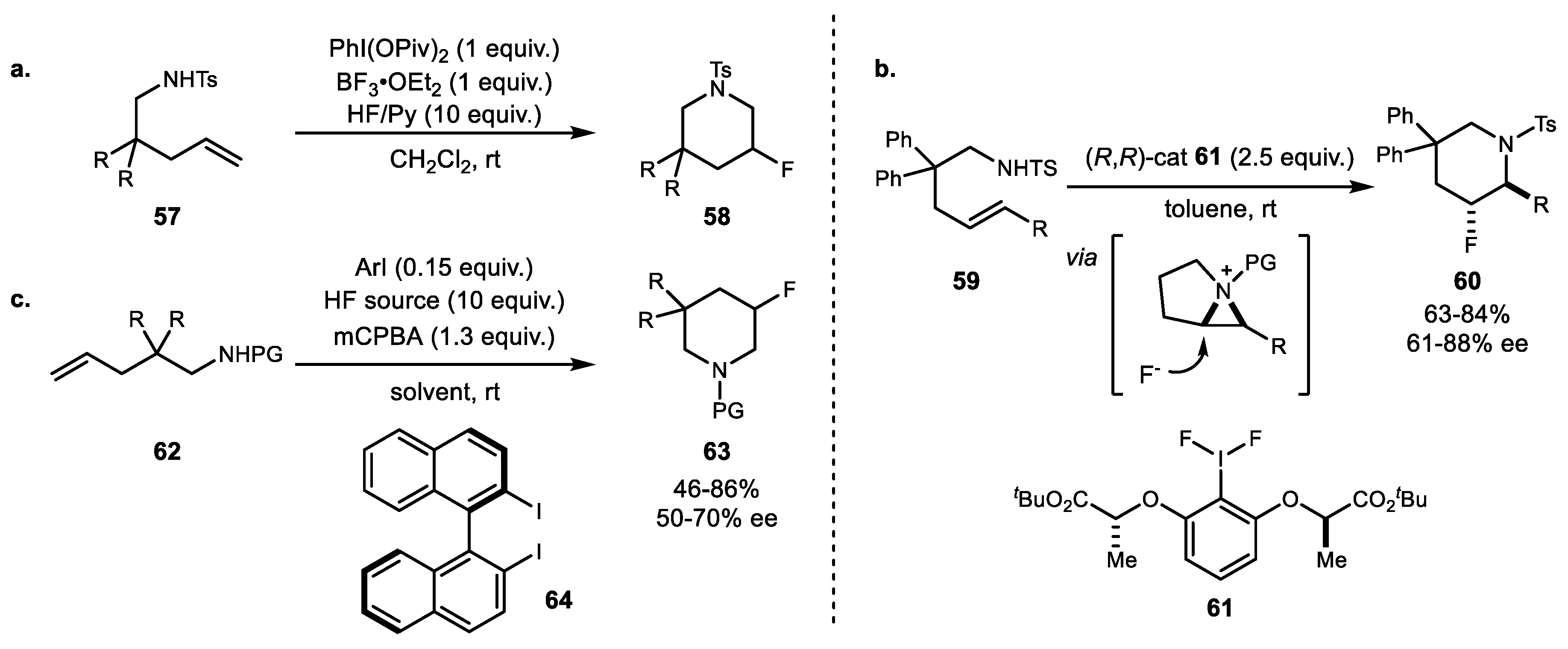
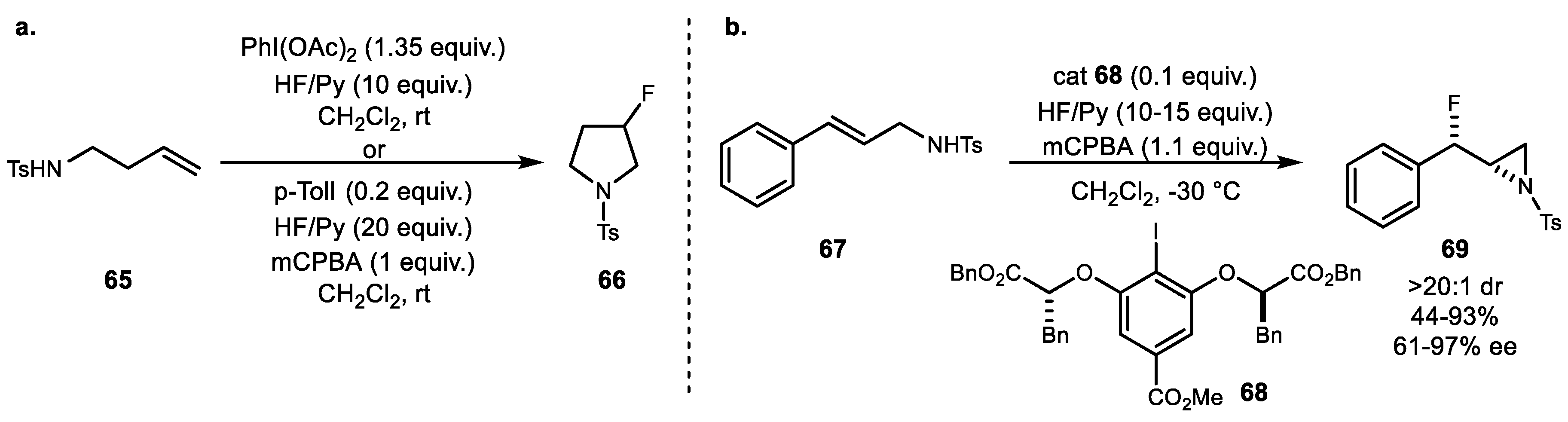
A series of metal-free intramolecular aminofluorination of alkenes was reported in 2012. The first example of stoichiometric hypervalent iodine reagent variation was reported by Meng and Li in the presence of HF·Py and BF3·OEt2 as a promoter (Scheme 12a) (Scheme 12a) [55]. The reaction resulted in endo-selective formation of 3-fluoropiperidine derivatives 58 in one step under mild conditions. Subsequently, an asymmetric version of this protocol was reported by Nevado and coworkers using tert-butyl lactate-based iododifluoride reagent 61. This reaction also yielded highly selective 6-endo-cyclization product 60 (Scheme 12b) [56]. Another catalytic example was developed by Kita and Shibata using axially chiral bisiodine reagent 64 in the presence of mCPBA (3-Chloroperoxybenzoic acid, meta-Chloroperbenzoic acid) (Scheme 12c) [57]. However, this protocol exhibited a moderate degree of enantioselectivity compared to lactate-based catalysts.
Aside from six-membered ring formation, Zhang and coworkers disclosed the first metal-free intramolecular aminofluorination of homoallylamine 65 for the synthesis of 3-fluoropyrrolidine 66 with PhIO oxidant and BF3·OEt2 as the fluorine source [58]. A more practical method was reported by Kitamura and coworkers [59]. They developed both stoichiometric methods using a PIDA/HF (phenyliodine(III) diacetate/hydrogen fluoride) combination and a catalytic method using the p-iodotoluene/mCPBA combination for the synthesis of 3-fluoropyrrolidines 66 (Scheme 13a). Recently, Jacobsen et al. reported the synthesis of syn-β-fluoroaziridine via intramolecular aminofluorination of allylic amine 67 using dibenzyl lactate-based chiral hypervalent iodine 68. Both a high yield and excellent enantioselectivity were observed, and this fluoroaziridine 69 was treated with a diverse range of nucleophiles to generate unique chiral building blocks (Scheme 13b) [60].
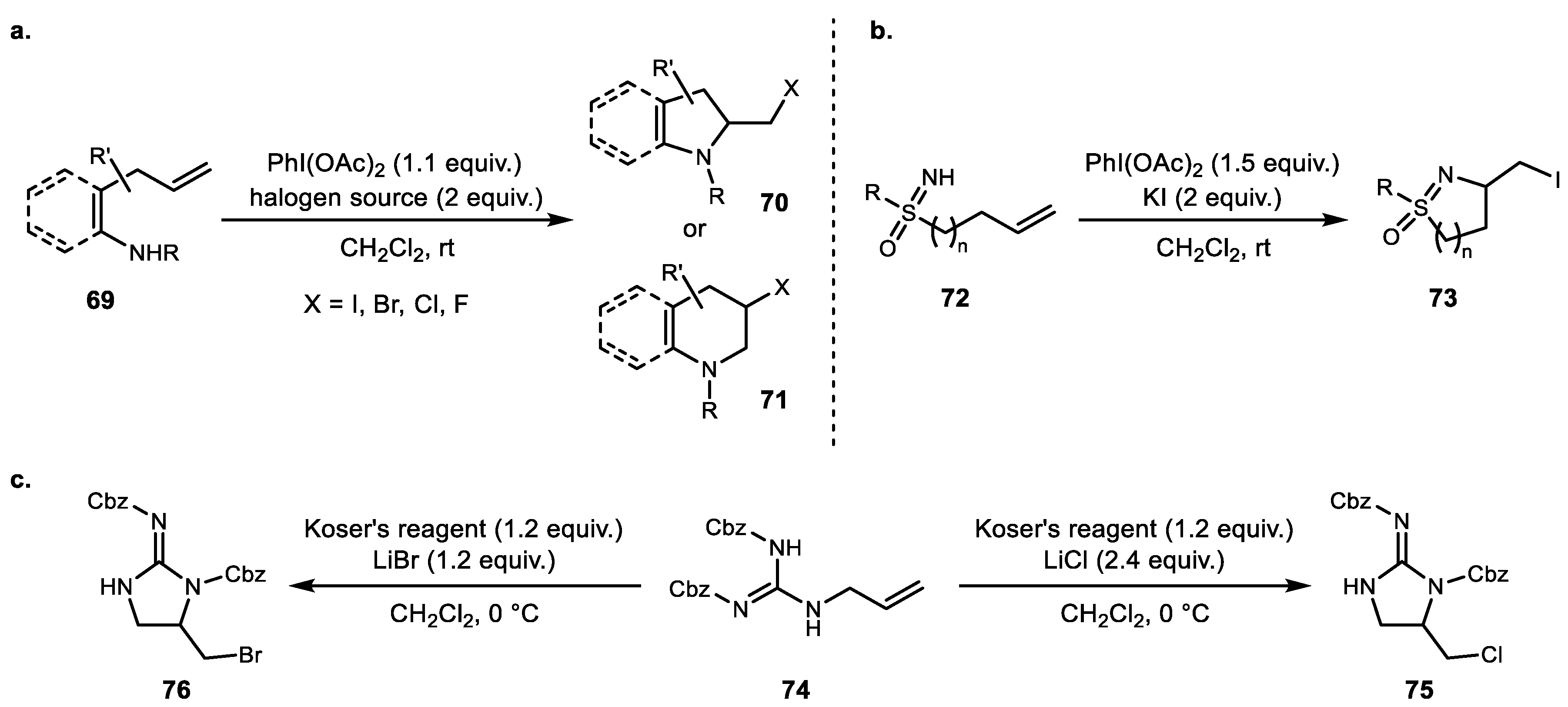
Aside from fluorine addition to alkene, other halogens have also been added using hypervalent iodine chemistry. In 2014, Li and Liu demonstrated PIDA-mediated regioselective aminohalogenation using all four halogen sources [61]. A diverse range of carbo- and heterocycles such as pyrrolidine, piperidine, indoline, and dihydrofuran were synthesized after forming new C–N and C–X bonds. Another interesting iodocyclization of sulfoximine was reported by Bolm in 2016 [62]. The use of PIDA/KI combination, a diverse range of sulfoximines 72 resulted in five- and six-membered isothiazole and thiazine derivatives. Dodd and Carious also developed hypervalent iodine-mediated umpolung halocyclization using halide salts [63]. Regioselective chloro- and bromocyclization were achieved using Koser’s reagent and the corresponding halide sources (Scheme 14).
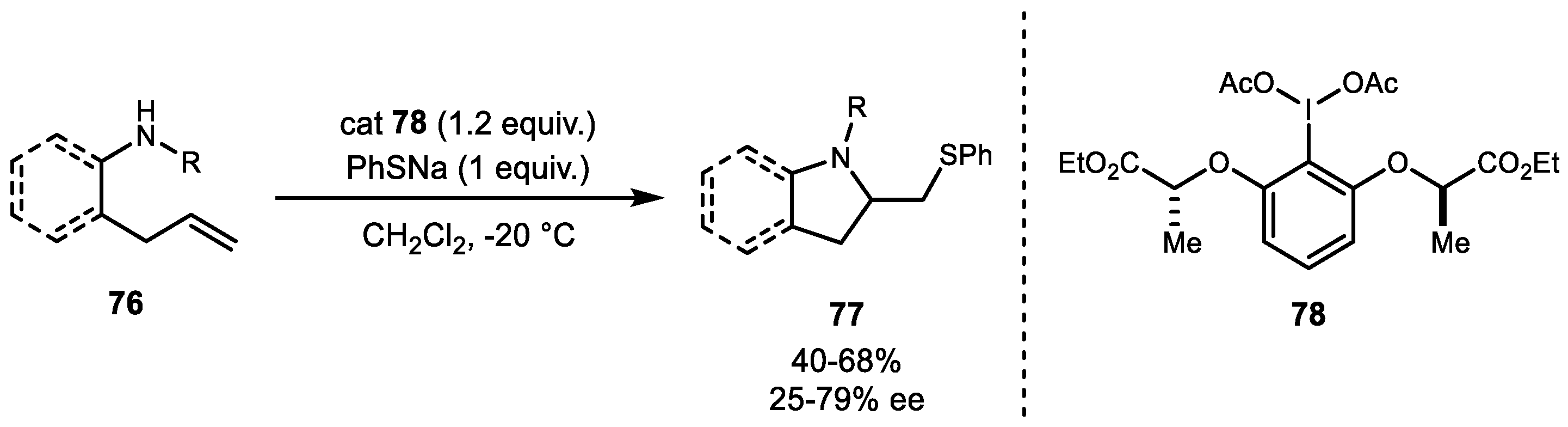
The final heteroatom example of aminofunctionalization of alkene is the thioamination reaction that was reported by Wirth and colleagues in 2016 [64]. Using PIFA and a sulfur nucleophile, direct thioamination resulted in various pyrrolidine and indoline ring systems 77. Additionally, lactate-based chiral hypervalent iodine 78 resulted in stereoselective formation of 1,2-aminothiols with up to 79% ee (Scheme 15).
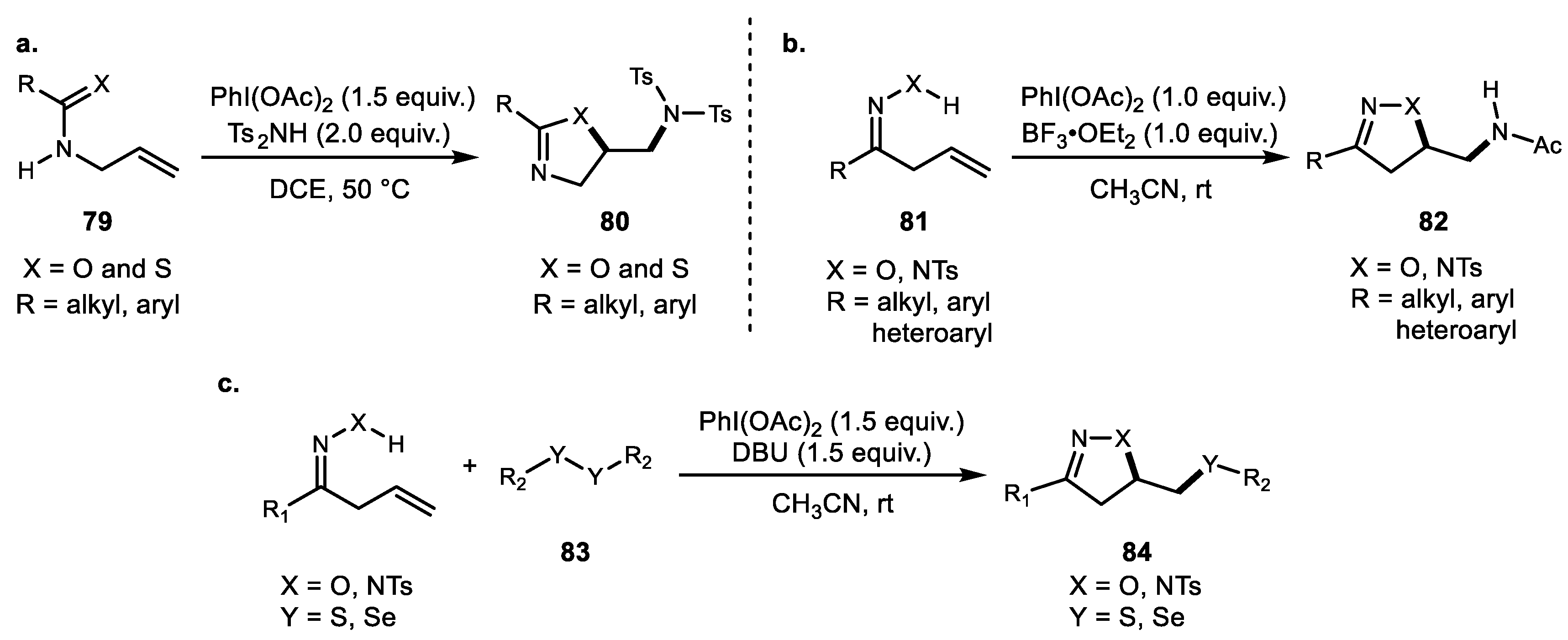
Other interesting heterocycle syntheses using alkene difunctionalization have recently been demonstrated. Using Muñiz’s protocol, Hong and coworkers reported oxazoline and thiazoline synthesis via alkene heterofunctionalization (Scheme 16a) [65]. Additionally, their systematic evaluation of amine functional groups provided evidence that the (benzene)sulfonyl group is the key moiety for the amination reaction. Another interesting Ritter-type amidation reaction was also reported by Hong and coworkers (Scheme 16b) [66]. The PIDA/BF3·OEt2 combination in CH3CN provided the unusual Ritter amidation adduct 82 after acetonitrile addition. Other heteroatoms such as sulfur and selenium were also incorporated with alkene in the presence of hypervalent iodine. Cai et al. developed efficient sulfeno- and selenofunctionalization of alkene to generate pyrazoline and isoxazoline in the presence of base (Scheme 16c) [67].
4. Alkene Diacetoxylation
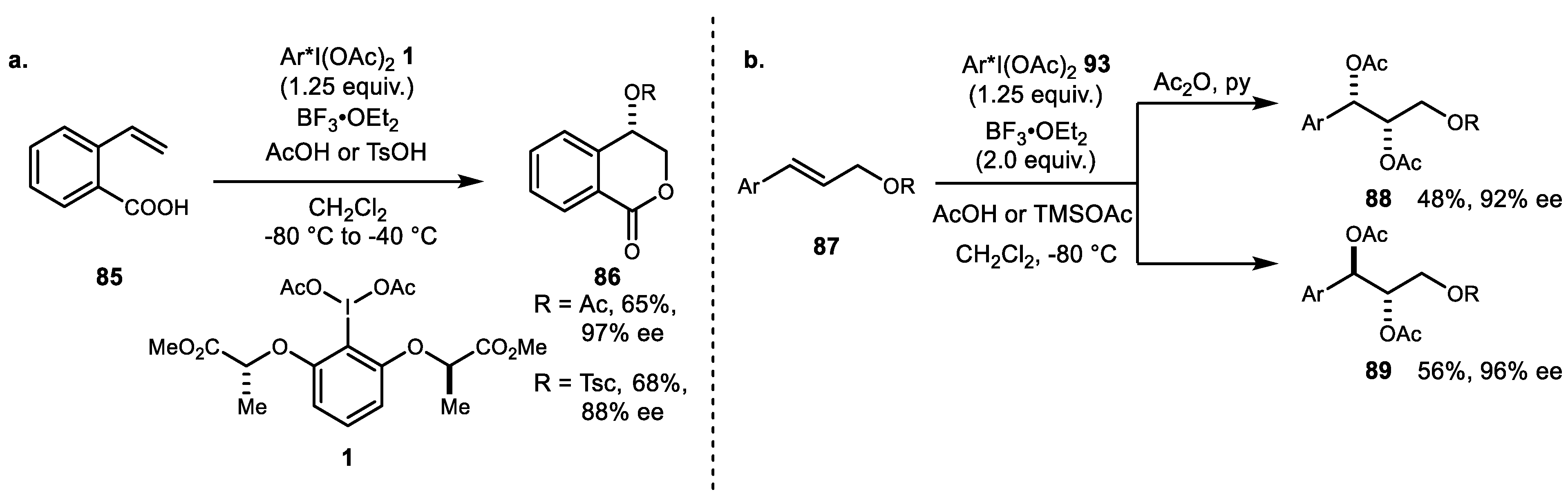
In terms of the dioxygenation reactions of alkene, recent advances have been mainly in regard to the reaction mechanism and synthetic applications with natural products, as reported by Fujita and coworkers in 2010. Initial screening of chiral lactate-based hypervalent iodine reagent 1 was reported in 2010 [68], and it resulted in both high enantioselectivities and endo-selective cyclization of 2-ethenylbezoic acid 85 into δ-lactone 86 (Scheme 17a). This protocol has been successfully applied to the enantioselective Prévost and Woodward reaction (Scheme 17b) [69]. The observed enantioselectivity switchover depended on the nucleophile and reaction temperature. The syn-product with retention of product was favored in the presence of water at low temperatures, while the anti-product was generated preferentially in the presence of acetic acid (or acetate).
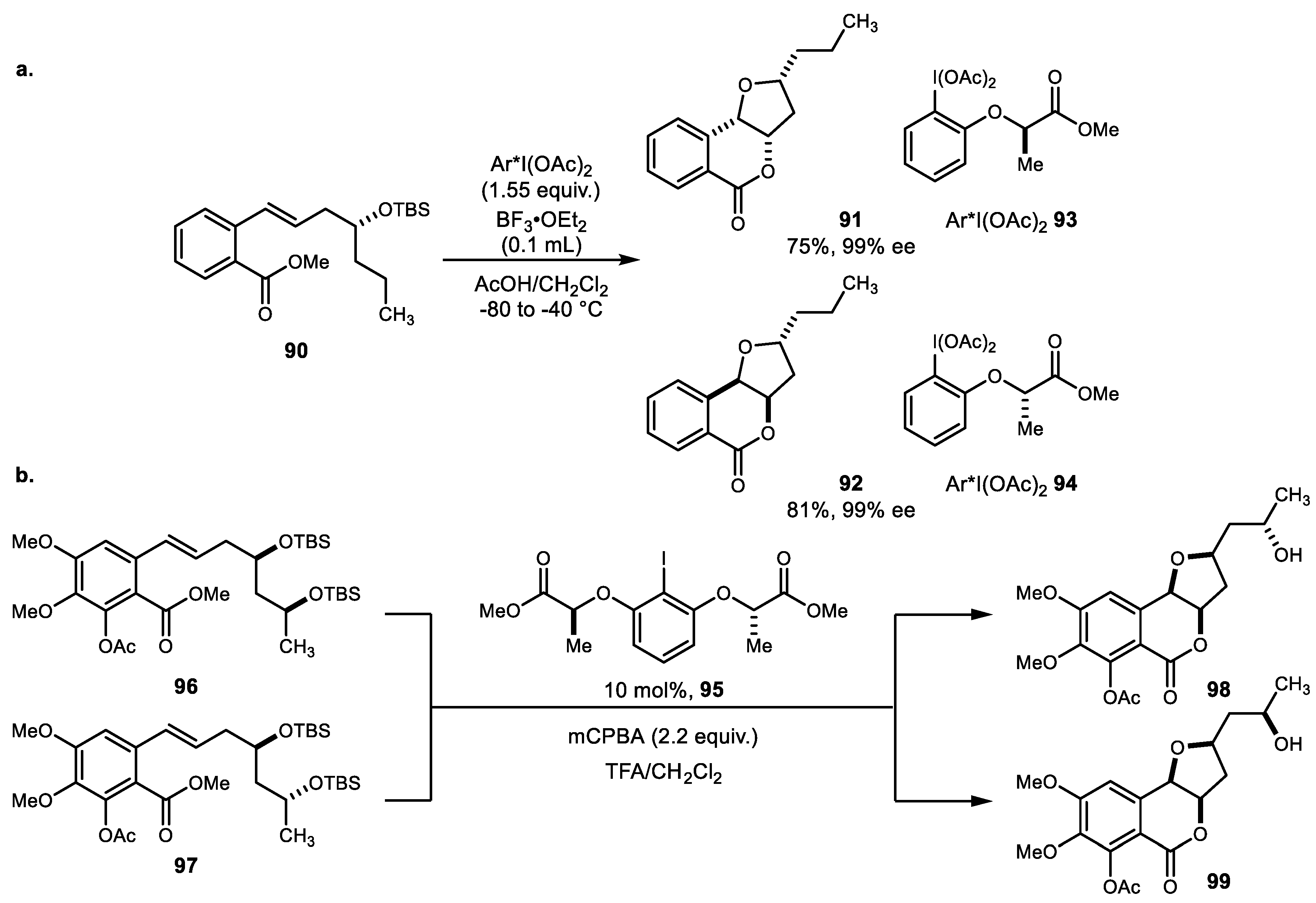
The same group also reported a series of 4-oxyisochroman-1-one polyketide natural products. A series of substrates was evaluated for selectivity. In case of silyloxy substrate 90, diastereoface selectivity was controlled by the chirality of the hypervalent iodine (Scheme 18a) [70]. The (R)-reagent 93 favored the Si-face attack to produce cyclized adduct 91, while the (S)-reagent 94 favored the Re-face. Catalytic variation of same reaction using catalyst 95 was subsequently developed using mCPBA as a stoichiometric oxidant and TFA as an activator. Both stoichiometric use of chiral hypervalent iodine and a catalytic version were successfully applied for the first total synthesis of (12S)-12-hydroxymonocerin and (12R)-12-hydroxymonocerin (Scheme 18b) [71]. This overall selectivity based on the lactate-side chain on the chiral iodoarene and in the catalytic variation study was also disclosed [72].
