Alcohol-Supported Cu-Mediated 18F-Fluorination of Iodonium Salts under “Minimalist” Conditions

 , ,
, ,
Abstract
:1. Introduction
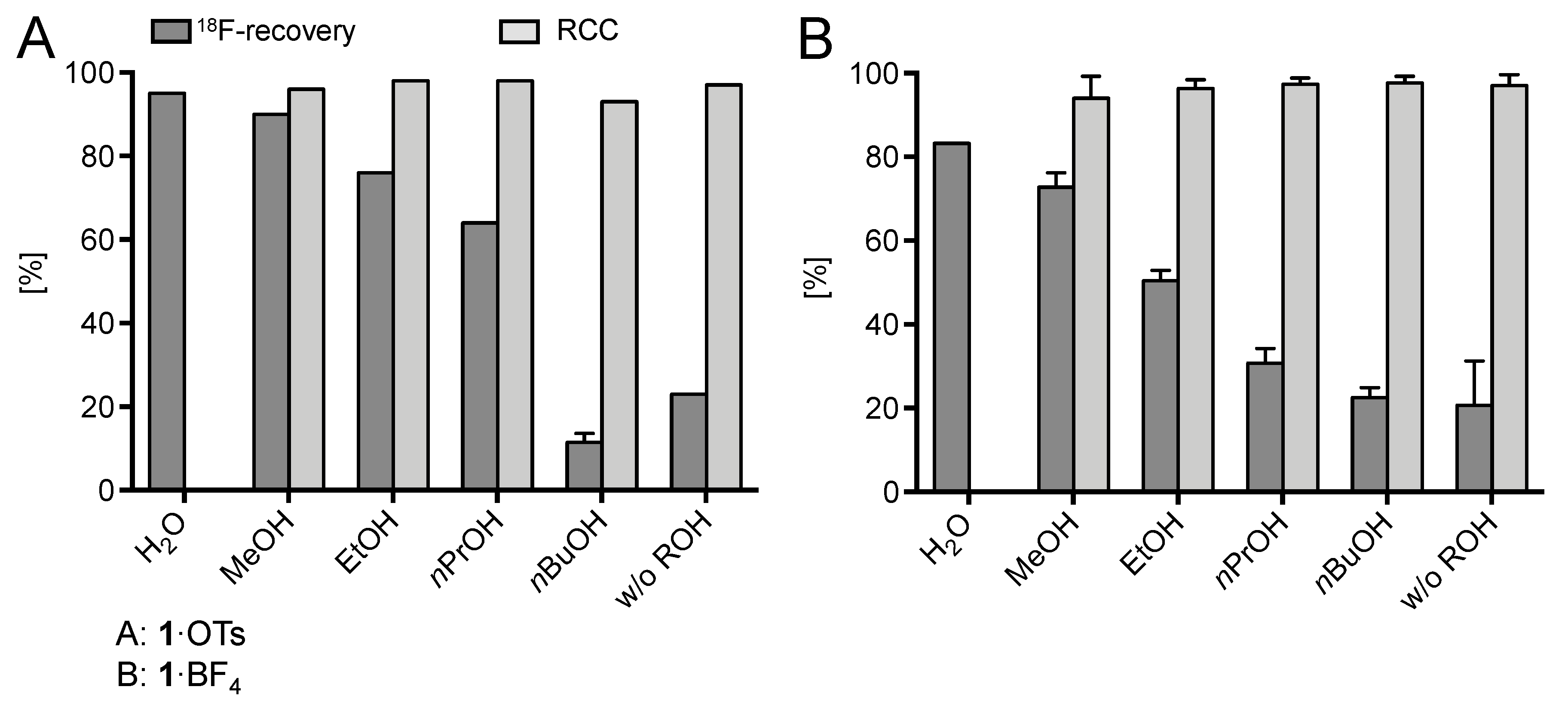
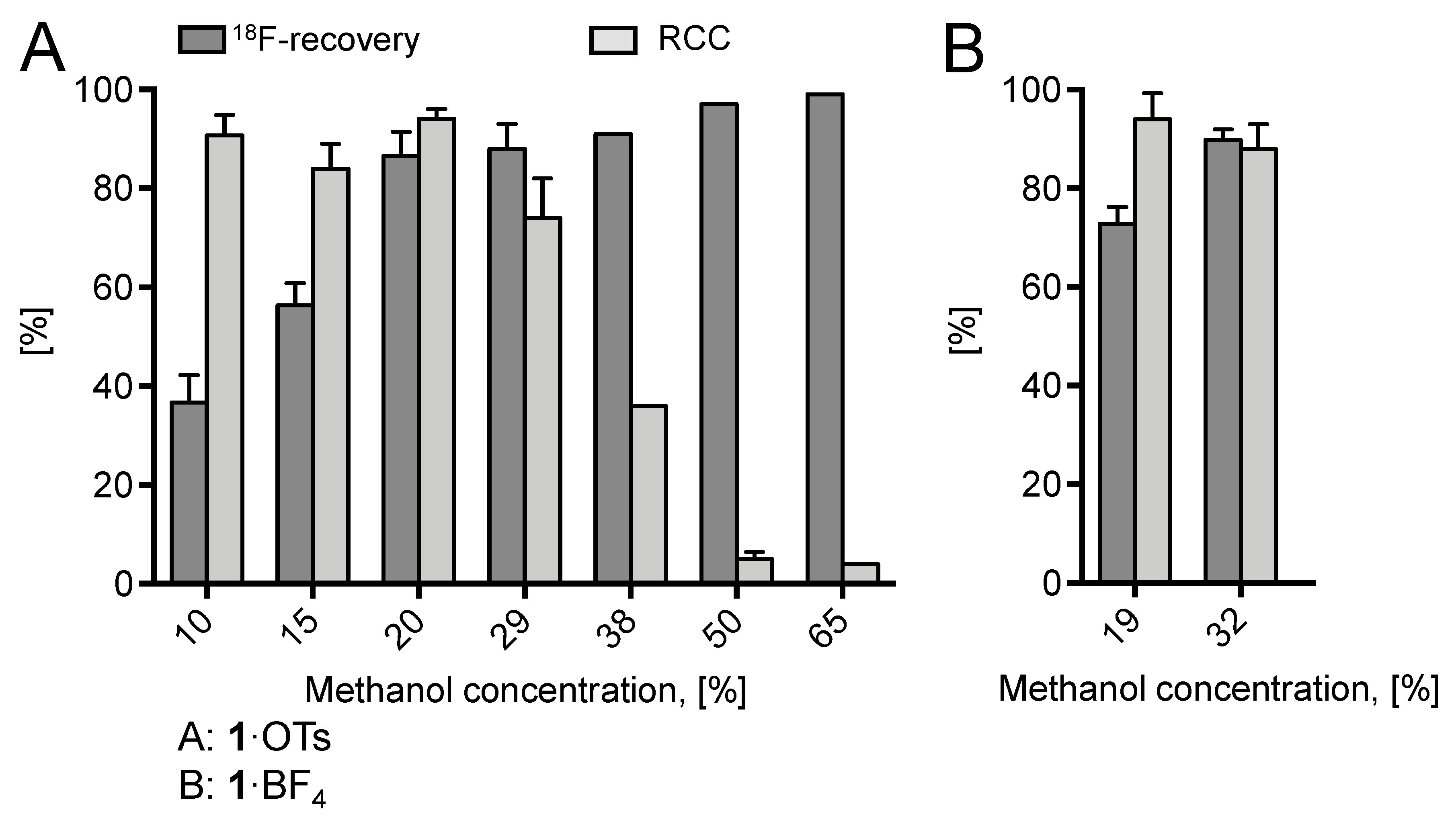
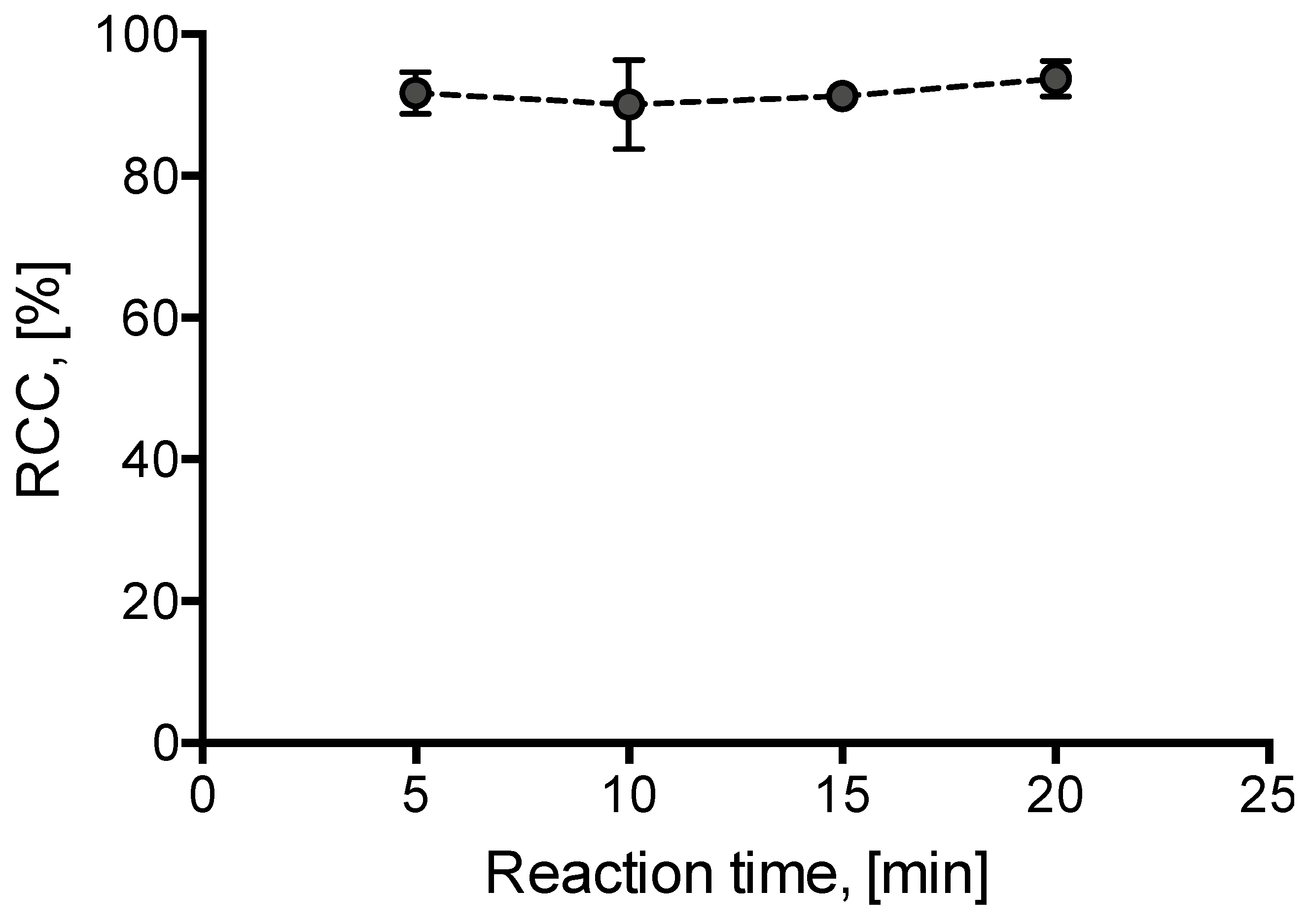
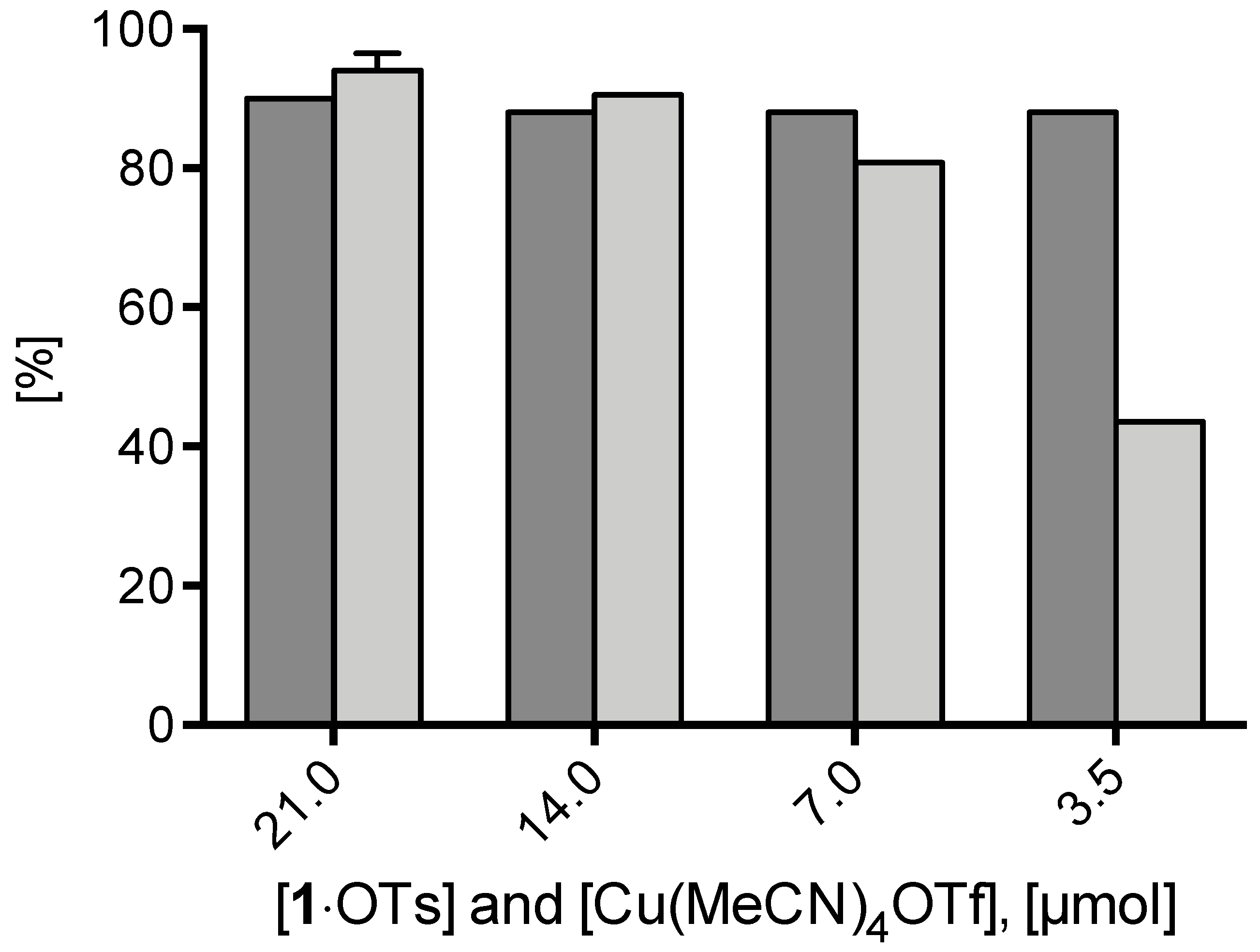
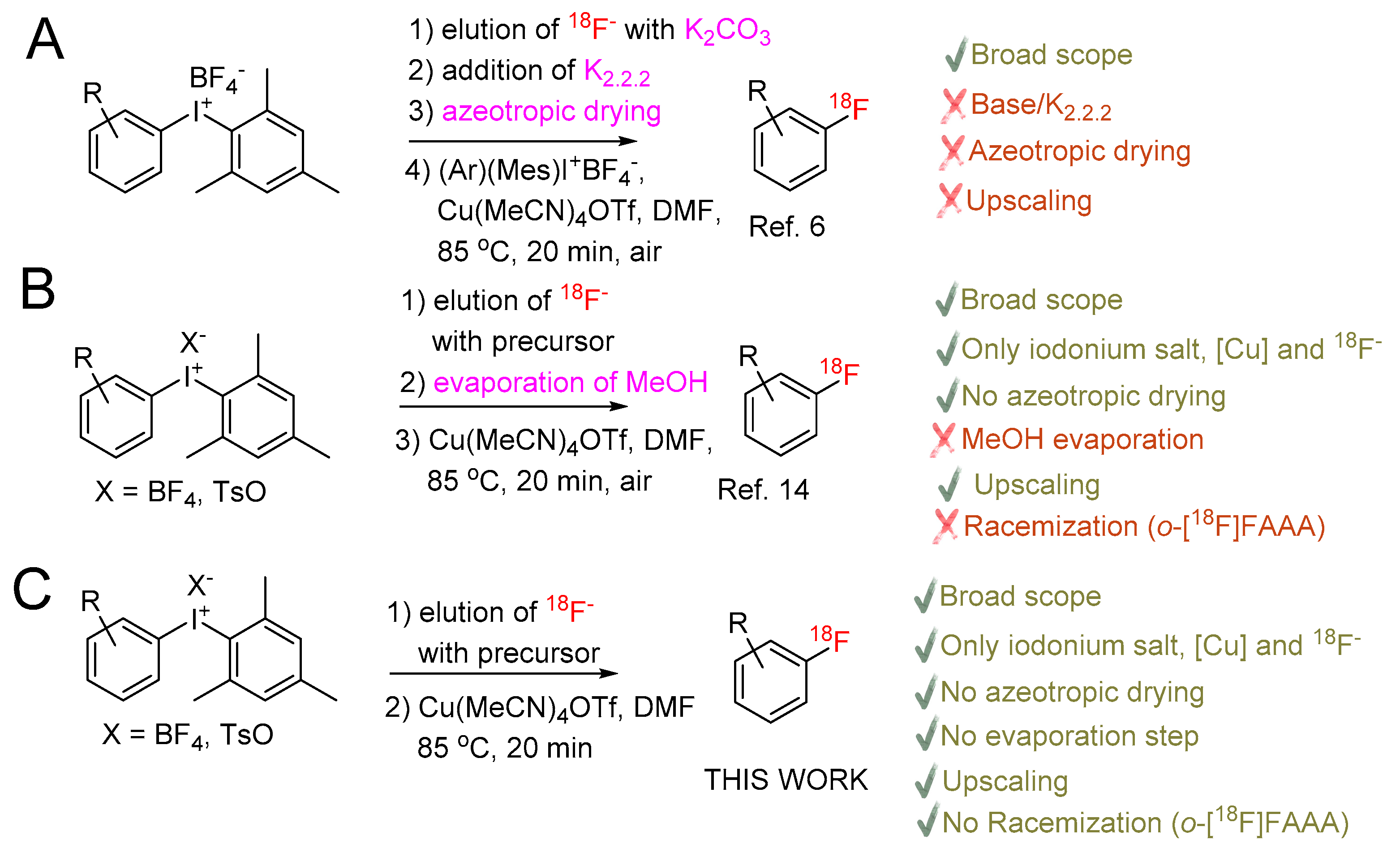
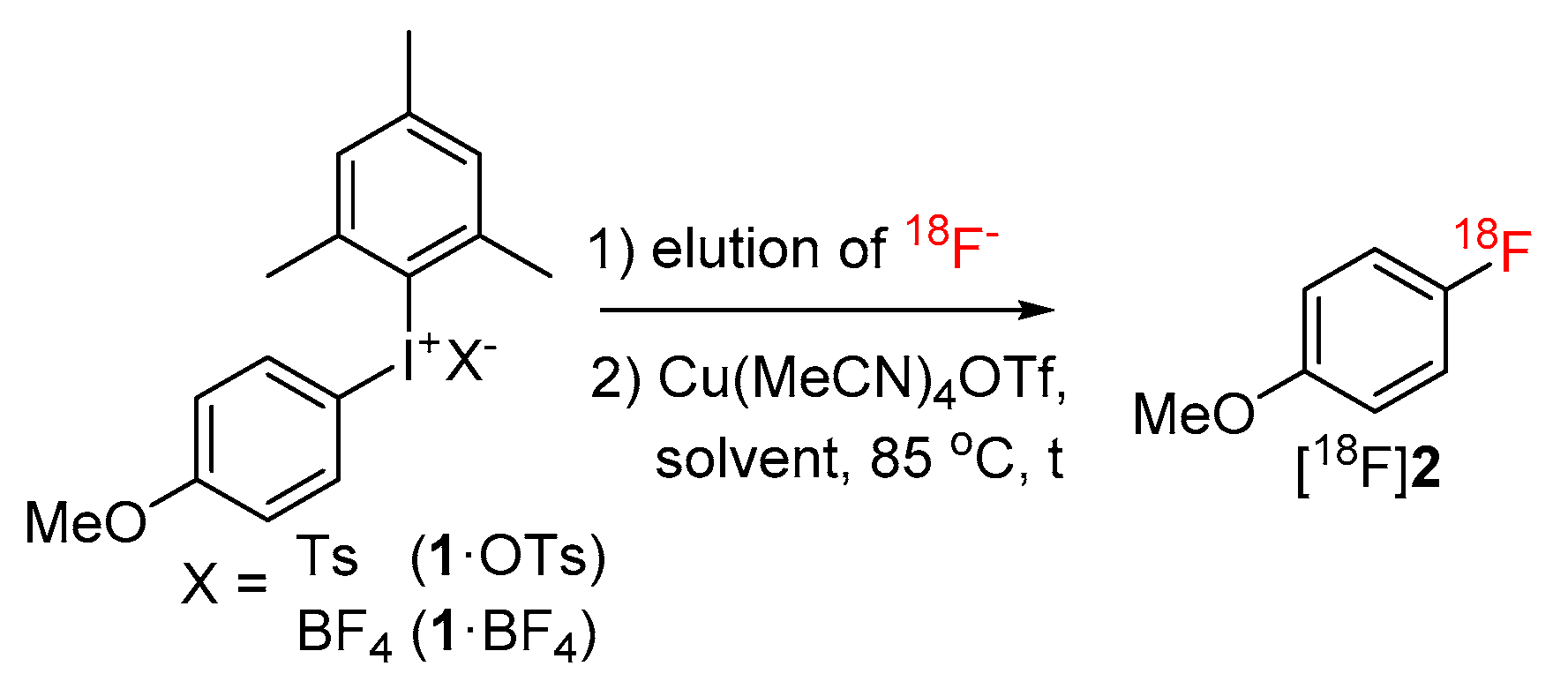
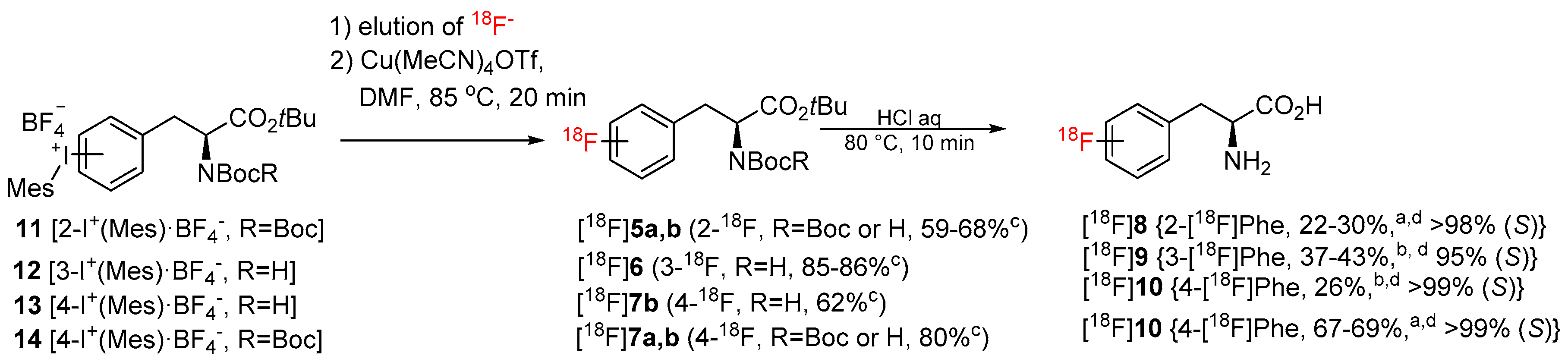
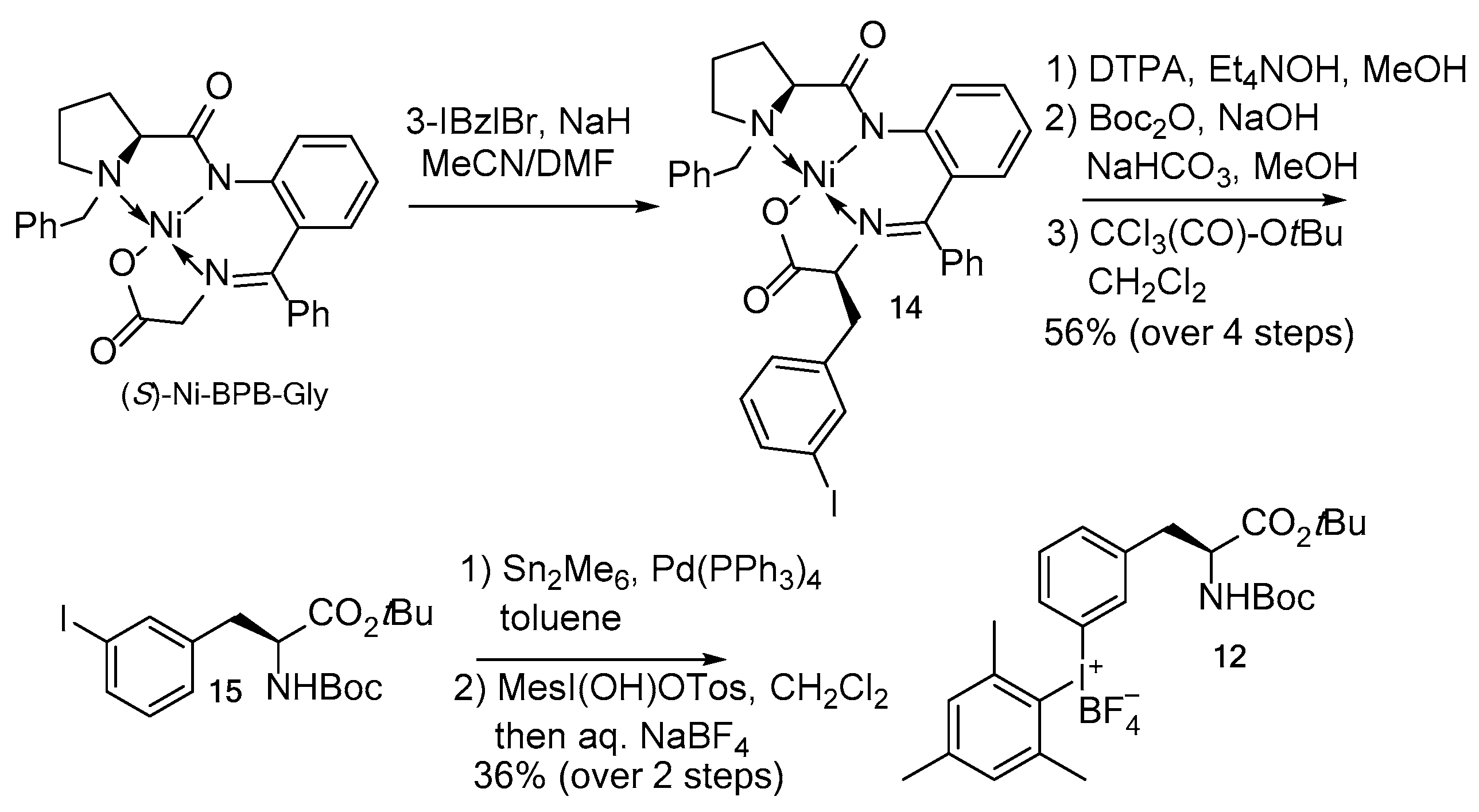
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Nuclear Magnetic Resonance
3.3. Mass Spectroscopy
3.4. Chemistry
3.4.1. (S,S)-Ni-BPB-3-IPhe (14)
3.4.2. Boc-3IPhe-OtBu (15)
3.4.3. 3-{2-[(2S)-2-{[(tert-Butoxy)carbonyl]amino}-3-(tert-butoxy)-3-oxopropyl]phenyl}(2,4,6-trime-thylphenyl)iodanium tetrafluoroborate (12)
3.5. Radiochemistry
3.5.1. General
3.5.2. TLC Analysis
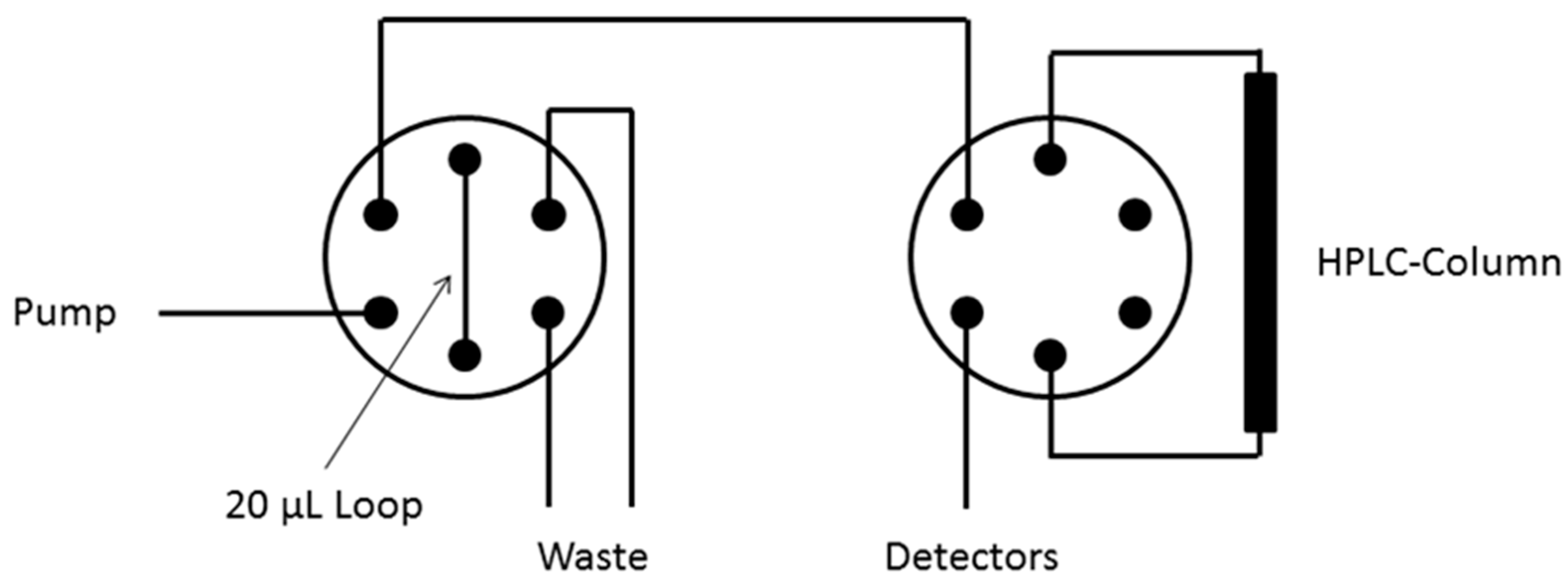
3.5.3. HPLC Analysis
3.5.4. Determination of Enantiomeric Purity
3.5.5. Manual Radiosynthesis. Screening of the Reaction Conditions and Reaction Scope—General Procedure 1 (GP 1)
3.5.6. Manual Radiosynthesis. Preparation of 2-[18F]FPhe ([18F]8) and 4-[18F]FPhe ([18F]10)–General Procedure 2 (GP 2)
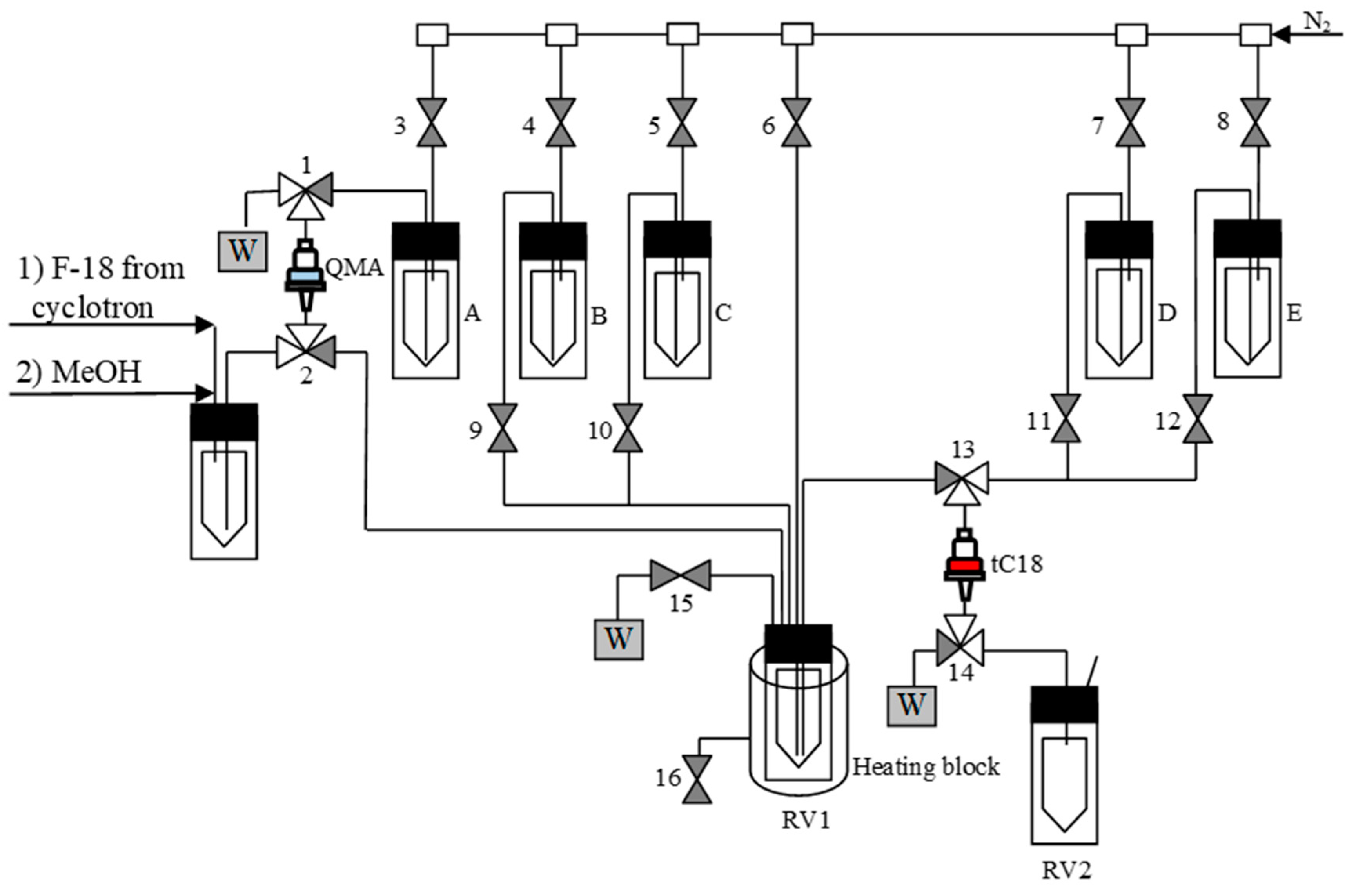
