
Gold Nanoparticles as Boron Carriers for Boron Neutron Capture Therapy: Synthesis, Radiolabelling and In Vivo Evaluation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of COSAN Derivatives
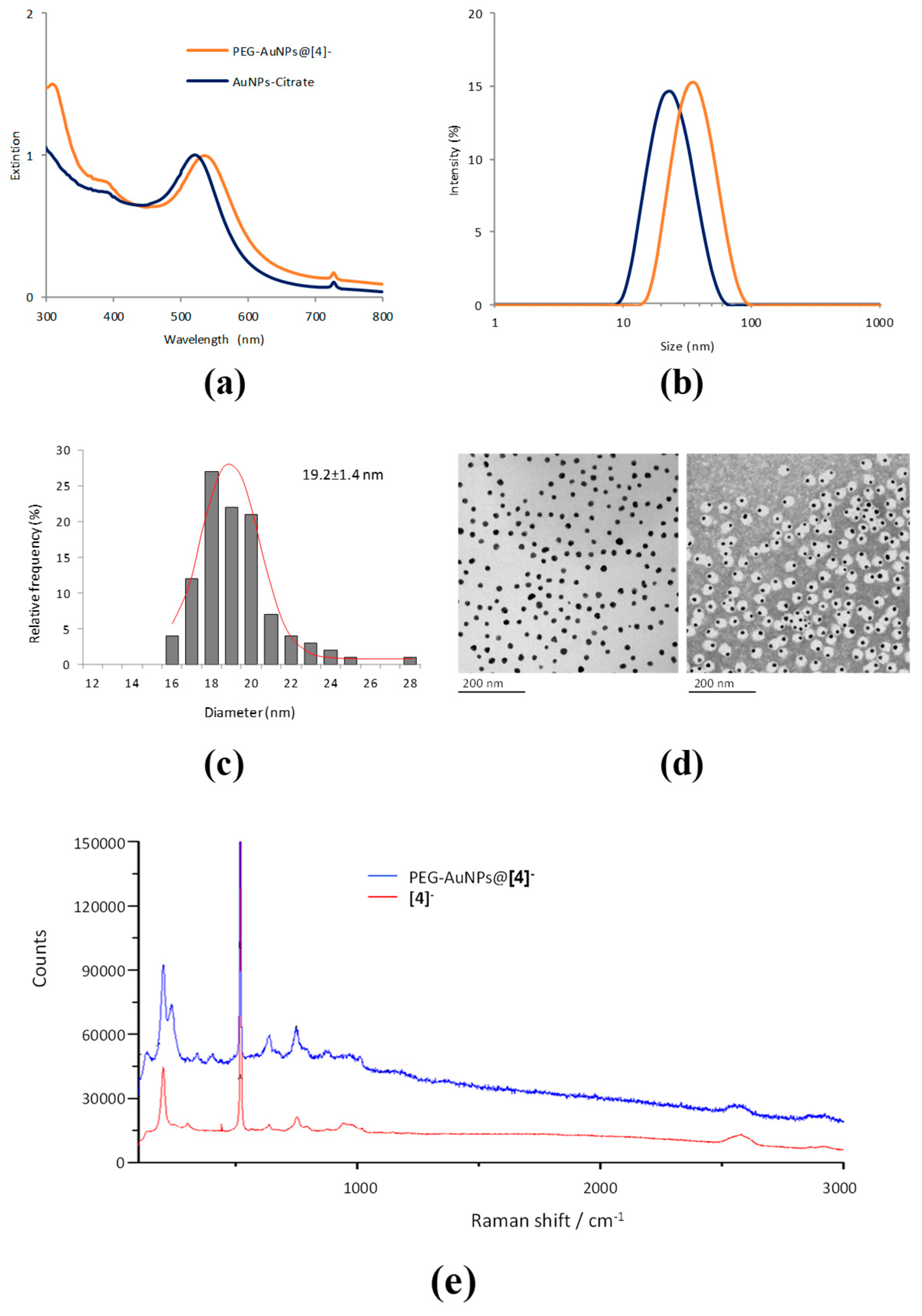
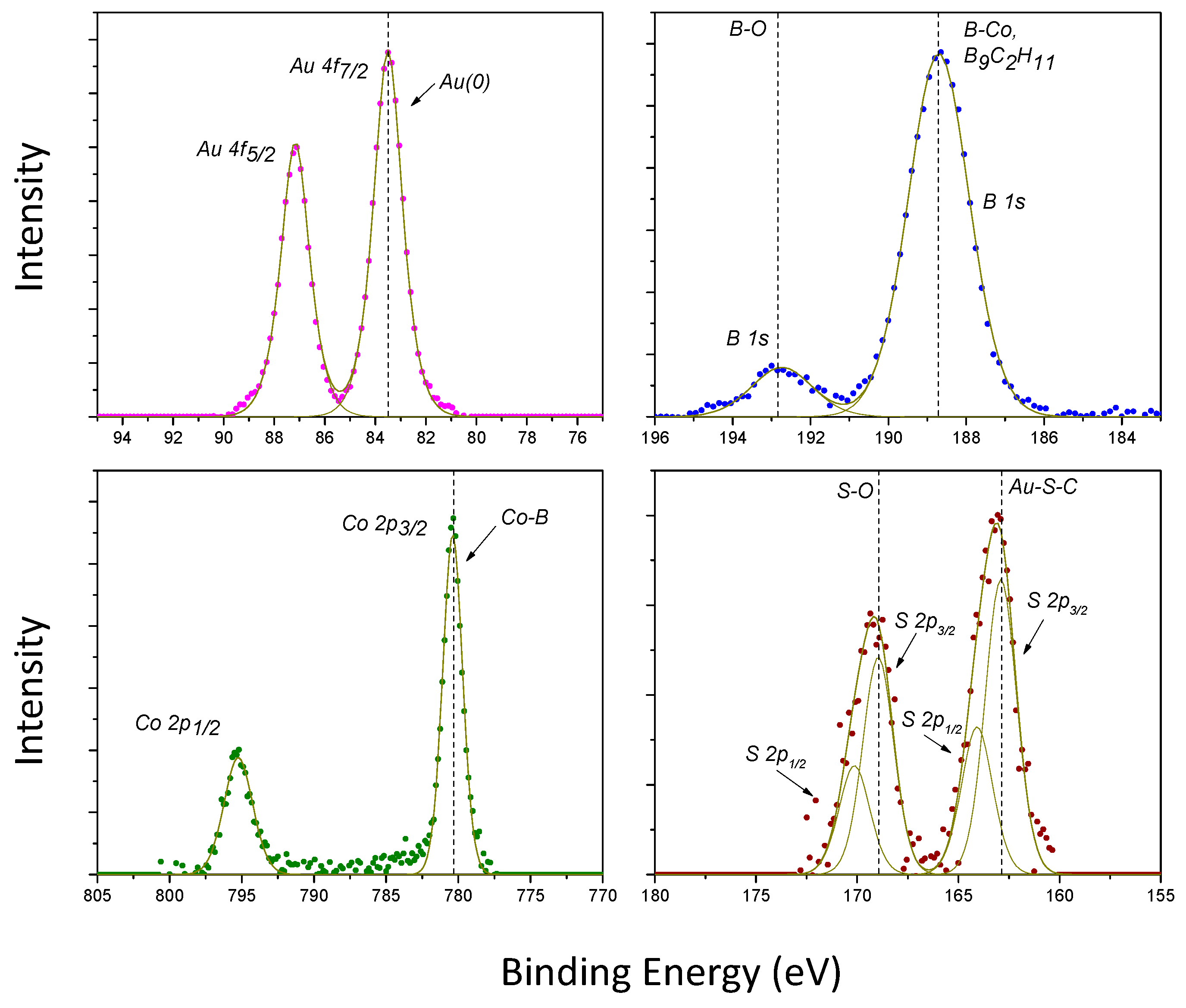
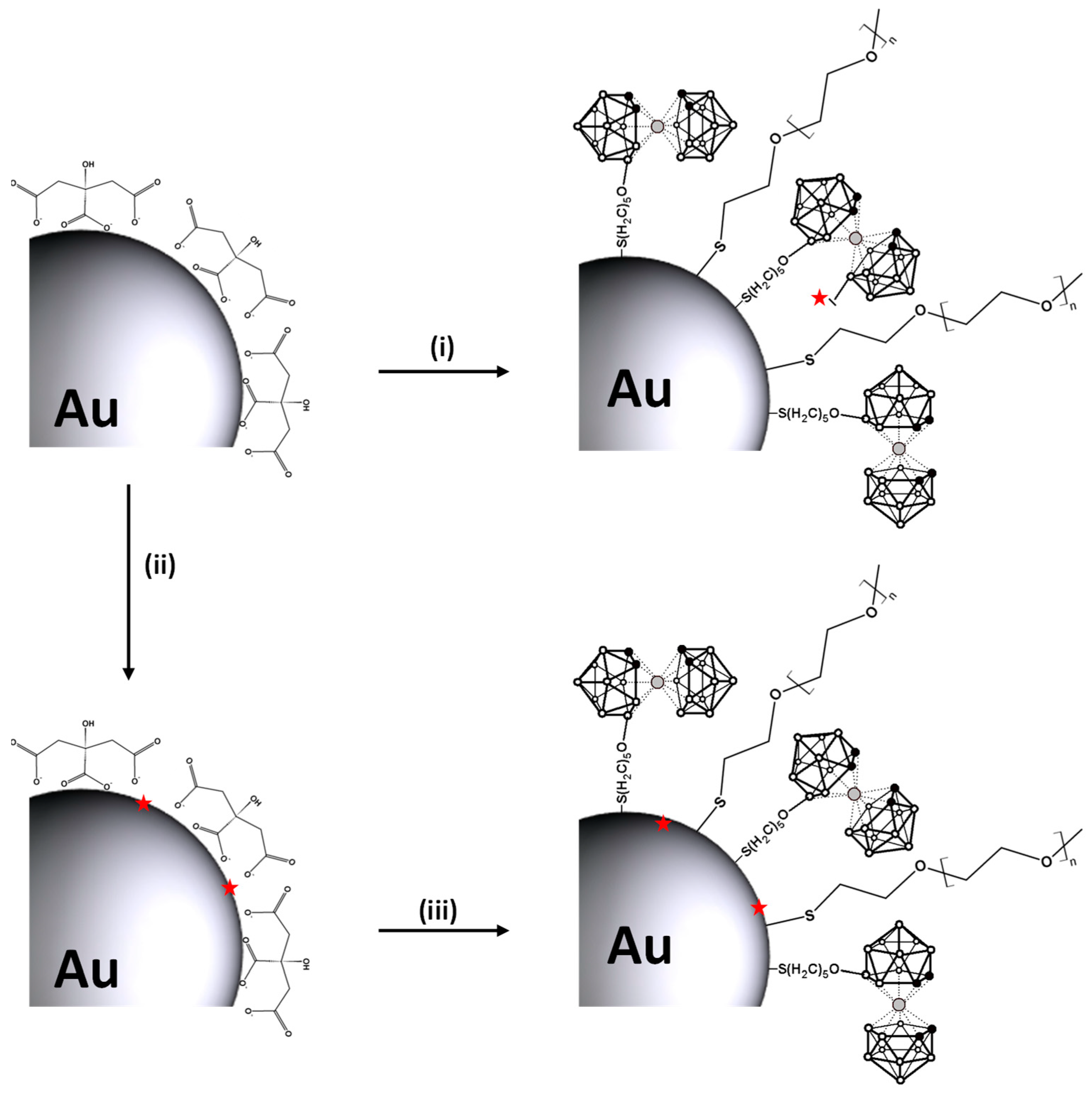
2.2. Synthesis of Functionalised AuNPs
2.3. Radiolabelling of AuNPs
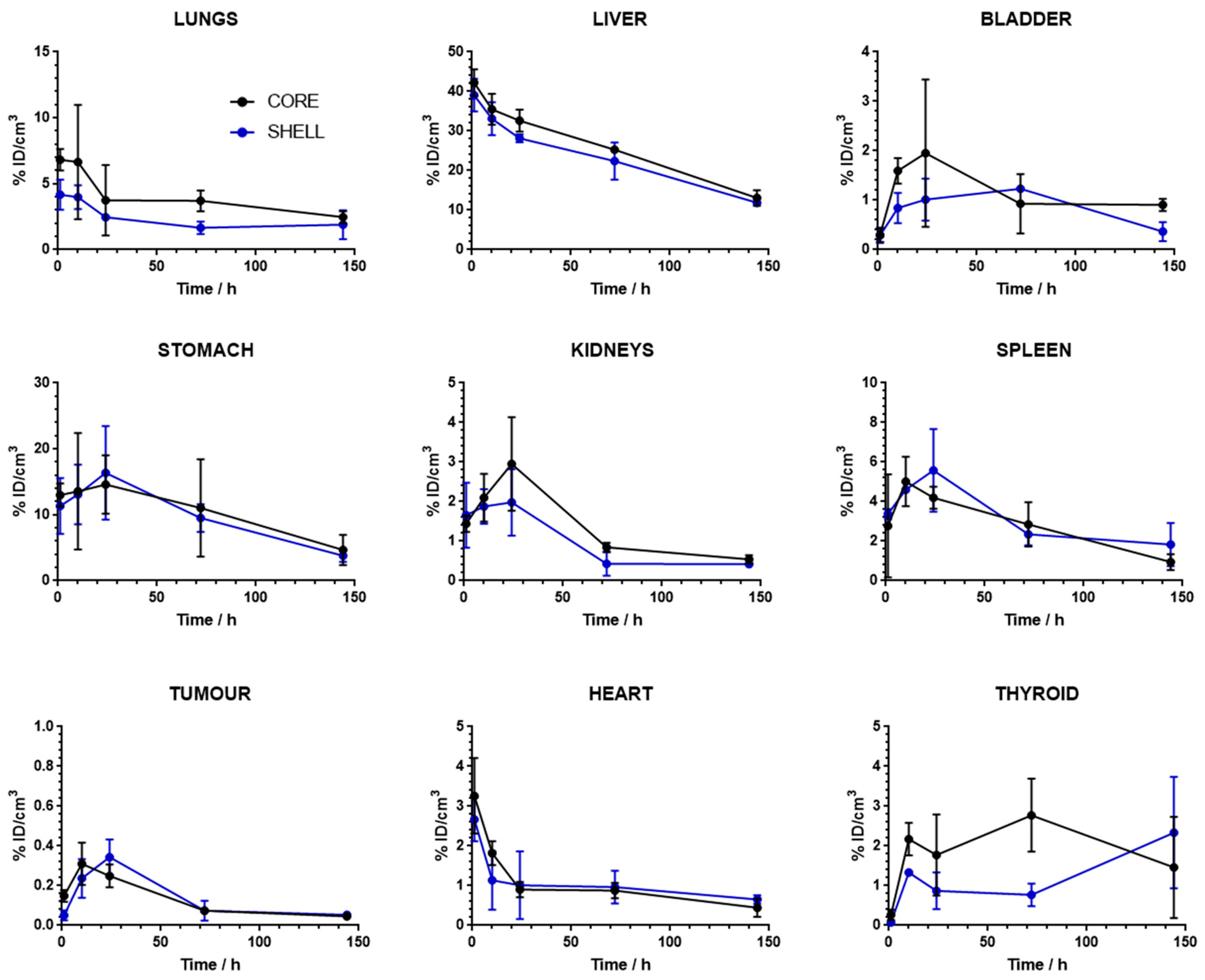
2.4. In Vivo Imaging Studies
3. Materials and Methods
3.1. Reagents
3.2. Instrumentation
3.3. Chemistry
3.3.1. Synthesis of [3]−
3.3.2. Synthesis of [4]−
3.3.3. Synthesis of [5]−
3.3.4. Synthesis of [6]−
3.4. Radiochemistry
3.4.1. Synthesis of 124I-[5]−
3.4.2. Synthesis of 124I-[6]−
3.5. Preparation of AuNPs
3.5.1. Synthesis of Citrate-Stabilized Gold NPs (CIT-AuNPs)
3.5.2. Synthesis of PEG-Stabilized, COSAN-Functionalized AuNPs (PEG-AuNPs@[4]−)
3.5.3. Synthesis of PEG-AuNPs@[4]− Labelled at the Core
3.5.4. Synthesis of PEG-AuNPs@[4]− Labelled at the Shell
3.6. In Vivo Studies
3.7. Ex Vivo Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0. Available online: http://globocan.iarc.fr. (accessed on 15 May 2013).
- Hally, C.; Rodríguez-Amigo, B.; Bresolí-Obach, R.; Planas, O.; Nos, J.; Boix-Garriga, E.; Ruiz-González, R.; Nonell, S. CHAPTER 4: Photodynamic Therapy. In RSC Drug Discovery Series; CRC Press: Boca Raton, FL, USA, 2018; Volume 2018, pp. 86–122. [Google Scholar]
- Nomoto, T.; Nishiyama, N. Photodynamic therapy. In Photochemistry for Biomedical Applications: From Device Fabrication to Diagnosis and Therapy; Springer: Berlin, Germany, 2018; pp. 301–313. [Google Scholar]
- England, C.G.; Cai, W. Theranostic nanoparticles for photothermal therapy of cancer. In Hybrid Nanomaterials: Design, Synthesis, and Biomedical Applications; CRC Press: Boca Raton, FL, USA, 2017; pp. 305–333. [Google Scholar]
- Liu, Z.; Chen, Z. Near infrared nanomaterials for photothermal therapy. In RSC Nanoscience and Nanotechnology; CRC Press: Boca Raton, FL, USA, 2016; Volume 2016, pp. 277–321. [Google Scholar]
- Locher, G.L. Biological effects and therapeutic possibilities of neutrons. Amer. J. Roentgenol. Radium Therapy Nucl. Med. 1936, 36, 1–13. [Google Scholar]
- Bregadze, V.I.; Sivaev, I.B. Polyhedral boron compounds for BNCT. In Boron Science: New Technologies and Applications; CRC Press: Boca Raton, FL, USA, 2016; pp. 181–207. [Google Scholar]