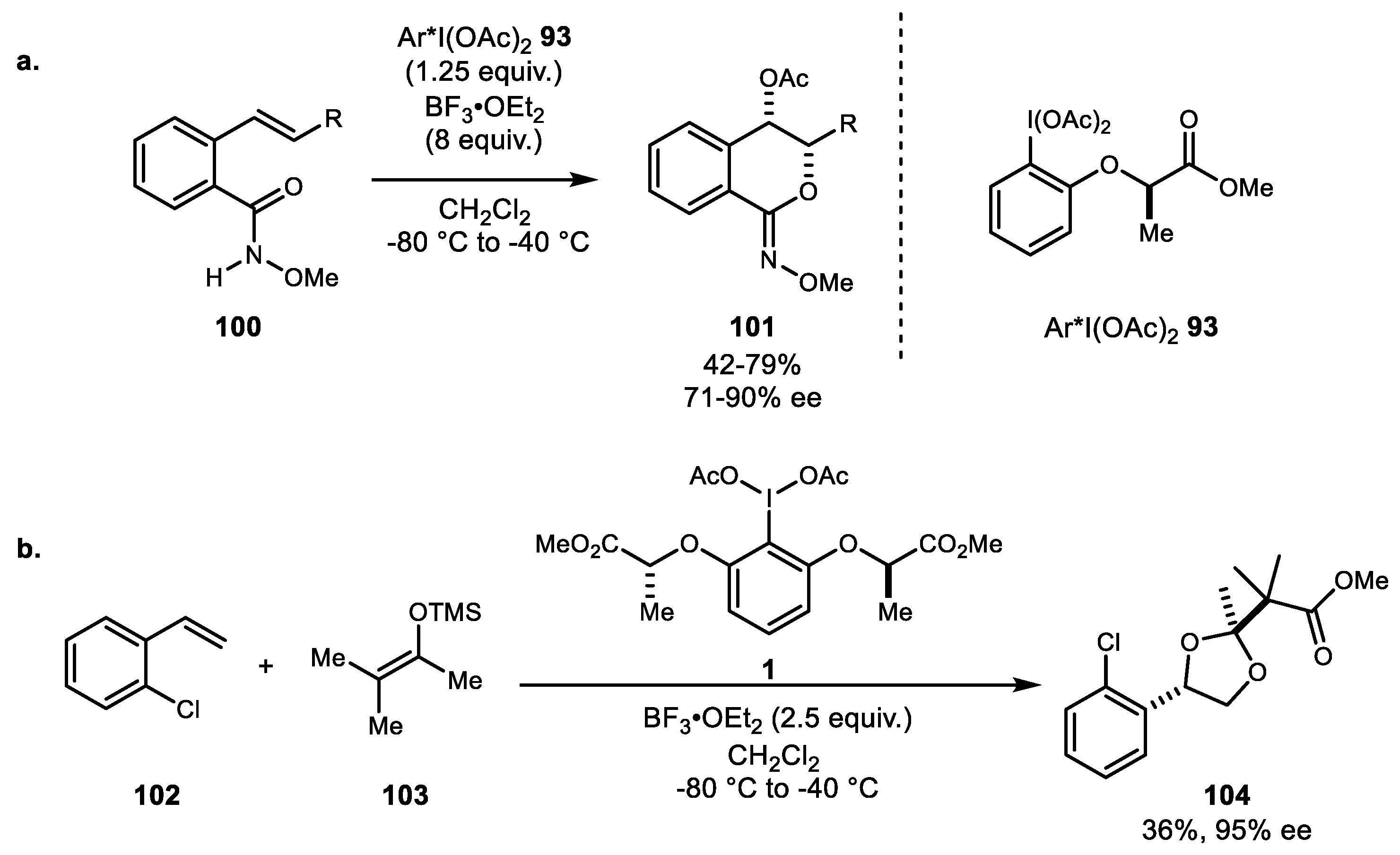
The development of a sequential oxidation reaction of alkenylbenzamide was applied for the synthesis of 4-hydroxymellein and related natural products (Scheme 19a) [73]. Oxidative cyclization of alkenylbenzamide 100 using chiral hypervalent iodine 93 yielded isochroman-1-imine, and palladium-catalyzed oxidative acetoxylation was subsequently shown to produce 4-hydroxymellein. Another interesting heterocycle family was obtained by alkene difunctionalization using a hypervalent iodine reagent. Using the external bidentate nucleophile 103, the intermediate dioxolanyl cation converted substituted dioxolane as a single diastereomer (Scheme 19b) [74]. Various carboxylic acids and other carbon nucleophiles were tested, and enantioselective variation was also successfully demonstrated.
Moran and colleagues continued to develop iodoarene-mediated and catalyzed oxidative cyclization reactions. In terms of alkene difunctionalization reactions, in 2015, Moran and coworkers reported cyclization of N-alkenylamide 105 to generate a diverse range of 5- to 7-membered ring systems 106 using catalytic amounts iodoarene and Selectfluor as the co-oxidant (Scheme 20a) [75]. Harned et al. had previously reported a similar reaction using a stoichiometric amount of the PIDA/ BF3·OEt2 combination [76]. However, their method only resulted in 5-membered ring systems with terminal alkene substrates. In Moran’s case, in addition to terminal alkenes, disubstituted alkenes could also readily be oxidized to the corresponding oxazoline derivatives 106. Additionally, a preliminary screen of the chiral iodoarene 109 catalyzed-reaction yielded γ-butyrolactone 108 with moderate enantioselectivity (Scheme 20b).
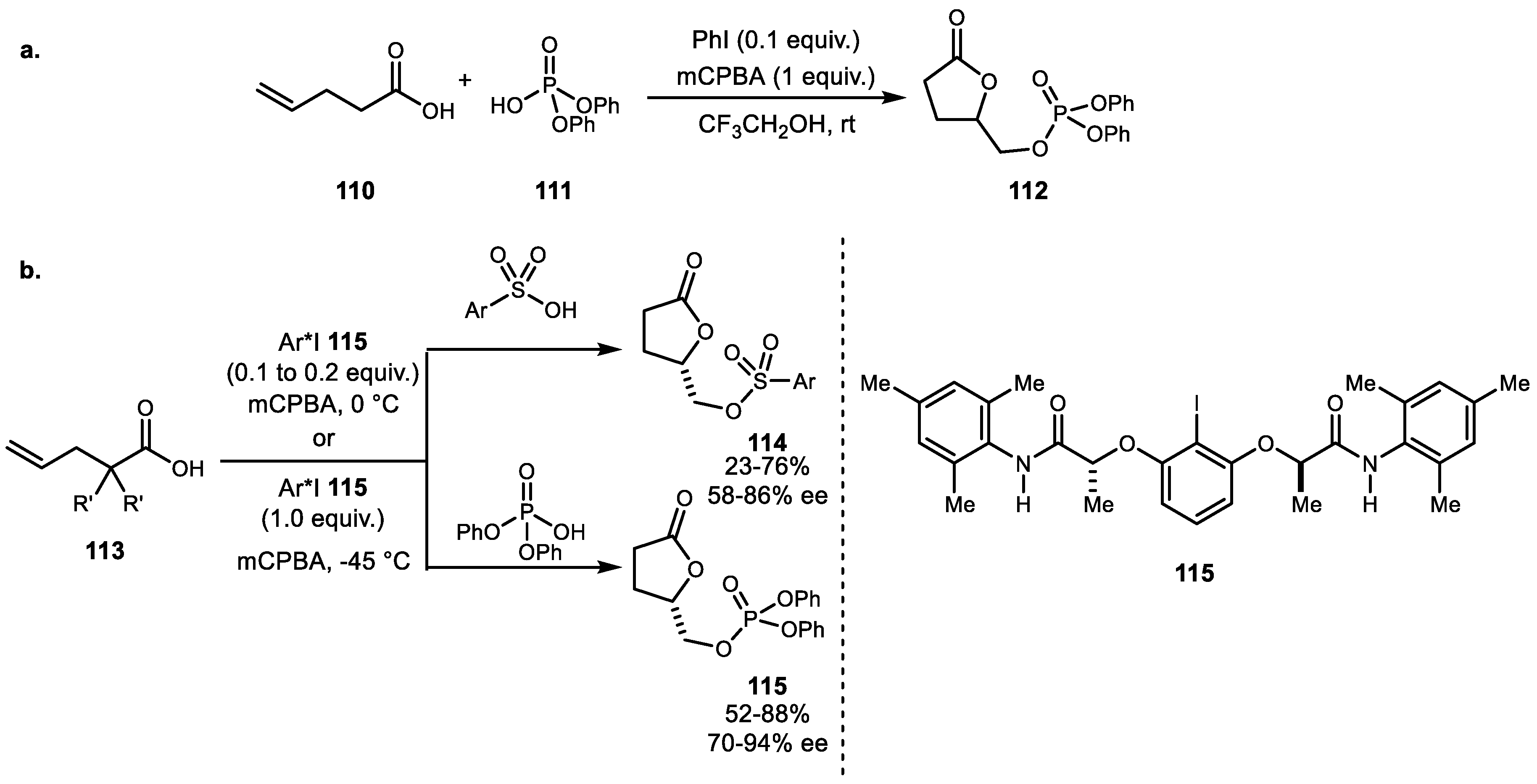
The first example of phosphoryloxylactonization was reported by Koser et al. in 1988 [77]. However, the first catalytic variation was reported by Zhou et al. in 2010 (Scheme 21a) [78]. A combination of a catalytic amount of PhI and mCPBA promoted alkenoic acid 110 cyclization with phosphate to yield lactone 112. Recently, Masson et al. developed the first enantioselective sufonyloxy- and phosphoryloxy-oxylactonization reaction using a catalytic lactamide-based chiral iodine and mCPBA combination (Scheme 21b) [79]. A diverse range of substrates 113 were subjected to either sulfonic acid or phosphoric acid nucleophile, resulting in cyclized adducts 114 and 115 with moderate to good enantioselectivity.
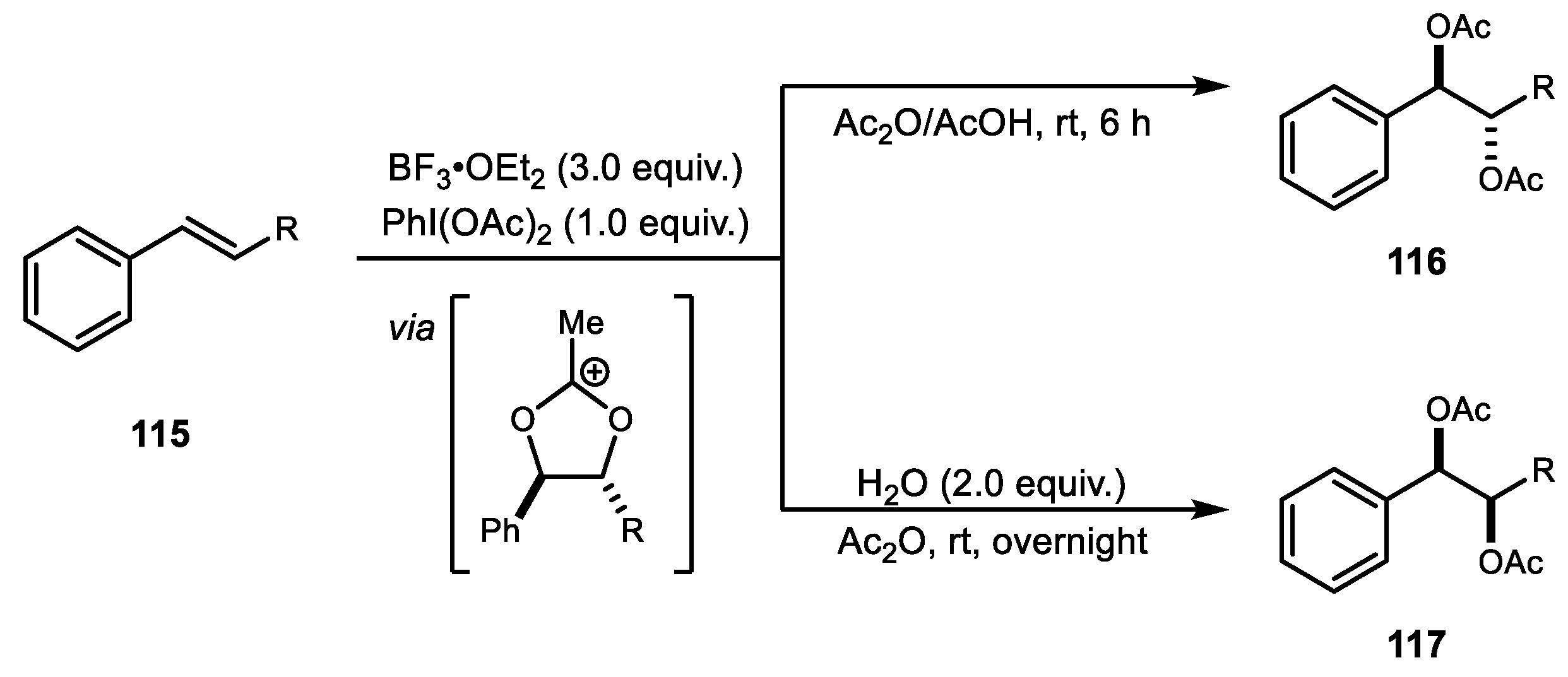
Another practical diastereoselective diacetoxylation of alkenes using PhI(OAc)2 was demonstrated by Li and Meng (Scheme 22) [80]. Using a PIDA/BF3·OEt2 combination, anti-selective diacetoxylation was achieved in Ac2O/AcOH. By contrast, syn-selective diacetoxylation was obtained in the presence of water in the same system. Various internal and terminal alkenes could be oxidized to generate the diacetate adducts. Notably, electron deficient alkenes were also tolerated under these reaction conditions and the efficiency of the reaction was maintained in a multi-gram scale reaction. Based on the Woodward–Prevost reaction mechanism [69], the BF3·OEt2/PIDA system activated alkene and formed the acetoxonium intermediate A. The syn-product was obtained after hydrolysis of acetoxonium A in the presence of water, while the anti-product was produced by ring opening of acetoxonium A. The same group subsequently reported syn-selective diacetoxylation of various alkenes using catalytic amounts of 4-MeC6H4I and BF3·OEt2/mCPBA in combination [81].
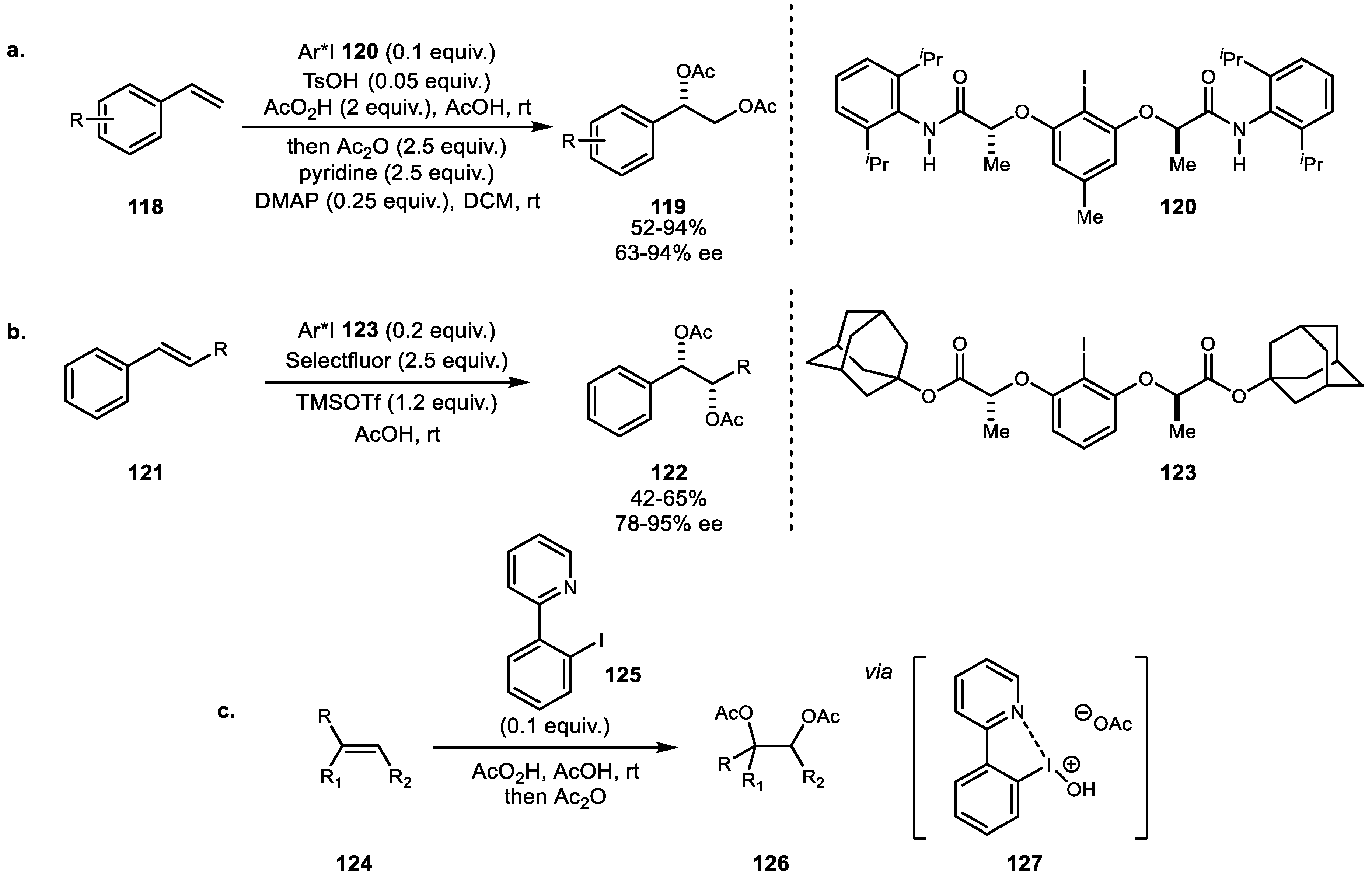
In 2016, Muñiz and colleagues developed a series of intermolecular diacetoxylation reactions. Muñiz and Ishihara elucidated the defined structural information of lactic amide-based chiral hypervalent iodine catalyst. Even though spatial information had been proposed previously [61], this work elucidated the structural information of the chiral iodine’s supramolecular helical chirality from the intramolecular hydrogen bonding network using X-ray analysis. Additionally, this work provided the first catalytic enantioselective diacetoxylation reaction (Scheme 23a) [82]. Along with the success of this new catalyst system, ongoing development of lactate-based chiral iodine chemistry was also developed by Muñiz et al. [83]. The newly synthesized adamantly substituted catalyst provided efficient diacetoxylation product up to 80% ee using Selectfluor as a promoter (Scheme 23b). Finally, a newly designed pyridine-coordinated iodine system was introduced by the same group (Scheme 23c) [84]. Pyridine moieties are commonly used for stabilization of the electrophilic iodine center. In this report, the catalyst structure revealed the coordination of the pyridine group to the iodine via a Lewis base-assisted activation.
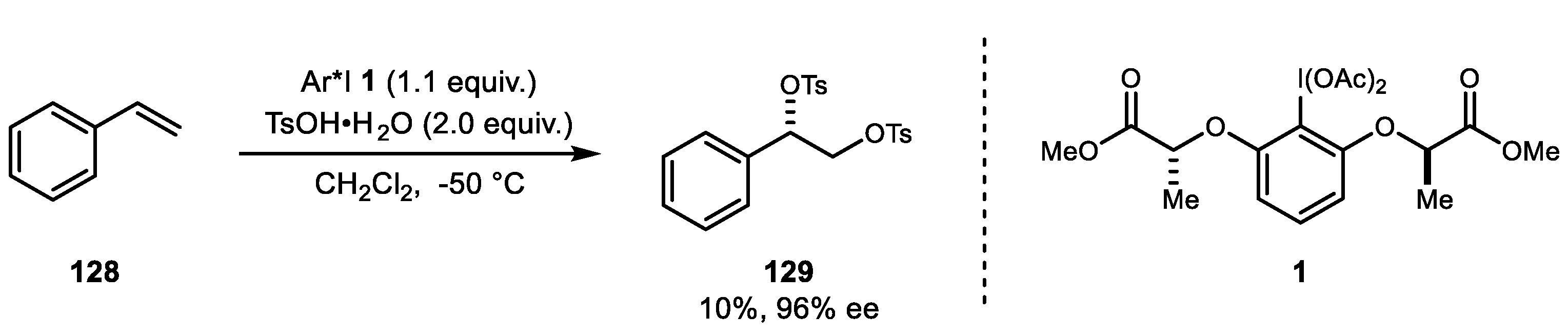
The most recent example of dioxytosylation of styrene was reported by Fujita et al. in 2018 (Scheme 24) [85]. The stoichiometric use of lactate-based chiral iodine reagent 1 yielded the desired dioxytosylated adducts 129 up to 96% ee in the presence of TsOH at low temperatures. However, the substrate scope and reaction optimization are needed for future development.
5. Alkene Oxyfunctionalization
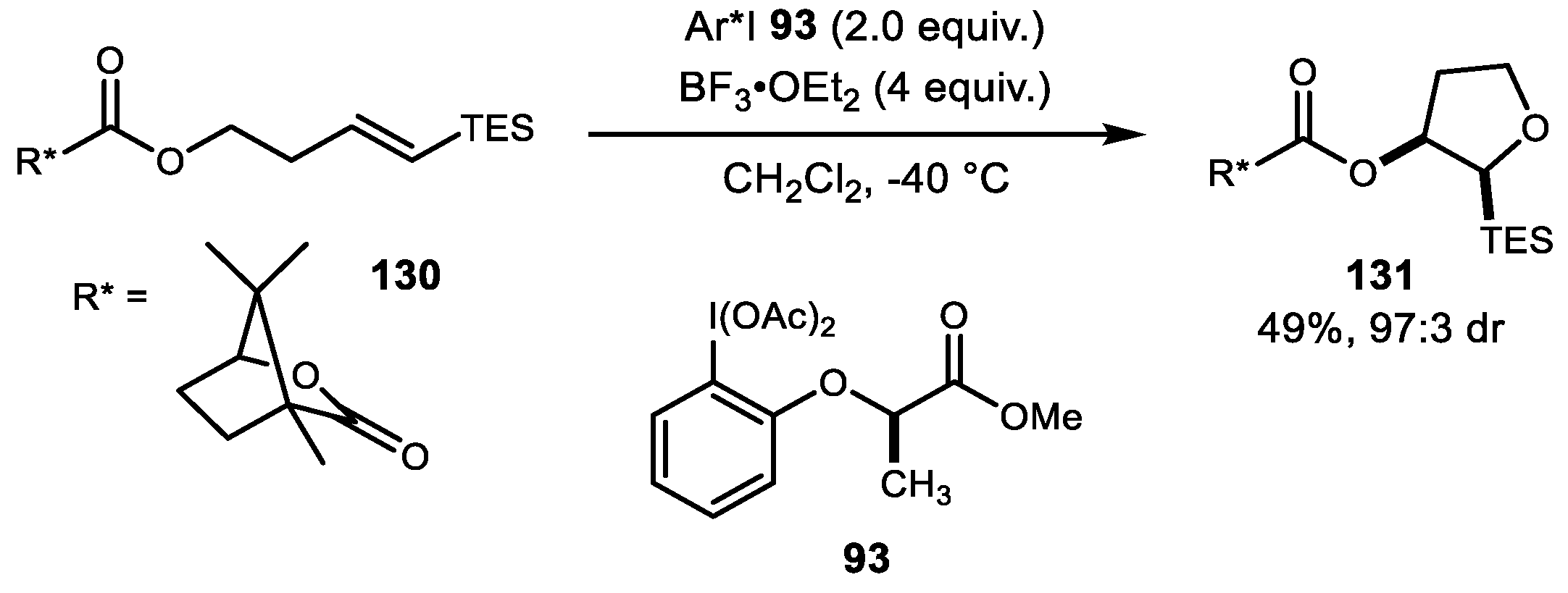
In 2009, Fujita et al. reported improved stereoselective cycloetherification using chiral auxiliary 130 and lactate-based chiral iodine reagent 93 (Scheme 25) [86]. The double asymmetric induction provided higher enantioselectivity compared to iodosylbenzene as a reagent.