3.5.7. Semi-Automated Synthesis of 18F-Labeled Products–General Procedure 3 (GP 3) (Figure 6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, E.; Kamlet, A.S.; Powers, D.C.; Neumann, C.N.; Boursalian, G.B.; Furuya, T.; Choi, D.C.; Hooker, J.M.; Ritter, T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamlet, A.S.; Neumann, C.N.; Lee, E.; Carlin, S.M.; Moseley, C.K.; Stephenson, N.; Hooker, J.M.; Ritter, T. Application of Palladium-Mediated 18F-Fluorination to PET Radiotracer Development: Overcoming Hurdles to Translation. PLoS ONE 2013, 8, e59187. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.J.; Lazari, M.; Ren, H.; Narayanam, M.K.; Murphy, J.M.; van Dam, R.M.; Hooker, J.M.; Ritter, T. A Transmetalation Reaction Enables the Synthesis of [18F]5-Fluorouracil from [18F]Fluoride for Human PET Imaging. Organometallics 2016, 35, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hooker, J.M.; Ritter, T. Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F] Fluoride. J. Am. Chem. Soc. 2012, 134, 17456–17458. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Urusova, E.A.; Endepols, H.; Kordys, E.; Frauendorf, H.; Mottaghy, F.M.; Neumaier, B. A Practical One-Pot Synthesis of Positron Emission Tomography (PET) Tracers via Nickel-Mediated Radiofluorination. ChemistryOpen 2015, 4, 457–462. [Google Scholar] [CrossRef]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F-Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Makaravage, K.J.; Miller, J.M.