- Nakamura, H.; Kirihata, M. Boron compounds: New candidates for boron carriers in BNCT. In Neutron Capture Therapy: Principles and Applications; Springer Science & Business Media: Berlin, Germany, 2012; Volume 9783642313349, pp. 99–116. [Google Scholar]
- Yokoyama, K.; Miyatake, S.I.; Kajimoto, Y.; Kawabata, S.; Doi, A.; Yoshida, T.; Asano, T.; Kirihata, M.; Ono, K.; Kuroiwa, T. Pharmacokinetic study of BSH and BPA in simultaneous use for BNCT. J. Neurooncol. 2006, 78, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Sauerwein, W.A.G.; Bet, P.M.; Wittig, A. Drugs for BNCT: BSH and BPA. In Neutron Capture Therapy: Principles and Applications; Springer Science & Business Media: Berlin, Germany, 2012; Volume 9783642313349, pp. 117–160. [Google Scholar]
- Kueffer, P.J.; Maitz, C.A.; Khan, A.A.; Schuster, S.A.; Shlyakhtina, N.I.; Jalisatgi, S.S.; Brockman, J.D.; Nigg, D.W.; Hawthorne, M.F. Boron neutron capture therapy demonstrated in mice bearing EMT6 tumors following selective delivery of boron by rationally designed liposomes. Proc. Natl. Acad. Sci. USA 2013, 110, 6512–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitz, C.A.; Khan, A.A.; Kueffer, P.J.; Brockman, J.D.; Dixson, J.; Jalisatgi, S.S.; Nigg, D.W.; Everett, T.A.; Hawthorne, M.F. Validation and Comparison of the Therapeutic Efficacy of Boron Neutron Capture Therapy Mediated By Boron-Rich Liposomes in Multiple Murine Tumor Models. Translational Oncol. 2017, 10, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, S.; Miyoshi, T.; Koganei, H.; El-Zaria, M.E.; Viñas, C.; Suzuki, M.; Ono, K.; Nakamura, H. Spermidinium closo-dodecaborate-encapsulating liposomes as efficient boron delivery vehicles for neutron capture therapy. Chem. Commun. 2014, 50, 12325–12328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, I.; Kishi, N.; Shiokawa, K.; Uchiro, H.; Makino, K. Polyborane encapsulated liposomes prepared using pH gradient and reverse-phase evaporation for boron neutron capture therapy: biodistribution in tumor-bearing mice. Colloid. Polym. Sci. 2018, 296, 1137–1144. [Google Scholar] [CrossRef]
- Takeuchi, I.; Tomoda, K.; Matsumoto, K.; Uchiro, H.; Makino, K. PEGylated liposomes prepared with polyborane instead of cholesterol for BNCT: characteristics and biodistribution evaluation. Colloid. Polym. Sci. 2016, 294, 1679–1685. [Google Scholar] [CrossRef]