A series of metal-free oxidative alkene heterofunctionalizations via an ortho-quinone methide intermediate leading to various heterocycles has been reported. In 2012, Ding et al. demonstrated a PIDA-mediated vinylphenol difunctionalization (Scheme 26a) [87]. Compared to previously reported metal-catalyzed reactions [88,89], this protocol resulted in good yields and excellent diastereoselectivity. Additionally, the carbon nucleophiles were not limited to indole derivatives, as other heterocycles were also investigated. Kita et al. also used highly reactive hypervalent iodine dimer for the oxidative couplings of phenols with styrenes to generate indoline adducts (Scheme 26b) [90]. Use of a PIPA dimer generally enhanced the phenolic oxidation process.
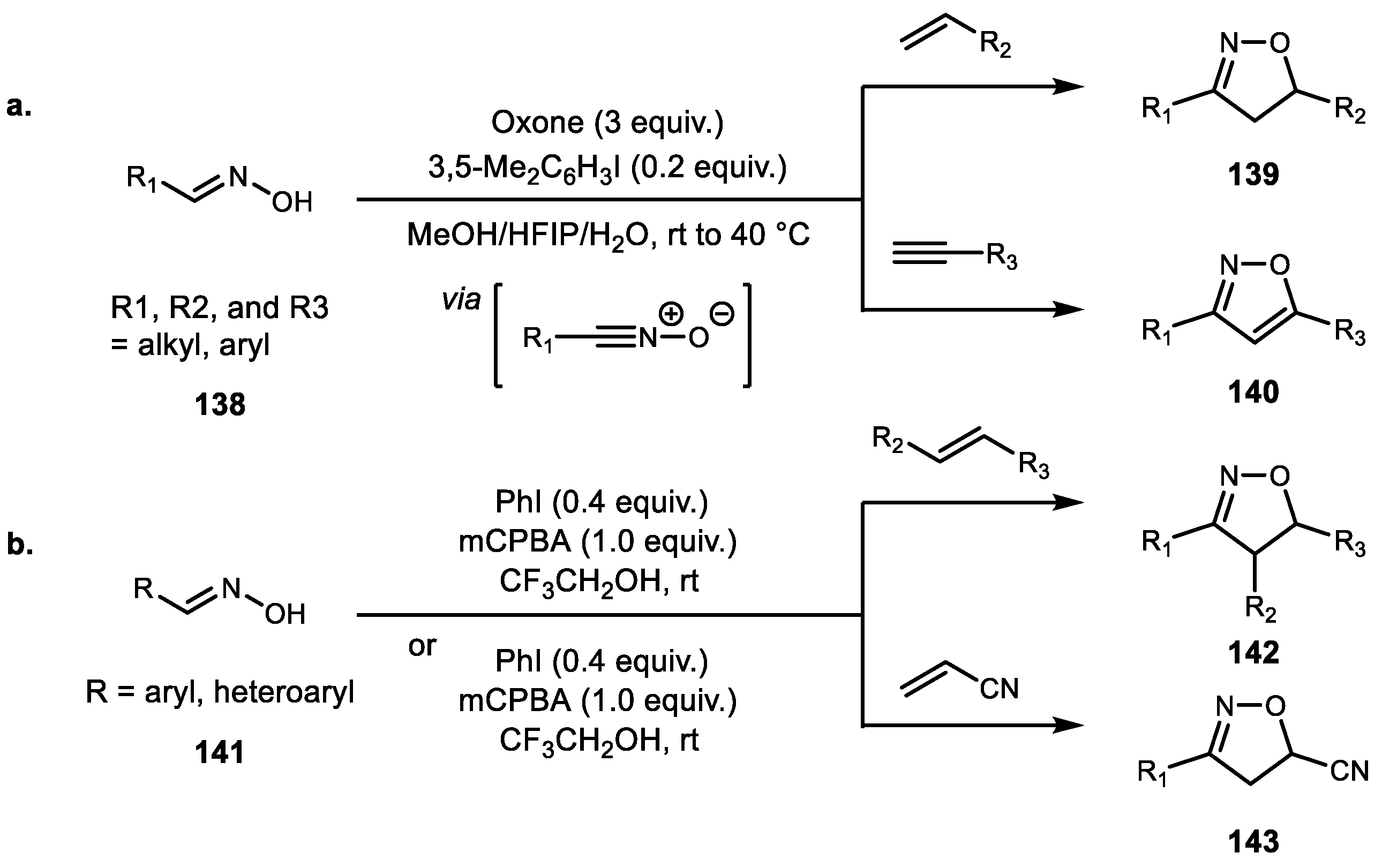
Several catalytic methods to generate an isoxazoline core have been reported. Zhdankin et al. demonstrated the first hypervalent iodine-catalyzed oxime cycloaddition with alkene using catalytic amounts of iodoarene and oxone as a terminal oxidant in aqueous HFIP. The overall efficiency was comparable to previously reported stoichiometric procedures, and alkynes are also applied to the synthesis of isoxazole (Scheme 27a) [91]. Similarly, Yan et al. reported a number of oxime cycloadditions with alkene using a substoichiometric amount of PhI and stoichiometric mCPBA oxidant. A diverse range of the corresponding isooxazolines were synthesized in moderate to good yields (Scheme 27b) [92]. Another cycloaddition using the hypervalent iodine-mediated process was reported by Prakash et al. for pyrazolyl isoxazoline derivatives [93].
Dodd and Cariou have continued to report hypervalent iodine-mediated oxyhalogenation reactions using the umpolung strategy. The combination of PIDA and inorganic bromide in EtOH (Ethyl alcohol) resulted in ethoxybromination reaction of enamides. The iodine promoted umpolung reactions of halide salt to generate electrophilic halide that can react with various double bonds (Scheme 28a) [94]. In case of the ethoxychlorination reaction, ZnCl2 was used as the halide source. Preliminary mechanistic studies have revealed that the reaction proceeds via in situ-generated chlorinated hypervalent iodine species (Scheme 28b) [95]. As an extension of this protocol, a pseudohalide source and TEMPO ((2,2,6,6-Tetramethylpiperidin-1-yl)oxyl) combination resulted in the oxyazidation reaction (Scheme 28c,d) [96].
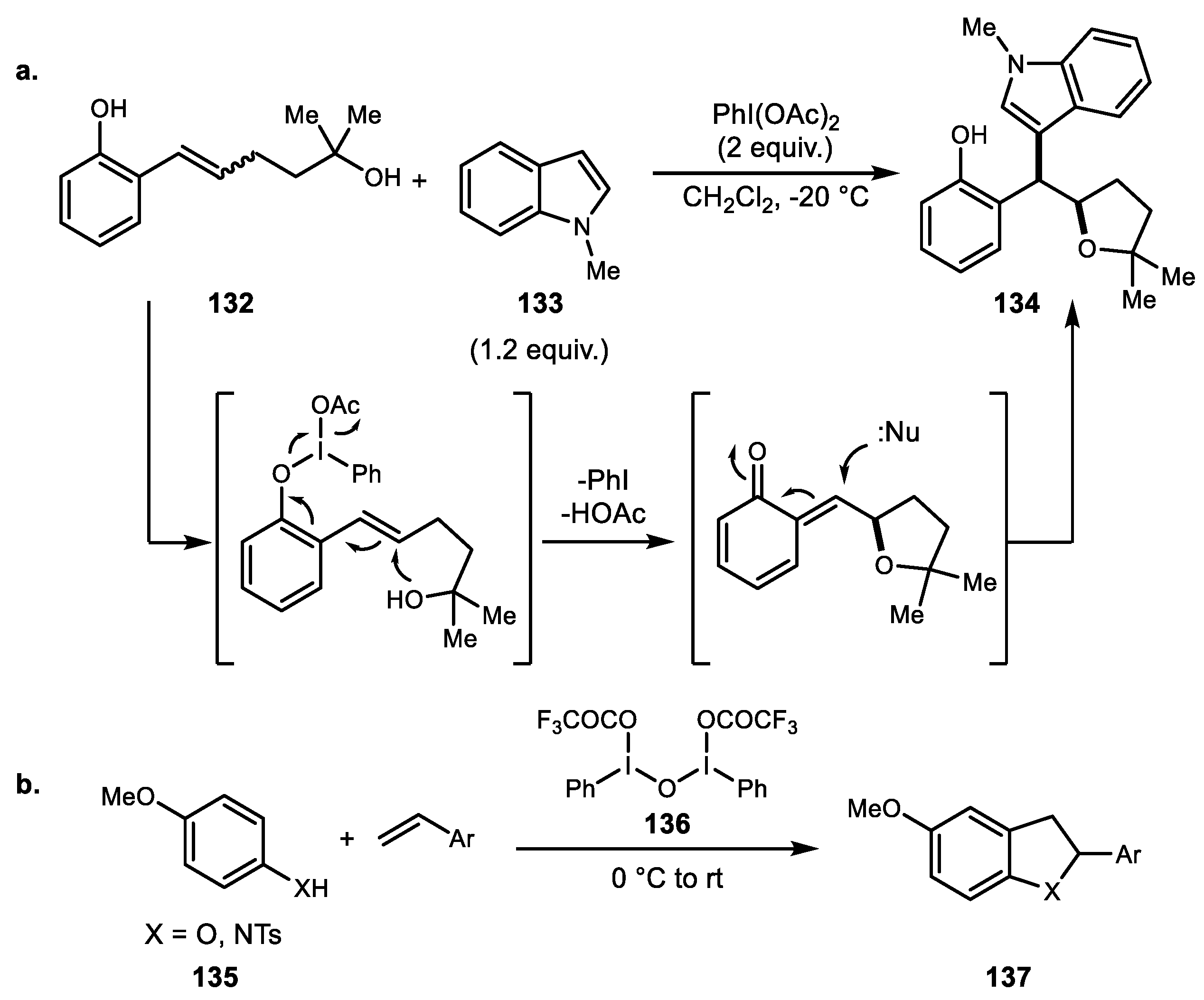
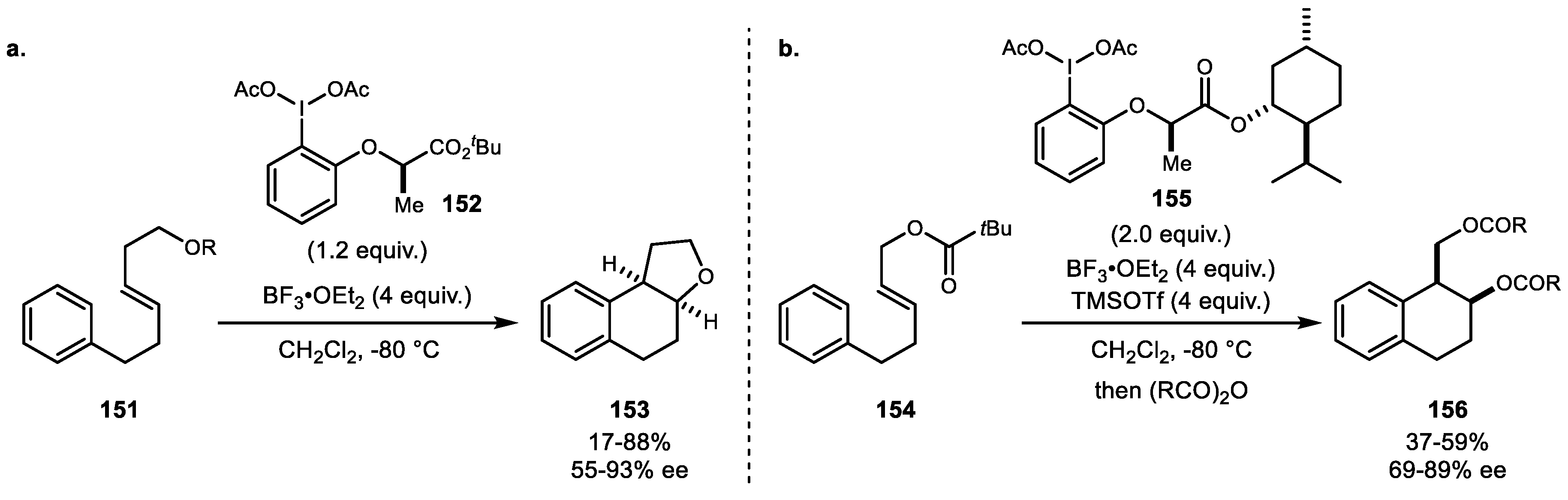
In 2016, Fujita and colleagues added a new version of chiral hypervalent iodine-mediated enantioselective oxidative C–C bond formation. The silyloxy-attached internal alkene 151 effectively yielded oxyarylation adduct in the presence of lactate-based chiral iodine 152 and BF3·OEt2 as a promoter (Scheme 29a) [97]. Another example was reported by Fujita et al. in 2017 (Scheme 29b) [98]. An acyloxy group was introduced for the alkene oxidative oxyarylation reaction. Neighboring nucleophilic acyloxy groups participated effectively in generating the desired transformation to yield the naphthalene structure that is found in serrulatane diterpene natural products.
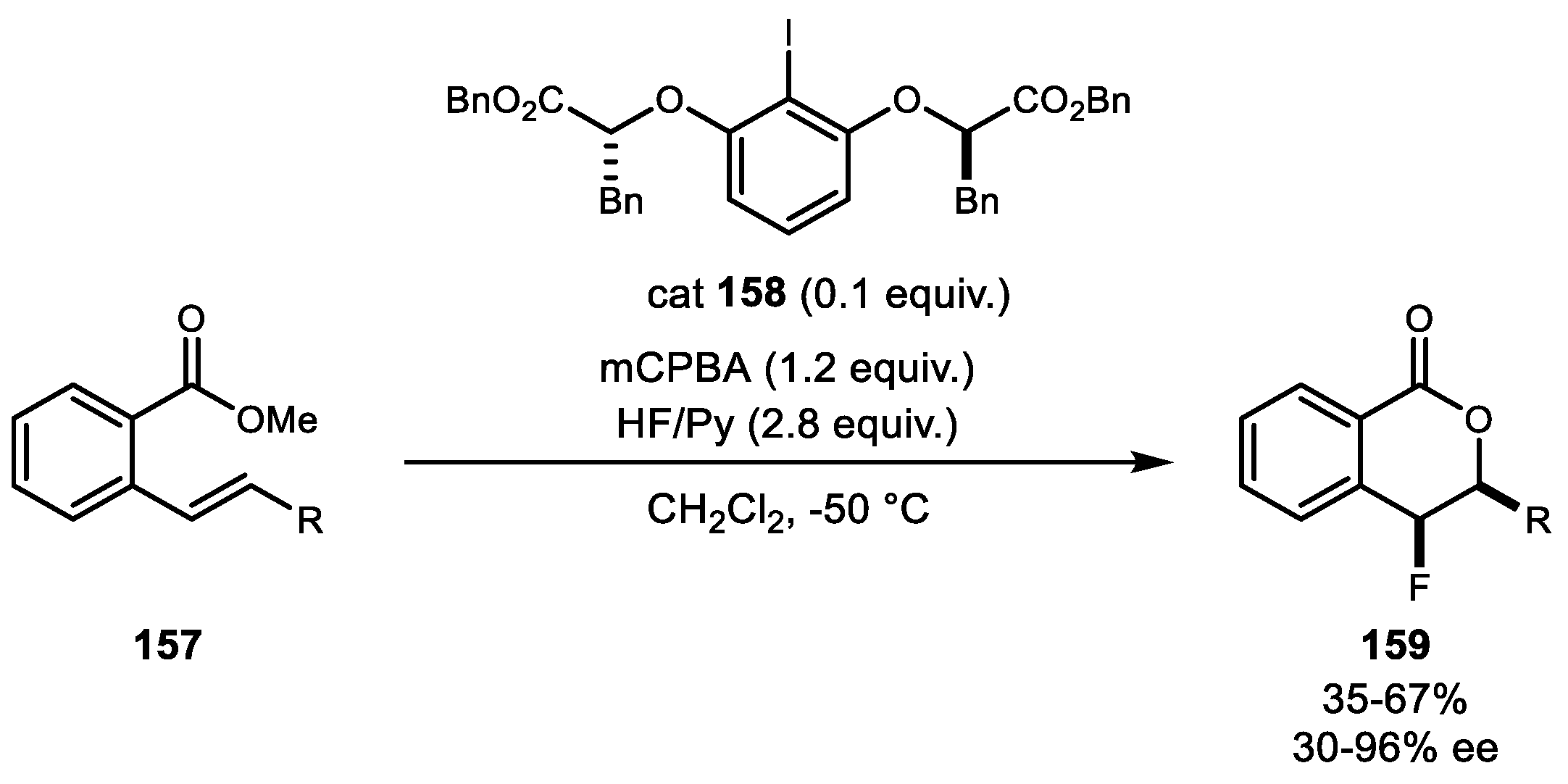
Recently, Jacobsen et al. reported an enantioselective, catalytic fluorolactonization reaction using HF/Py as a nucleophilic fluorine source, mCPBA as the terminal oxidant, and lactate-based chiral iodine 158 (Scheme 30) [99]. A diverse range of terminal and internal styrenes with ortho carboxylic esters were converted to the enantioenriched 4-fluoroisochromanone scaffold 159.
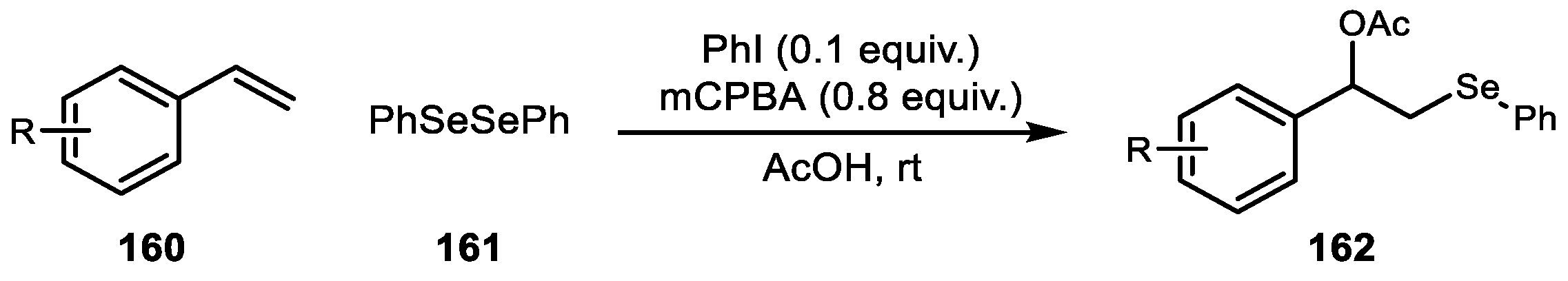
Lastly, Yan et al. reported an iodine-catalyzed acetoxyselenylation reaction via in situ generated electrophilic selenium species (Scheme 31) [100]. Although electrophilic selenium has been used for other hypervalent iodine reactions, the catalytic oxyselenylation has gained limited interest to date.
6. Alkene Dihalogenation
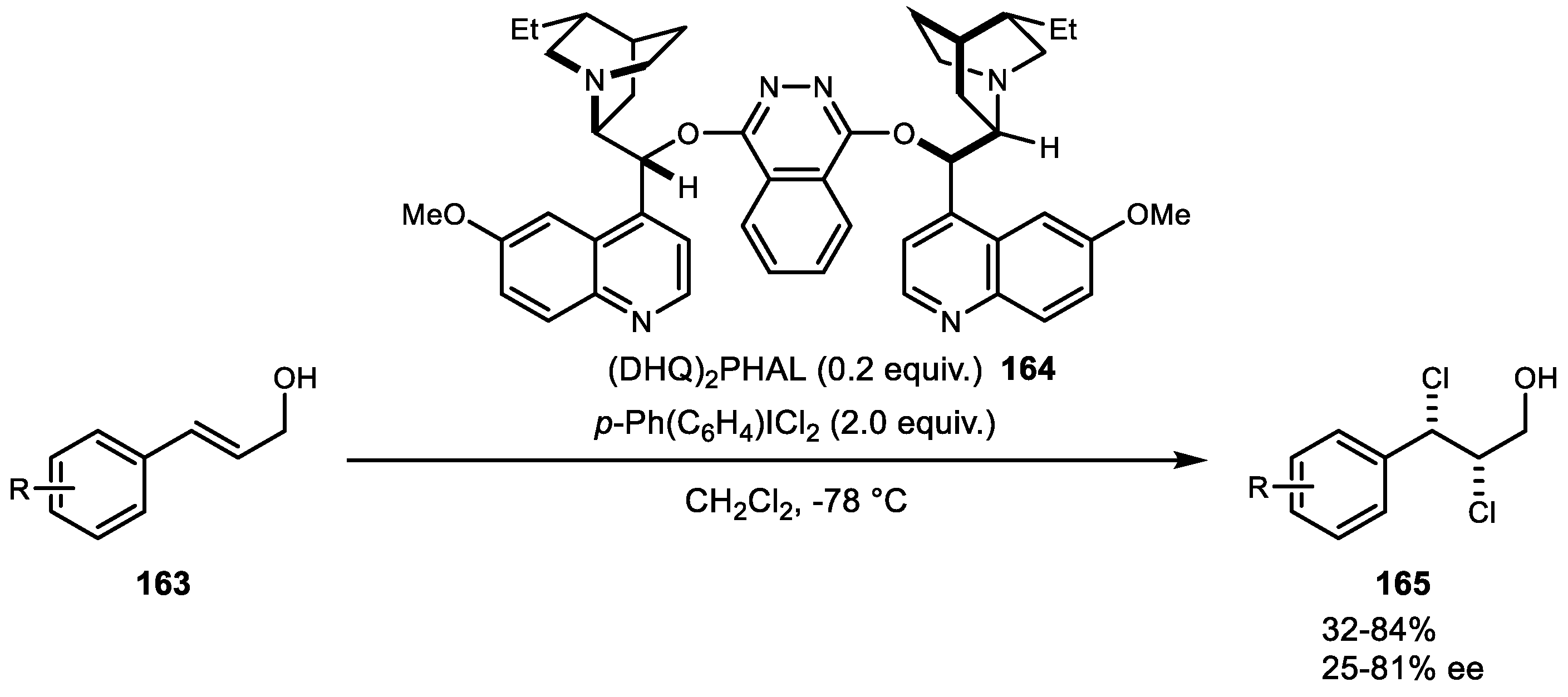
Alkene dihalogenation reactions using hypervalent iodine chemistry have recently attracted increasing attention. Seminal work in this regard was reported by Lupton et al. in 2009, but the overall process failed to achieve good selectivity [101]. In 2011, Nicolaou and coworkers developed an enantioselective alkene dichlorination reaction using cinchona alkaloid and p-Ph(C6H4)ICl2 as a chlorine source (Scheme 32) [102]. Although the substrates were limited to allyl alcohol with moderate enantioselection, this report provided insights regarding chlorenium ion formation by electrophilic iodine and a chiral catalyst.
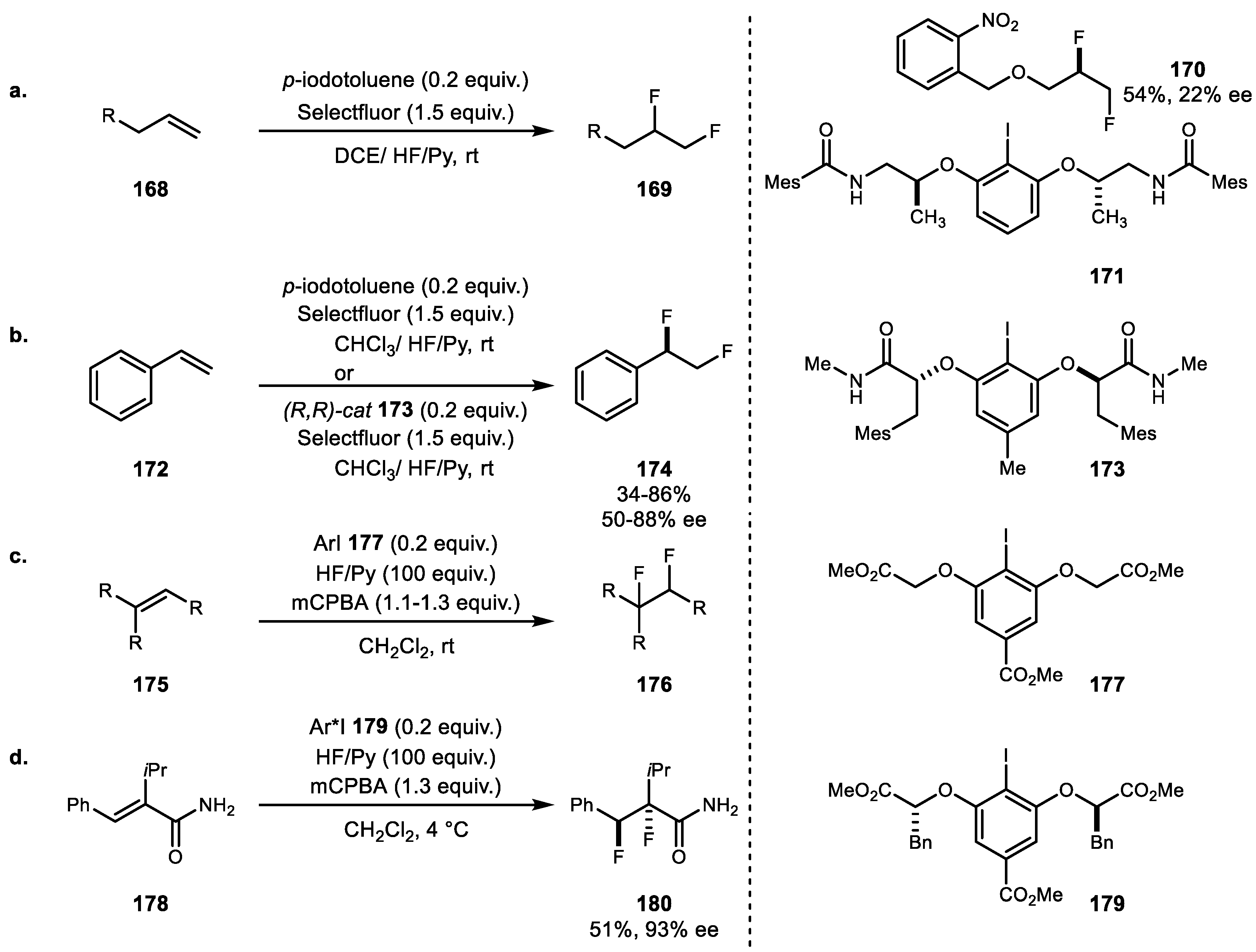
Although stoichiometric variation of alkene difluorination using p-iodotoluene difluoride was reported in 1998 by Hara and coworker [103], catalytic variation was recently reported by Gilmour et al. (Scheme 33a) [104]. Based on the combination of p-iodotoluene and Selectfluor, terminal alkene difluorination reaction was achieved using HF/Py as a halogen source. However, initial attempts at enantioselective variation using chiral iodine 171 only yielded 22% ee of 170. The same group subsequently reported catalytic enantioselective difluorination using newly designed chiral iodine 173 [105]. This study revealed not only excellent new catalyst for enantioselective difluorination but also the correlation of ee values and the substrate electronic effect for determination of both the reaction course and the selectivity of the reaction pathway. Jacobsen et al. concurrently reported catalytic, diasteroselective vicinal difluorination using aryl iodide 174 and mCPBA oxidant (Scheme 33c) [106]. In addition to terminal alkenes, a diverse range of internal alkenes and electron-deficient alkenes were tolerated. Moreover, chiral iodine 177 yielded 93% ee of 176 adduct (Scheme 33d).