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- McCammant, M.S.; Thompson, S.; Brooks, A.F.; Krska, S.W.; Scott, P.J.H.; Sanford, M.S. Cu-Mediated C–H 18F-Fluorination of Electron-Rich (Hetero)arenes. Org. Lett. 2017, 19, 3939–3942. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Makaravage, K.J.; Brooks, A.F.; Scott, P.J.H.; Sanford, M.S. Copper-Mediated Aminoquinoline-Directed Radiofluorination of Aromatic C−H Bonds with K18F. Angew. Chem. Int. Ed. 2019, 58, 3119–3122. [Google Scholar] [CrossRef] [PubMed]
- Antuganov, D.; Zykov, M.; Timofeev, V.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Fedorova, O.; Krasikova, R. Copper-Mediated Radiofluorination of Aryl Pinacolboronate Esters: A Straightforward Protocol by Using Pyridinium Sulfonates. Eur. J. Org. Chem. 2019, 2019, 918–922. [Google Scholar] [CrossRef]
- Antuganov, D.; Zykov, M.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Krasikova, R. Effect of Pyridine Addition on the Efficiency of Copper-Mediated Radiofluorination of Aryl Pinacol Boronates. ChemistrySelect 2017, 2, 7909–7912. [Google Scholar] [CrossRef]
- Preshlock, S.; Calderwood, S.; Verhoog, S.; Tredwell, M.; Huiban, M.; Hienzsch, A.; Gruber, S.; Wilson, T.C.; Taylor, N.J.; Cailly, T.; et al. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun. 2016, 52, 8361–8364. [Google Scholar] [CrossRef]