- Yannopoulos, S.N.; Zouganelis, G.D.; Nurmohamed, S.; Smith, J.R.; Bouropoulos, N.; Calabrese, G.; Fatouros, D.G.; Tsibouklis, J. Physisorbed o-carborane onto lyso-phosphatidylcholine-functionalized, single-walled carbon nanotubes: A potential carrier system for the therapeutic delivery of boron. Nanotechnology 2010, 21, 085101. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.H.; Miranda, M.C.; Rocha, Z.; Leal, A.S.; Gomes, D.A.; Sousa, E.M.B. An assessment of the potential use of BNNTs for boron neutron capture therapy. Nanomaterials 2017, 7. [Google Scholar] [CrossRef]
- da Silva, W.M.; Hilário Ferreira, T.; de Morais, C.A.; Soares Leal, A.; Barros Sousa, E.M. Samarium doped boron nitride nanotubes. Appl. Radiat. Isot. 2018, 131, 30–35. [Google Scholar] [CrossRef]
- Iizumi, Y.; Okazaki, T.; Zhang, M.; Yuge, R.; Ichihashi, T.; Nakamura, M.; Ikehara, Y.; Iijima, S.; Yudasaka, M. Preparation and functionalization of boron nitride containing carbon nanohorns for boron neutron capture therapy. Carbon 2015, 93, 595–603. [Google Scholar] [CrossRef]
- Singh, B.; Kaur, G.; Singh, P.; Singh, K.; Kumar, B.; Vij, A.; Kumar, M.; Bala, R.; Meena, R.; Singh, A.; et al. Nanostructured Boron Nitride with High Water Dispersibility for Boron Neutron Capture Therapy. Sci. Rep. 2016, 6, 35535. [Google Scholar] [CrossRef] [PubMed]
- Tietze, R.; Unterweger, H.; Dürr, S.; Lyer, S.; Canella, L.; Kudejova, P.; Wagner, F.M.; Petry, W.; Taccardi, N.; Alexiou, C. Boron containing magnetic nanoparticles for neutron capture therapy-an innovative approach for specifically targeting tumors. Appl. Radiat. Isot. 2015, 106, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, J.; Xing, C.; Chai, R.; Abbas, S.; Song, T.; Tang, C.; Huang, Y. Fe3O4 nanoparticle-coated boron nitride nanospheres: Synthesis, magnetic property and biocompatibility study. Ceram. Int. 2017, 43, 6371–6376. [Google Scholar] [CrossRef]