Lastly, Cariou and coworkers recently demonstrated a chemodivergent, tunable, and selective method for bromofunctionalization of polyprenoids (Scheme 34) [107,108]. Depending on the combination of a hypervalent iodine source, inorganic salt, solvent, and temperature, the reaction produced selective dibromination, oxy-bromination, and bromocyclization reactions.
7. Conclusions
In this review, we provided an overview of the advances regarding hypervalent iodine-mediated and catalyzed alkene difunctionalization reactions in the past 10 years. Although we only focused on diamination, aminofunctionalization, diacetoxylation, oxyfunctionalization, and dihalogenation reactions, numerous advances and improvements have been observed regarding new reactions, reagent developments, and enantioselective transformations using chiral iodine reagents. While some of the aforementioned methodologies are needed to improve enantioselectivity and to diversify the range of the substrate scope, hypervalent iodine reagents and the catalysts themselves still exhibit complementary reactivity compared to transition metal-catalyzed organic transformations. Owing to their transition metal-like properties and environmentally benign nature, it is likely that more advances in this area of synthetic chemistry will be forthcoming in the near future.
Author Contributions
S.C. and K.B.H. conceived and designed the overall structure of the review. J.H.L., S.C., and K.B.H. wrote the manuscript. All the authors approved the final version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2018R1D1A1B07048454 to J.H.L. and NRF-2018R1D1A3B07046990 to S.C.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wirth, T.; Hirt, U.H. Hypervalent iodine compounds. Recent advances in synthetic applications. Synthesis 1999, 1999, 1271–1287. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Ladziata, U.; Zhdankin, V.V. Hypervalent iodine(V) reagents in organic synthesis. Arkivoc 2006, 9, 26–58. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohi, T.; Kita, Y. Hypervalent iodine reagents as a new entrance to organocatalysts. Chem. Commun. 2009, 2073–2085. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Hypervalent iodine(III) reagents in organic synthesis. Arkivoc 2009, 1–62. [Google Scholar]
- Farid, U.; Wirth, T. Stereoselective Synthesis with Hypervalent Iodine Reagents; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 197–203. [Google Scholar]
- Brown, M.; Farid, U.; Wirth, T. Hypervalent iodine reagents as powerful electrophiles. Synlett 2013, 24, 424–431. [Google Scholar]
- Singh, F.V.; Wirth, T. Oxidative Functionalization with Hypervalent Halides; Elsevier B.V.: Amsterdam, The Netherlands, 2014; pp. 880–933. [Google Scholar]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Quideau, S. Hypervalent iodine chemistry: Recent advances and applications; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Zhdankin, V.V. Hypervalent Iodine Chemistry Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds; John Wiley & Sons: Chichester/West Sussex, UK, 2014. [Google Scholar]
- Wirth, T. Hypervalent Iodine Chemistry; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Moriarty, R.M.; Prakash, O. Oxidation of phenolic compounds with organohypervalent iodine reagents. Org. React. 2001, 57, 327–415. [Google Scholar]
- Rodriguez, S.; Wipf, P. Oxidative spiroacetalizations and spirolactonizations of arenes. Synthesis 2004, 2767–2783. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Benziodoxole-based hypervalent iodine reagents in organic synthesis. Curr. Org. Synth. 2005, 2, 121–145. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Braun, N.A.; Canesi, S.; Ousmer, M.; Chang, J.; Chai, D. Oxidative amidation of phenols through the use of hypervalent iodine reagents. Development and applications. Synthesis 2007, 3759–3772. [Google Scholar] [CrossRef]
- Pouysegu, L.; Deffieux, D.; Quideau, S. Hypervalent iodine-mediated phenol dearomatization in natural product synthesis. Tetrahedron 2010, 66, 2235–2261. [Google Scholar] [CrossRef]
- Fernandez Gonzalez, D.; Benfatti, F.; Waser, J. Asymmetric Organocatalysis Meets Hypervalent Iodine Chemistry for the α-Functionalization of Carbonyl Compounds. ChemCatChem 2012, 4, 955–958. [Google Scholar] [CrossRef]
- Parra, A.; Reboredo, S. Chiral Hypervalent Iodine Reagents: Synthesis and Reactivity. Chem. Eur. J. 2013, 19, 17244–17260. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.V.; Wirth, T. Oxidative rearrangements with hypervalent iodine reagents. Synthesis 2013, 45, 2499–2511. [Google Scholar]
- Romero, R.M.; Woeste, T.H.; Muniz, K. Vicinal Difunctionalization of Alkenes with Iodine (III) Reagents and Catalysts. Chem. Asian J. 2014, 9, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, J.; Fruh, N.; Togni, A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 2015, 115, 650–682. [Google Scholar] [CrossRef]
- Kumar, R.; Wirth, T. Asymmetric synthesis with hypervalent iodine reagents. Top. Curr. Chem. 2016, 373, 243–262. [Google Scholar]
- Muniz, K. Aminations with hypervalent iodine. Top. Curr. Chem. 2016, 373, 105–134. [Google Scholar]
- Waser, J. Alkynylation with hypervalent iodine reagents. Top. Curr. Chem. 2016, 373, 187–222. [Google Scholar] [PubMed]
- Boelke, A.; Finkbeiner, P.; Nachtsheim, B.J. Atom-economical group-transfer reactions with hypervalent iodine compounds. Beilstein J. Org. Chem. 2018, 14, 1263–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy Kandimalla, S.; Prathima Parvathaneni, S.; Sabitha, G.; Subba Reddy, B.V. Recent Advances in Intramolecular Metal-Free Oxidative C-H Bond Aminations Using Hypervalent Iodine (III) Reagents. Eur. J. Org. Chem. 2019, 2019, 1687–1714. [Google Scholar] [CrossRef]
- Xing, L.; Zhang, Y.; Du, Y. Hypervalent Iodine-Mediated Synthesis of Spiroheterocycles via Oxidative Cyclization. Curr. Org. Chem. 2019, 23, 14–37. [Google Scholar] [CrossRef]