- Zarrad, F.; Zlatopolskiy, B.D.; Krapf, P.; Zischler, J.; Neumaier, B. A Practical Method for the Preparation of 18F-Labeled Aromatic Amino Acids from Nucleophilic [18F]Fluoride and Stannyl Precursors for Electrophilic Radiohalogenation. Molecules 2017, 22, 2231. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Bernard-Gauthier, V.; Bailey, J.J.; Ichiishi, N.; Schirrmacher, R.; Sanford, M.S.; Scott, P.J.H. Automated synthesis of PET radiotracers by copper-mediated 18F-fluorination of organoborons: Importance of the order of addition and competing protodeborylation. J. Labelled Compd. Radiopharm. 2018, 61, 228–236. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Ichiishi, N.; Makaravage, K.J.; Sanford, M.S.; Scott, P.J.H. Development of Customized [18F]Fluoride Elution Techniques for the Enhancement of Copper-Mediated Late-Stage Radiofluorination. Sci. Rep. 2017, 7, 233. [Google Scholar] [CrossRef]
- Richarz, R.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Neumaier, B.; Zlatopolskiy, B.D. Neither azeotropic drying, nor base nor other additives: A minimalist approach to 18F-labeling. Org. Biomol. Chem. 2014, 12, 8094–8099. [Google Scholar] [CrossRef]