- Mortensen, M.W.; Björkdahl, O.; Sørensen, P.G.; Hansen, T.; Jensen, M.R.; Gundersen, H.J.G.; Bjørnholm, T. Functionalization and cellular uptake of boron carbide naraoparticles. The first step toward T cell-guided boron neutron capture therapy. Bioconjugate Chem. 2006, 17, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.W.; Sørensen, P.G.; Björkdahl, O.; Jensen, M.R.; Gundersen, H.J.G.; Bjørnholm, T. Preparation and characterization of Boron carbide nanoparticles for use as a novel agent in T cell-guided boron neutron capture therapy. Appl. Radiat. Isot. 2006, 64, 315–324. [Google Scholar] [CrossRef]
- Grandi, S.; Spinella, A.; Tomasi, C.; Bruni, G.; Fagnoni, M.; Merli, D.; Mustarelli, P.; Guidetti, G.F.; Achilli, C.; Balduini, C. Synthesis and characterisation of functionalized borosilicate nanoparticles for boron neutron capture therapy applications. J. Sol-Gel Sci. Technol. 2012, 64, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Vigderman, L.; Zubarev, E.R. Therapeutic platforms based on gold nanoparticles and their covalent conjugates with drug molecules. Adv. Drug Del. Rev. 2013, 65, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Mahato, K.; Nagpal, S.; Shah, M.A.; Srivastava, A.; Maurya, P.K.; Roy, S.; Jaiswal, A.; Singh, R.; Chandra, P. Gold nanoparticle surface engineering strategies and their applications in biomedicine and diagnostics. 3 Biotech 2019, 9, 57. [Google Scholar] [CrossRef]
- Panahi, Y.; Mohammadhosseini, M.; Nejati-Koshki, K.; Abadi, A.J.N.; Moafi, H.F.; Akbarzadeh, A.; Farshbaf, M. Preparation, Surface Properties, and Therapeutic Applications of Gold Nanoparticles in Biomedicine. Drug Res. 2017, 67, 77–87. [Google Scholar] [CrossRef]
- Versiani, A.F.; Andrade, L.M.; Martins, E.M.N.; Scalzo, S.; Geraldo, J.M.; Chaves, C.R.; Ferreira, D.C.; Ladeira, M.; Guatimosim, S.; Ladeira, L.O.; et al. Gold nanoparticles and their applications in biomedicine. Future Virol. 2016, 11, 293–309. [Google Scholar] [CrossRef]
- Schmid, G.; Pugin, R.; Meyer-Zaika, A.W.; Simon, U. Clusters on clusters: closo-dodecaborate as a ligand for Au55 clusters. Eur. J. Inorg. Chem. 1999, 2051–2055. [Google Scholar] [CrossRef]
- Liang, L.; Rapakousiou, A.; Salmon, L.; Ruiz, J.; Astruc, D.; Dash, B.P.; Satapathy, R.; Sawicki, J.W.; Hosmane, N.S. “Click” assembly of carborane-appended polymers and stabilization of gold and palladium nanoparticles. Eur. J. Inorg. Chem. 2011, 3043–3049. [Google Scholar] [CrossRef]
- Cioran, A.M.; Teixidor, F.; Krpetić, Ž.; Brust, M.; Viñas, C. Preparation and characterization of Au nanoparticles capped with mercaptocarboranyl clusters. Dalton Trans. 2014, 43, 5054–5061. [Google Scholar] [CrossRef] [PubMed]