- Röben, C.; Souto, J.A.; González, Y.; Lishchynskyi, A.; Muñiz, K. Enantioselective Metal-Free Diamination of Styrenes. Angew. Chem. Int. Ed. 2011, 50, 9478–9482. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K.; Barreiro, L.; Romero, R.M.; Martínez, C. Catalytic Asymmetric Diamination of Styrenes. J. Am. Chem. Soc. 2017, 139, 4354–4357. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.A.; González, Y.; Iglesias, A.; Zian, D.; Lishchynskyi, A.; Muñiz, K. Iodine (III)-Promoted Intermolecular Diamination of Alkenes. Chem. Asian J. 2012, 7, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.A.; Martínez, C.; Velilla, I.; Muñiz, K. Defined Hypervalent Iodine (III) Reagents Incorporating Transferable Nitrogen Groups: Nucleophilic Amination through Electrophilic Activation. Angew. Chem. Int. Ed. 2013, 52, 1324–1328. [Google Scholar] [CrossRef]
- Romero, R.M.; Souto, J.A.; Muñiz, K. Substitution Effects of Hypervalent Iodine (III) Reagents in the Diamination of Styrene. J. Org. Chem. 2016, 81, 6118–6122. [Google Scholar] [CrossRef]
- Röben, C.; Souto, J.A.; Escudero-Adán, E.C.; Muñiz, K. Oxidative Diamination Promoted by Dinuclear Iodine (III) Reagents. Org. Lett. 2013, 15, 1008–1011. [Google Scholar] [CrossRef]
- Kong, A.; Blakey, S.B. Intramolecular Olefin Diamination for the Stereoselective Synthesis of 3-Aminopiperidines. Synthesis 2012, 44, 1190–1198. [Google Scholar]
- Kim, H.J.; Cho, S.H.; Chang, S. Intramolecular Oxidative Diamination and Aminohydroxylation of Olefins under Metal-Free Conditions. Org. Lett. 2012, 14, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K. Advancing Palladium-Catalyzed C–N Bond Formation: Bisindoline Construction from Successive Amide Transfer to Internal Alkenes. J. Am. Chem. Soc. 2007, 129, 14542–14543. [Google Scholar] [CrossRef] [PubMed]
- Mizar, P.; Laverny, A.; El-Sherbini, M.; Farid, U.; Brown, M.; Malmedy, F.; Wirth, T. Enantioselective Diamination with Novel Chiral Hypervalent Iodine Catalysts. Chem. Eur. J. 2014, 20, 9910–9913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-T.; Lu, X.-W.; Xing, M.-L.; Sun, X.-Q.; Miao, C.-B. Hypervalent Iodine Reagent Mediated Diamination of [60] Fullerene with Sulfamides or Phosphoryl Diamides. Org. Lett. 2014, 16, 5882–5885. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.B.; Johnston, J.N. Alkene Diamination Using Electron-Rich Amines: Hypervalent Iodine-Promoted Inter-/Intramolecular C–N Bond Formation. Org. Lett. 2014, 16, 3804–3807. [Google Scholar] [CrossRef]
- Danneman, M.W.; Hong, K.B.; Johnston, J.N. Oxidative Inter-/Intermolecular Alkene Diamination of Hydroxy Styrenes with Electron-Rich Amines. Org. Lett. 2015, 17, 2558–2561. [Google Scholar] [CrossRef]
- Danneman, M.W.; Hong, K.B.; Johnston, J.N. A Unified Approach to the Four Azaindoline Families by Inter-/Intramolecular Annulative Diamination of Vinylpyridines. Org. Lett. 2015, 17, 3806–3809. [Google Scholar] [CrossRef]
- Wu, X.-L.; Wang, G.-W. Hypervalent iodine-mediated aminobromination of olefins in water. Tetrahedron 2009, 65, 8802–8807. [Google Scholar] [CrossRef]
- Lovick, H.M.; Michael, F.E. Metal-Free Highly Regioselective Aminotrifluoroacetoxylation of Alkenes. J. Am. Chem. Soc. 2010, 132, 1249–1251. [Google Scholar] [CrossRef]
- Farid, U.; Wirth, T. Highly Stereoselective Metal-Free Oxyaminations Using Chiral Hypervalent Iodine Reagents. Angew. Chem. Int. Ed. 2012, 51, 3462–3465. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kaga, A.; Chiba, S. Diastereoselective Aminooxygenation and Diamination of Alkenes with Amidines by Hypervalent Iodine (III) Reagents. Org. Lett. 2014, 16, 6136–6139. [Google Scholar] [CrossRef] [PubMed]
- Sanjaya, S.; Chiba, S. Copper-Catalyzed Aminooxygenation of N-Allylamidines with PhI(OAc)2. Org. Lett. 2012, 14, 5342–5345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Zhu, X.; Chiba, S. Copper-Catalyzed Aerobic [3 + 2]-Annulation of N-Alkenyl Amidines. J. Am. Chem. Soc. 2012, 134, 3679–3682. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kaga, A.; Chiba, S. Anti-Selective aminofluorination of alkenes with amidines mediated by hypervalent iodine (iii) reagents. Org. Biomol. Chem. 2016, 14, 5481–5485. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.J.; Bowen, E.G.; Forslund, R.E.; Sussman, A.D.; Weerasekera, S.L. Intramolecular Oxamidation of Unsaturated O-Alkyl Hydroxamates: A Remarkably Versatile Entry to Hydroxy Lactams. J. Am. Chem. Soc. 2010, 132, 1188–1189. [Google Scholar] [CrossRef] [PubMed]
- Bowen, E.G.; Wardrop, D.J. Diastereoselective Nitrenium Ion-Mediated Cyclofunctionalization: Total Synthesis of (+)-Castanospermine. Org. Lett. 2010, 12, 5330–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardrop, D.J.; Bowen, E.G. Nitrenium Ion-Mediated Alkene Bis-Cyclofunctionalization: Total Synthesis of (−)-Swainsonine. Org. Lett. 2011, 13, 2376–2379. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Gerasimov, M.V.; DeJong, S.; Wardrop, D.J. Oxamidation of Unsaturated O-Alkyl Hydroxamates: Synthesis of the Madangamine Diazatricylic (ABC Rings) Skeleton. Org. Lett. 2017, 19, 6570–6573. [Google Scholar] [CrossRef]
- Wang, Q.; Zhong, W.; Wei, X.; Ning, M.; Meng, X.; Li, Z. Metal-free intramolecular aminofluorination of alkenes mediated by PhI(OPiv)2/hydrogen fluoride–pyridine system. Org. Biomol. Chem. 2012, 10, 8566–8569. [Google Scholar] [CrossRef]
- Kong, W.; Feige, P.; de Haro, T.; Nevado, C. Regio- and Enantioselective Aminofluorination of Alkenes. Angew. Chem. Int. Ed. 2013, 52, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kamo, T.; Fukushi, K.; Hiramatsu, T.; Tokunaga, E.; Dohi, T.; Kita, Y.; Shibata, N. Iodoarene-catalyzed fluorination and aminofluorination by an Ar-I/HF·pyridine/mCPBA system. Chem. Sci. 2014, 5, 2754–2760. [Google Scholar] [CrossRef]