- Zischler, J.; Krapf, P.; Richarz, R.; Zlatopolskiy, B.D.; Neumaier, B. Automated synthesis of 4-[18F]fluoroanisole, [18F]DAA1106 and 4-[18F]FPhe using Cu-mediated radiofluorination under “minimalist” conditions. Appl. Radiat. Isot. 2016, 115, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Schäfer, D.; Urusova, E.A.; Guliyev, M.; Bannykh, O.; Endepols, H.; Neumaier, B. Discovery of 7-[18F]Fluorotryptophan as a Novel Positron Emission Tomography (PET) Probe for the Visualization of Tryptophan Metabolism in Vivo. J. Med. Chem. 2018, 61, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Richarz, R.; Lauchner, K.; Neumaier, B. Minimalist approach meets green chemistry: Synthesis of 18F- labeled (hetero)aromatics in pure ethanol. J. Labelled Compd. Radiopharm. 2019, 62, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Corbett, C.A.; Martínez, T.J.; Lisy, J.M. Solvation of the Fluoride Anion by Methanol. J. Phys. Chem. A 2002, 106, 10015–10021. [Google Scholar] [CrossRef]
- Ichiishi, N.; Canty, A.J.; Yates, B.F.; Sanford, M.S. Mechanistic Investigations of Cu-Catalyzed Fluorination of Diaryliodonium Salts: Elaborating the CuI/CuIII Manifold in Copper Catalysis. Organometallics 2014, 33, 5525–5534. [Google Scholar] [CrossRef] [PubMed]
- Belokon, Y.N.; Tararov, V.I.; Maleev, V.I.; Savel’eva, T.F.; Ryzhov, M.G. Improved procedures for the synthesis of (S)-2-[N-(N′-benzylprolyl)amino]benzophenone (BPB) and Ni(II) complexes of Schiff’s bases derived from BPB and amino acids. Tetrahedron: Asymmetry 1998, 9, 4249–4252. [Google Scholar] [CrossRef]