- Cioran, A.M.; Musteti, A.D.; Teixidor, F.; Krpetić, Z.; Prior, I.A.; He, Q.; Kiely, C.J.; Brust, M.; Viñas, C. Mercaptocarborane-capped gold nanoparticles: Electron pools and ion traps with switchable hydrophilicity. J. Am. Chem. Soc. 2012, 134, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Ciani, L.; Bortolussi, S.; Postuma, I.; Cansolino, L.; Ferrari, C.; Panza, L.; Altieri, S.; Ristori, S. Rational design of gold nanoparticles functionalized with carboranes for application in Boron Neutron Capture Therapy. Int. J. Pharm. 2013, 458, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhao, P.; Salmon, L.; Ruiz, J.; Zabawa, M.; Hosmane, N.S.; Astruc, D. “Click” star-shaped and dendritic PEGylated gold nanoparticle-carborane assemblies. Inorg. Chem. 2013, 52, 11146–11155. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Lin, J.-J.; Chang, W.-Y.; Hsieh, C.-Y.; Wu, C.-C.; Chen, H.-S.; Hsu, H.-J.; Yang, A.-S.; Hsu, M.-H.; Kuo, W.-Y. Development of theranostic active-targeting boron-containing gold nanoparticles for boron neutron capture therapy (BNCT). Colloids Surf. B Biointerfaces 2019, 183, 110387. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Ye, J.; Li, Z.; Jiang, H.; Yan, H.; Stogniy, M.Y.; Sivaev, I.B.; Bregadze, V.I.; Wang, X. Carborane Derivative Conjugated with Gold Nanoclusters for Targeted Cancer Cell Imaging. Biomacromolecules 2017, 18, 1466–1472. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Swain, B.R.; Mahanta, C.S.; Jena, B.B.; Hosmane, N.S. Cobalt bis(dicarbollide) anion and its derivatives. J. Organomet. Chem. 2017, 849–850, 170–194. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Chemistry of cobalt bis(dicarbollides). A review. Collect. Czech. Chem. Commun. 1999, 64, 783–805. [Google Scholar] [CrossRef]
- Llop, J.; Masalles, C.; Viñas, C.; Teixidor, F.; Sillanpää, R.; Kivekäs, R. The [3,3′-Co(1,2-C2B9H11)2]-anion as a platform for new materials: Synthesis of its functionalized monosubstituted derivatives incorporating synthons for conducting organic polymers. J. Chem. Soc. Dalton Trans. 2003, 556–561. [Google Scholar] [CrossRef]