- Cui, J.; Jia, Q.; Feng, R.-Z.; Liu, S.-S.; He, T.; Zhang, C. Boron Trifluoride Etherate Functioning as a Fluorine Source in an Iodosobenzene-Mediated Intramolecular Aminofluorination of Homoallylic Amines. Org. Lett. 2014, 16, 1442–1445. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Miyake, A.; Muta, K.; Oyamada, A.J. Hypervalent Iodine/HF Reagents for the Synthesis of 3-Fluoropyrrolidines. J. Org. Chem. 2017, 82, 11721–11726. [Google Scholar] [CrossRef] [PubMed]
- Mennie, K.M.; Banik, S.M.; Reichert, E.C.; Jacobsen, E.N. Catalytic Diastereo- and Enantioselective Fluoroamination of Alkenes. J. Am. Chem. Soc. 2018, 140, 4797–4802. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-Q.; Li, Y.-M. Regioselective (Diacetoxyiodo)benzene-Promoted Halocyclization of Unfunctionalized Olefins. J. Org. Chem. 2014, 79, 10094–10109. [Google Scholar] [CrossRef]
- Wang, H.; Frings, M.; Bolm, C. Halocyclizations of Unsaturated Sulfoximines. Org. Lett. 2016, 18, 2431–2434. [Google Scholar] [CrossRef]
- Daniel, M.; Blanchard, F.; Nocquet-Thibault, S.; Cariou, K.; Dodd, R.H. Halocyclization of Unsaturated Guanidines Mediated by Koser’s Reagent and Lithium Halides. J. Org. Chem. 2015, 80, 10624–10633. [Google Scholar] [CrossRef]
- Mizar, P.; Niebuhr, R.; Hutchings, M.; Farooq, U.; Wirth, T. Thioamination of Alkenes with Hypervalent Iodine Reagents. Chem. Eur. J. 2016, 22, 1614–1617. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, D.; Lee, J.H.; Song, J.; Lee, W.S.; Kang, D.W.; Kang, S.; Lee, S.B.; Choi, S.; Hong, K.B. Hypervalent Iodine-Mediated Alkene Functionalization: Oxazoline and Thiazoline Synthesis via Inter-/Intramolecular Aminohydroxylation and Thioamination. Adv. Synth. Catal. 2018, 360, 779–783. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, S.-H.; Song, J.; Park, G.Y.; Kim, D.; Nam, T.-G.; Hong, K.B. Hypervalent iodine-mediated Ritter-type amidation of terminal alkenes: The synthesis of isoxazoline and pyrazoline cores. Beilstein J. Org. Chem. 2018, 14, 1028–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.-M.; Cai, C. Iodine(iii)-mediated intramolecular sulfeno- and selenofunctionalization of β,γ-unsaturated tosyl hydrazones and oximes. Org. Biomol. Chem. 2018, 16, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yoshida, Y.; Miyata, K.; Wakisaka, A.; Sugimura, T. Enantiodifferentiating endo-Selective Oxylactonization of ortho-Alk-1-enylbenzoate with a Lactate-Derived Aryl-λ3-Iodane. Angew. Chem. Int. Ed. 2010, 49, 7068–7071. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Wakita, M.; Sugimura, T. Enantioselective Prévost and Woodward reactions using chiral hypervalent iodine(iii): Switchover of stereochemical course of an optically active 1,3-dioxolan-2-yl cation. Chem. Commun. 2011, 47, 3983–3985. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. Asymmetric Synthesis of 4,8-Dihydroxyisochroman-1-one Polyketide Metabolites Using Chiral Hypervalent Iodine(III). Org. Lett. 2012, 14, 1294–1297. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. Total synthesis of (12R)- and (12S)-12-hydroxymonocerins: Stereoselective oxylactonization using a chiral hypervalent iodine(iii) species. RSC Adv. 2013, 3, 17717–17725. [Google Scholar] [CrossRef]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Enantioselective Oxidation of Alkenylbenzoates Catalyzed by Chiral Hypervalent Iodine(III) To Yield 4-Hydroxyisochroman-1-ones. Eur. J. Org. Chem. 2013, 2013, 7128–7138. [Google Scholar] [CrossRef]
- Takesue, T.; Fujita, M.; Sugimura, T.; Akutsu, H. A Series of Two Oxidation Reactions of ortho-Alkenylbenzamide with Hypervalent Iodine(III): A Concise Entry into (3R,4R)-4-Hydroxymellein and (3R,4R)-4-Hydroxy-6-methoxymellein. Org. Lett. 2014, 16, 4634–4637. [Google Scholar] [CrossRef]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Stereoselective Formation of Substituted 1,3-Dioxolanes through a Three-Component Assembly during the Oxidation of Alkenes with Hypervalent Iodine(III). Molecules 2015, 20, 17041. [Google Scholar] [CrossRef]
- Alhalib, A.; Kamouka, S.; Moran, W.J. Iodoarene-Catalyzed Cyclizations of Unsaturated Amides. Org. Lett. 2015, 17, 1453–1456. [Google Scholar] [CrossRef] [Green Version]
- Moon, N.G.; Harned, A.M. Iodine(III)-promoted synthesis of oxazolines from N-allylamides. Tetrahedron Lett. 2013, 54, 2960–2963. [Google Scholar] [CrossRef]
- Koser, G.F.; Lodaya, J.S.; Ray, D.G.; Kokil, P.B. Direct. alpha.-phosphoryloxylation of ketones and (phosphoryloxy) lactonization of pentenoic acids with [hydroxy[(bis(phenyloxy)phosphoryl)oxy]iodo]benzene. J. Am. Chem. Soc. 1988, 110, 2987–2988. [Google Scholar] [CrossRef]
- Zhou, Z.-S.; He, X.-H. A convenient phosphoryloxylactonization of pentenoic acids with catalytic hypervalent iodine(III) reagent. Tetrahedron Lett. 2010, 51, 2480–2482. [Google Scholar] [CrossRef]
- Gelis, C.; Dumoulin, A.; Bekkaye, M.; Neuville, L.; Masson, G. Chiral Hypervalent Iodine(III) Catalyst Promotes Highly Enantioselective Sulfonyl- and Phosphoryl-oxylactonizations. Org. Lett. 2017, 19, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Yang, J.; Meng, X.; Li, Z. BF3·OEt2-Promoted Diastereoselective Diacetoxylation of Alkenes by PhI(OAc)2. J. Org. Chem. 2011, 76, 9997–10004. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Liu, S.; Yang, J.; Meng, X.; Li, Z. Metal-Free, Organocatalytic Syn Diacetoxylation of Alkenes. Org. Lett. 2012, 14, 3336–3339. [Google Scholar] [CrossRef]
- Haubenreisser, S.; Wöste, T.H.; Martínez, C.; Ishihara, K.; Muñiz, K. Structurally Defined Molecular Hypervalent Iodine Catalysts for Intermolecular Enantioselective Reactions. Angew. Chem. Int. Ed. 2016, 55, 413–417. [Google Scholar] [CrossRef]
- Wöste, T.H.; Muñiz, K. Enantioselective Vicinal Diacetoxylation of Alkenes under Chiral Iodine(III) Catalysis. Synthesis 2016, 48, 816–827. [Google Scholar] [Green Version]