- Modemann, D.J.; Zlatopolskiy, B.D.; Urusova, E.A.; Zischler, J.; Craig, A.; Ermert, J.; Guliyev, M.; Endepols, H.; Neumaier, B. 2-[18F]Fluorophenylalanine: Synthesis by Nucleophilic 18F-Fluorination and Preliminary Biological Evaluation. Synthesis 2019, 51, 664–676. [Google Scholar] [CrossRef]
- Lindstedt, E.; Reitti, M.; Olofsson, B. One-Pot Synthesis of Unsymmetric Diaryliodonium Salts from Iodine and Arenes. J. Org. Chem. 2017, 82, 11909–11914. [Google Scholar] [CrossRef]
- Ichiishi, N.; Canty, A.J.; Yates, B.F.; Sanford, M.S. Cu-Catalyzed Fluorination of Diaryliodonium Salts with KF. Org. Lett. 2013, 15, 5134–5137. [Google Scholar] [CrossRef] [Green Version]
- Merritt, E.A.; Carneiro, V.M.T.; Silva, L.F.; Olofsson, B. Facile Synthesis of Koser’s Reagent and Derivatives from Iodine or Aryl Iodides. J. Org. Chem. 2010, 75, 7416–7419. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Murai, Y.; Yoshida, T.; Ishida, A.; Masuda, K.; Sakihama, Y.; Hashidoko, Y.; Hatanaka, Y.; Hashimoto, M. Alternative One-Pot Synthesis of (Trifluoromethyl)phenyldiazirines from Tosyloxime Derivatives: Application for New Synthesis of Optically Pure Diazirinylphenylalanines for Photoaffinity Labeling. Org. Lett. 2015, 17, 616–619. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all compounds are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Product | 18F-Recovery (%) | RCC (TLC) (%) | RCC (HPLC) (%) |