- Gabel, D.; Foster, S.; Fairchild, R.G. The Monte Carlo simulation of the biological effect of the 10B(n,α)7Li reaction in cells and tissue and its implication for boron neutron capture therapy. Radiat. Res. 1987, 111, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Fairchild, R.G.; Bond, V.P. Current status of 10B-neutron capture therapy: Enhancement of tumor dose via beam filtration and dose rate, and the effects of these parameters on minimum boron content: A theoretical evaluation. Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 831–840. [Google Scholar] [CrossRef]
- Stylianopoulos, T. EPR-effect: Utilizing size-dependent nanoparticle delivery to solid tumors. Ther. Deliv. 2013, 4, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Homolka, T.; Bastús, N.G.; Puntes, V.; Weller, H.; Vossmeyer, T. Little adjustments significantly improve the Turkevich synthesis of gold nanoparticles. Langmuir 2014, 30, 10779–10784. [Google Scholar] [CrossRef]
- Wagner, C.D.; Naumkin, A.V.; Kraut-Vass, A.; Allison, J.W.; Powell, C.J.; Rumble, J.R.J. NIST X-ray Photoelectron Spectroscopy Database National Institute of Standards and Technology. Available online: https://srdata.nist.giv/xps/ (accessed on 25 May 2019).
- Di Mauro, P.P.; Gómez-Vallejo, V.; Baz Maldonado, Z.; Llop Roig, J.; Borrós, S. Novel 18F Labeling Strategy for Polyester-Based NPs for In Vivo PET-CT Imaging. Bioconjugate Chem. 2015, 26, 582–592. [Google Scholar] [CrossRef]
- Royo, F.; Cossío, U.; Ruiz De Angulo, A.; Llop, J.; Falcon-Perez, J.M. Modification of the glycosylation of extracellular vesicles alters their biodistribution in mice. Nanoscale 2019, 11, 1531–1537. [Google Scholar] [CrossRef] [Green Version]
- Frigell, J.; García, I.; Gómez-Vallejo, V.; Llop, J.; Penadés, S. 68Ga-labeled gold glyconanoparticles for exploring blood-brain barrier permeability: Preparation, biodistribution studies, and improved brain uptake via neuropeptide conjugation. J. Am. Chem. Soc. 2014, 136, 449–457. [Google Scholar] [CrossRef]
- Llop, J.; Gómez-Vallejo, V.; Gibson, N. Quantitative determination of the biodistribution of nanoparticles: Could radiolabeling be the answer? Nanomedicine 2013, 8, 1035–1038. [Google Scholar] [CrossRef]
- Pérez-Campaña, C.; Gómez-Vallejo, V.; Puigivila, M.; Martín, A.; Calvo-Fernández, T.; Moya, S.E.; Ziolo, R.F.; Reese, T.; Llop, J. Biodistribution of different sized nanoparticles assessed by positron emission tomography: A general strategy for direct activation of metal oxide particles. ACS Nano 2013, 7, 3498–3505. [Google Scholar] [CrossRef] [PubMed]