- Aertker, K.; Rama, R.J.; Opalach, J.; Muñiz, K. Vicinal Difunctionalization of Alkenes under Iodine(III) Catalysis involving Lewis Base Adducts. Adv. Synth. Catal. 2017, 359, 1290–1294. [Google Scholar] [CrossRef]
- Fujita, M.; Miura, K.; Sugimura, T. Enantioselective dioxytosylation of styrenes using lactate-based chiral hypervalent iodine(III). Beilstein J. Org. Chem. 2018, 14, 659–663. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Ookubo, Y.; Sugimura, T. Asymmetric cycloetherification based on a chiral auxiliary for 4-acyloxy-1-butene substrates during oxidation with iodosylbenzene via a 1,3-dioxan-2-yl cation. Tetrahedron Lett. 2009, 50, 1298–1300. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, H.; Zheng, J.; Yu, L.; Ding, H. Hypervalent iodine mediated alkene difunctionalization of vinylphenols: Diastereoselective synthesis of substituted indoles and indolizines. Chem. Commun. 2015, 51, 6399–6402. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.H.; Pathak, T.P.; Zhang, Y.; Sigman, M.S. Palladium-Catalyzed Enantioselective Addition of Two Distinct Nucleophiles across Alkenes Capable of Quinone Methide Formation. J. Am. Chem. Soc. 2009, 131, 17074–17075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.H.; Webb, J.D.; Sigman, M.S. Advancing the Mechanistic Understanding of an Enantioselective Palladium-Catalyzed Alkene Difunctionalization Reaction. J. Am. Chem. Soc. 2010, 132, 17471–17482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohi, T.; Toyoda, Y.; Nakae, T.; Koseki, D.; Kubo, H.; Kamitanaka, T.; Kita, Y. Phenol and aniline oxidative coupling with alkenes by using hypervalent iodine dimer for the rapid access to dihydrobenzofurans and indolines. Heterocylces 2015, 90, 631–644. [Google Scholar]
- Yoshimura, A.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Koski, S.R.; Maskaev, A.V.; Zhdankin, V.V. Hypervalent Iodine Catalyzed Generation of Nitrile Oxides from Oximes and their Cycloaddition with Alkenes or Alkynes. Org. Lett. 2013, 15, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Li, T.; Yan, J. Hypervalent Iodine–Catalyzed Cycloaddition of Nitrile Oxides to Alkenes. Synth. Commun. 2014, 44, 682–688. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, M.; Prakash, O. A Simple and Efficient Method for the Synthesis of Novel Pyrazolyl Isoxazoline Derivatives Using Hypervalent Iodine (III) Reagent. Heteroat. Chem. 2016, 27, 228–234. [Google Scholar] [CrossRef]
- Nocquet-Thibault, S.; Retailleau, P.; Cariou, K.; Dodd, R.H. Iodine(III)-Mediated Umpolung of Bromide Salts for the Ethoxybromination of Enamides. Org. Lett. 2013, 15, 1842–1845. [Google Scholar] [CrossRef]
- Nocquet-Thibault, S.; Minard, C.; Retailleau, P.; Cariou, K.; Dodd, R.H. Iodine(III)-mediated ethoxychlorination of enamides with iron(III) chloride. Tetrahedron 2014, 70, 6769–6775. [Google Scholar] [CrossRef]
- Nocquet-Thibault, S.; Rayar, A.; Retailleau, P.; Cariou, K.; Dodd, R.H. Iodine(III)-Mediated Diazidation and Azido-oxyamination of Enamides. Chem. Eur. J. 2015, 21, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Metal-Free Enantioselective Oxidative Arylation of Alkenes: Hypervalent-Iodine-Promoted Oxidative C–C Bond Formation. Angew. Chem. Int. Ed. 2016, 55, 15797–15801. [Google Scholar] [CrossRef] [PubMed]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Enantioselective C-C Bond Formation during the Oxidation of 5-Phenylpent-2-enyl Carboxylates with Hypervalent Iodine(III). J. Org. Chem. 2017, 82, 11836–11840. [Google Scholar] [CrossRef] [PubMed]
- Woerly, E.M.; Banik, S.M.; Jacobsen, E.N. Enantioselective, Catalytic Fluorolactonization Reactions with a Nucleophilic Fluoride Source. J. Am. Chem. Soc. 2016, 138, 13858–13861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, S.; Yan, J. PhI Catalyzed Acetoxyselenylation and Formyloxyselenylation of Alkenes. Helv. Chim. Acta 2017, 100, e1600306. [Google Scholar] [CrossRef]
- Ngatimin, M.; Gartshore, C.J.; Kindler, J.P.; Naidu, S.; Lupton, D.W. The α-halogenation of α,β-unsaturated carbonyls and dihalogenation of alkenes using bisacetoxyiodobenzene/pyridine hydrohalides. Tetrahedron Lett. 2009, 50, 6008–6011. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Simmons, N.L.; Ying, Y.; Heretsch, P.M.; Chen, J.S. Enantioselective Dichlorination of Allylic Alcohols. J. Am. Chem. Soc. 2011, 133, 8134–8137. [Google Scholar] [CrossRef] [Green Version]
- Hara, S.; Nakahigashi, J.; Ishi-i, K.; Sawaguchi, M.; Sakai, H.; Fukuhara, T.; Yoneda, N. Difluorination of Alkenes with Iodotoluene Difluoride. Synlett 1998, 1998, 495–496. [Google Scholar] [CrossRef]
- Molnár, I.G.; Gilmour, R. Catalytic Difluorination of Olefins. J. Am. Chem. Soc. 2016, 138, 5004–5007. [Google Scholar] [CrossRef]
- Scheidt, F.; Schäfer, M.; Sarie, J.C.; Daniliuc, C.G.; Molloy, J.J.; Gilmour, R. Enantioselective, Catalytic Vicinal Difluorination of Alkenes. Angew. Chem. Int. Ed. 2018, 57, 16431–16435. [Google Scholar] [CrossRef]
- Banik, S.M.; Medley, J.W.; Jacobsen, E.N. Catalytic, Diastereoselective 1,2-Difluorination of Alkenes. J. Am. Chem. Soc. 2016, 138, 5000–5003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grayfer, T.D.; Retailleau, P.; Dodd, R.H.; Dubois, J.; Cariou, K. Chemodivergent, Tunable, and Selective Iodine(III)-Mediated Bromo-Functionalizations of Polyprenoids. Org. Lett. 2017, 19, 4766–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peilleron, L.; Grayfer, T.D.; Dubois, J.; Dodd, R.H.; Cariou, K. Iodine(III)-mediated halogenations of acyclic monoterpenoids. Beilstein J. Org. Chem. 2018, 14, 1103–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Scheme 1.
(a) Stoichiometric diamination with chiral hypervalent iodine and (b) catalytic diamination with chiral iodide.
Scheme 1.
(a) Stoichiometric diamination with chiral hypervalent iodine and (b) catalytic diamination with chiral iodide.