|---|---|---|---|---|
| 1*a |  | 73 ± 3 (n = 3) | 94±3 (n = 3) | >99 (n = 3) |
| 2**b |  | 94 ± 2 (n = 3) | – | 94 ± 5 (n = 3) |
| 3**b |  | – | >99 (n = 3) | |
| 4**b |  | 72 ± 9 (n = 3) | 89 ± 5 (n = 3) | |
| 5**a,b |  | 87, 96 (n = 2) | 59, 68 (n = 2) | – |
| 6**a,b |  | 89, 92 (n = 2) | 85, 86 (n = 2) | – |
| 7**b |  | 92 (n = 1) | 62, 80 (n = 2) | 66 (n=1) |
| 8**a,b |  | 86 ± 6 (n = 3) | 80, 98 (n = 2) | 99 |
| Compound | Rf |
|---|---|
| [18F]2 | 0.4a |
| [18F]4 | 0.3a |
| [18F]5a | 0.5b |
| [18F]5b | 0.4b |
| [18F]6 | 0.4b |
| [18F]7a | 0.5b |
| [18F]7b | 0.5b |
| HPLC-Method | HPLC-Conditions |
|---|---|
| A | X-Bridge C18 column 5 µm, 150 × 4.6 mm (Waters), 0–2 min: 5% MeCN, 2–13 min: 5→90% MeCN (2.0 mL/min), UV detection: 210 nm, injection loop: 20 µL |
| B | Hydro-RP 4 µm 80 A 4.6 × 250 mm (Phenomenex), 2% EtOH (1.0 mL/min), UV detection: 210 nm, injection loop: 5 mL |
| C | Hydro-RP 4 µm 80 A 4.6 × 250 mm (Phenomenex), 6% EtOH aq + 0.1% TFA (1.0 mL/min), UV detection: 210 nm, injection loop: 20 µL |
| D | Crownpak (+) 5 μm 4.0 × 150 mm (Daicel Corporation), 0.025 M HClO4 (1.0 mL/min), UV detection: 210 nm, injection loop: 20 µL |