- Polyak, A.; Ross, T.L. Nanoparticles for SPECT and PET imaging: Towards personalized medicine and theranostics. Curr. Med. Chem. 2018, 25, 4328–4353. [Google Scholar] [CrossRef] [PubMed]
- Varani, M.; Galli, F.; Auletta, S.; Signore, A. Radiolabelled nanoparticles for cancer diagnosis. Clin. Transl. Imaging 2018, 6, 271–292. [Google Scholar] [CrossRef]
- Llop, J.; Jiang, P.; Marradi, M.; Gómez-Vallejo, V.; Echeverría, M.; Yu, S.; Puigivila, M.; Baz, Z.; Szczupak, B.; Pérez-Campaña, C.; et al. Visualisation of dual radiolabelled poly(lactide-co-glycolide) nanoparticle degradation In Vivo using energy-discriminant SPECT. J. Mater. Chem. B 2015, 3, 6293–6300. [Google Scholar] [CrossRef]
- Gona, K.B.; Zaulet, A.; Gómez-Vallejo, V.; Teixidor, F.; Llop, J.; Viñas, C. COSAN as a molecular imaging platform: Synthesis and “In Vivo” imaging. Chem. Commun. 2014, 50, 11415–11417. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Zhang, H.; Rajian, J.R.; Chamberland, D.L.; Sherman, P.S.; Quesada, C.A.; Koch, A.E.; Kotov, N.A.; Wang, X. 125I-labeled gold nanorods for targeted imaging of inflammation. ACS Nano 2011, 5, 8967–8973. [Google Scholar] [CrossRef]
- Agarwal, A.; Shao, X.; Rajian, J.R.; Zhang, H.; Chamberland, D.L.; Kotov, N.A.; Wang, X. Dual-mode imaging with radiolabeled gold nanorods. J. Biomed. Opt. 2011, 16, 051307. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M. Boron neutron capture therapy (BNCT): a unique role in radiotherapy with a view to entering the accelerator-based BNCT era. Int. J. Clin. Oncol. 2019, 1–8. [Google Scholar] [CrossRef]
- Kaushal, A.; Citrin, D. The Role of Radiation Therapy in the Management of Sarcomas. Surg. Clin. N. Am. 2008, 88, 629–646. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.Y.; Dorn, M.; Vogt, J.; Spemann, D.; Yu, W.; Mao, Z.W.; Estrela-Lopis, I.; Donath, E.; Gao, C.Y. A quantitative study of the intracellular concentration of graphene/noble metal nanoparticle composites and their cytotoxicity. Nanoscale 2014, 6, 8535–8542. [Google Scholar] [CrossRef]