Scheme 2.
(a) Muñiz’s general combination of reagent for alkene diamination; (b) electronic effect on the hypervalent iodine; and (c) dinuclear iodine reagent for diamination.
Scheme 2.
(a) Muñiz’s general combination of reagent for alkene diamination; (b) electronic effect on the hypervalent iodine; and (c) dinuclear iodine reagent for diamination.

Scheme 3.
(a) Blakey’s alkene diamination protocol and (b) Chang’s intramolecular diamination.

Scheme 4.
Wirth’s first catalytic intramolecular diamination.

Scheme 5.
Miao’s C60 diamination.

Scheme 6.
(a) Alkene diamination—indoline synthesis; (b) intermolecular diamination; and (c) azaindoline synthesis.
Scheme 6.
(a) Alkene diamination—indoline synthesis; (b) intermolecular diamination; and (c) azaindoline synthesis.

Scheme 7.
The first PhI(OAc)2 catalyzed intermolecular aminofunctionalization.

Scheme 8.
FIFA-mediated (Bis(trifluoroacetoxy)iodo)benzene) endo-selective aminooxygenation.

Scheme 9.
The first example of stereoselective aminooxygenation.

Scheme 10.
(a) Diastereoselective alkene aminooxygenation and (b) anti-selective aminofluorination.

Scheme 11.
(a) Intramolecular oxamidation and (b) application to the synthesis of natural product.

Scheme 12.
(a) First aminofluorination using stoichiometric iodine reagent; (b) asymmetric aminofluorination; and (c) catalytic aminofluorination.
Scheme 12.
(a) First aminofluorination using stoichiometric iodine reagent; (b) asymmetric aminofluorination; and (c) catalytic aminofluorination.

Scheme 13.
(a) Aminofluorination of homoallylamine and (b) catalytic enantioselective aminofluorination.
Scheme 13.
(a) Aminofluorination of homoallylamine and (b) catalytic enantioselective aminofluorination.

Scheme 14.
(a) Haloamination (F, Cl, Br, and I) of alkene; (b) iodoamination; and (c) chloro(bromo)amination.
Scheme 14.
(a) Haloamination (F, Cl, Br, and I) of alkene; (b) iodoamination; and (c) chloro(bromo)amination.

Scheme 15.
Wirth’s enantioselective thioamination.

Scheme 16.
(a) Oxazoline and thiazoline synthesis; (b) Ritter-type amidation; and (c) sulfeno- and selenofunctionalization.
Scheme 16.
(a) Oxazoline and thiazoline synthesis; (b) Ritter-type amidation; and (c) sulfeno- and selenofunctionalization.

Scheme 17.
(a) Enantioseleictive oxylactonization and (b) enantioselective Prévost and Woodward reaction.
Scheme 17.
(a) Enantioseleictive oxylactonization and (b) enantioselective Prévost and Woodward reaction.

Scheme 18.
(a) Synthesis of 4-hydroxymellein, monocerin and (b) total synthesis of (12S)-12-hydroxymonocerin and (12R)-12-hydroxymonocerin.
Scheme 18.
(a) Synthesis of 4-hydroxymellein, monocerin and (b) total synthesis of (12S)-12-hydroxymonocerin and (12R)-12-hydroxymonocerin.

Scheme 19.
(a)Total synthesis of 4-hydroxymellein and 4-hydroxy-6-methoxymellein and (b) substituted 1,3-diozolane synthesis.
Scheme 19.
(a)Total synthesis of 4-hydroxymellein and 4-hydroxy-6-methoxymellein and (b) substituted 1,3-diozolane synthesis.

Scheme 20.
(a) N-Alkenylamide oxidative cyclization and (b) enantioselective variation.

Scheme 21.
(a) First catalytic phosphoryloxylactonization and (b) enantioselective sufonyloxylactonizaiton and phosphoryloxylactonization.
Scheme 21.
(a) First catalytic phosphoryloxylactonization and (b) enantioselective sufonyloxylactonizaiton and phosphoryloxylactonization.

Scheme 22.
Diastereoselective diacetoxylation.

Scheme 23.
(a) First catalytic enantioselective diacetoxylation; (b) newly designed adamantly based chiral iodine; and (c) coordinating Lewis base-assisted iodine catalyst.
Scheme 23.
(a) First catalytic enantioselective diacetoxylation; (b) newly designed adamantly based chiral iodine; and (c) coordinating Lewis base-assisted iodine catalyst.

Scheme 24.
Enantioselective dioxytosylation.

Scheme 25.
Fujita’s cycloetherification.

Scheme 26.
(a) Vinylphenol difunctionalization via quinone methide and (b) oxidative coupling via quinonium intermediate.
Scheme 26.
(a) Vinylphenol difunctionalization via quinone methide and (b) oxidative coupling via quinonium intermediate.

Scheme 27.
(a) Zhdankin’s cycloaddition and (b) Yan’s cycloaddition.

Scheme 28.
(a) Ethoxybromination; (b) ethoxychlorination; (c) diazidation; and (d) oxiazidation.

Scheme 29.
(a) Enantioselective oxyarylation and (b) acyloxy C–C bond formation.

Scheme 30.
Catalytic enantioselective fluorolactonization.

Scheme 31.
Catalytic oxyselenylation.

Scheme 32.
Nicolaou’s enantioselective dichlorination.

Scheme 33.
(a) Gilmour’s initial difluorination; (b) Gilmour’s enantioselective variation; (c) Jacobsen’s difluorination; and (d) Jacobsen’s enantioselective variation.
Scheme 33.
(a) Gilmour’s initial difluorination; (b) Gilmour’s enantioselective variation; (c) Jacobsen’s difluorination; and (d) Jacobsen’s enantioselective variation.

Scheme 34.
Cariou’s selective dibromination of polyprenoids.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, J.H.; Choi, S.; Hong, K.B. Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years. Molecules 2019, 24, 2634. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24142634
AMA Style
Lee JH, Choi S, Hong KB. Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years. Molecules. 2019; 24(14):2634. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24142634
Chicago/Turabian StyleLee, Ji Hoon, Sungwook Choi, and Ki Bum Hong. 2019. "Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years" Molecules 24, no. 14: 2634. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24142634