| Compound | HPLC-Method | k’a |
|---|---|---|
| [18F]2 | A | 9.8 |
| [18F]3 | A | 9.6 |
| [18F]4 | A | 8.0 |
| 2-FPhe | B | 5.1 |
| (R)-2-FPhe | C | 2.4 |
| (S)-2-FPhe | C | 3.7 |
| 4-FPhe | B | 6.3 |
| (R)-4-FPhe | C | 3.0 |
| (S)-4-FPhe | C | 4.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlovskaya, V.V.; Modemann, D.J.; Kuznetsova, O.F.; Fedorova, O.S.; Urusova, E.A.; Kolks, N.; Neumaier, B.; Krasikova, R.N.; Zlatopolskiy, B.D. Alcohol-Supported Cu-Mediated 18F-Fluorination of Iodonium Salts under “Minimalist” Conditions. Molecules 2019, 24, 3197. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173197
Orlovskaya VV, Modemann DJ, Kuznetsova OF, Fedorova OS, Urusova EA, Kolks N, Neumaier B, Krasikova RN, Zlatopolskiy BD. Alcohol-Supported Cu-Mediated 18F-Fluorination of Iodonium Salts under “Minimalist” Conditions. Molecules. 2019; 24(17):3197. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173197
Chicago/Turabian StyleOrlovskaya, Victoriya V., Daniel J. Modemann, Olga F. Kuznetsova, Olga S. Fedorova, Elizaveta A. Urusova, Niklas Kolks, Bernd Neumaier, Raisa N. Krasikova, and Boris D. Zlatopolskiy. 2019. "Alcohol-Supported Cu-Mediated 18F-Fluorination of Iodonium Salts under “Minimalist” Conditions" Molecules 24, no. 17: 3197. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173197