- Veith, L.; Bottner, J.; Vennemann, A.; Breitenstein, D.; Engelhard, C.; Meijer, J.; Estrela-Lopis, I.; Wiemann, M.; Hagenhoff, B. Detection of ZrO(2) Nanoparticles in Lung Tissue Sections by Time-of-Flight Secondary Ion Mass Spectrometry and Ion Beam Microscopy. Nanomaterials 2018, 8. [Google Scholar] [CrossRef]
- Meyer, T.; Venus, T.; Sieg, H.; Bohmert, L.; Kunz, B.M.; Krause, B.; Jalili, P.; Hogeveen, K.; Chevance, S.; Gauffre, F.; et al. Simultaneous Quantification and Visualization of Titanium Dioxide Nanomaterial Uptake at the Single Cell Level in an In Vitro Model of the Human Small Intestine. Small Methods 2019, 3, 18500540. [Google Scholar] [CrossRef]
- Lichtenstein, D.; Meyer, T.; Bohmert, L.; Juling, S.; Fahrenson, C.; Selve, S.; Thunemann, A.; Meijer, J.; Estrela-Lopis, I.; Braeuning, A.; et al. Dosimetric Quantification of Coating-Related Uptake of Silver Nanoparticles. Langmuir 2017, 33, 13087–13097. [Google Scholar] [CrossRef] [PubMed]
- Estrela-Lopis, I.; Romero, G.; Rojas, E.; Moya, S.E.; Donath, E. Nanoparticle uptake and their co-localization with cell compartments—A confocal Raman microscopy study at single cell level. J. Phys. CS 2011, 304, 1–10. [Google Scholar] [CrossRef]
- Romero, G.; Estrela-Lopis, I.; Castro-Hartmann, P.; Rojas, E.; Llarena, I.; Sanz, D.; Donath, E.; Moya, S.E. Stepwise surface tailoring of carbon nanotubes with polyelectrolyte brushes and lipid layers to control their intracellular distribution and “in vitro” toxicity. Soft Matt. 2011, 7, 6883–6890. [Google Scholar] [CrossRef]
- Romero, G.; Estrela-Lopis, I.; Zhou, J.; Rojas, E.; Franco, A.; Espinel, C.S.; Fernandez, A.G.; Gao, C.Y.; Donath, E.; Moya, S.E. Surface Engineered Poly(lactide-co-glycolide) Nanoparticles for Intracellular Delivery: Uptake and Cytotoxicity—A Confocal Raman Microscopic Study. Biomacromolecules 2010, 11, 2993–2999. [Google Scholar] [CrossRef] [PubMed]
- Llop, J.; Estrela-Lopis, I.; Ziolo, R.F.; Gonzalez, A.; Fleddermann, J.; Dorn, M.; Vallejo, V.G.; Simon-Vazquez, R.; Donath, E.; Mao, Z.G.; et al. Uptake, Biological Fate, and Toxicity of Metal Oxide Nanoparticles. Part. Part. Syst. Charact. 2014, 31, 24–35. [Google Scholar] [CrossRef]
- Blucher, C.; Zilberfain, C.; Venus, T.; Spindler, N.; Dietrich, A.; Burkhardt, R.; Stadler, S.C.; Estrela-Lopis, I. Single cell study of adipose tissue mediated lipid droplet formation and biochemical alterations in breast cancer cells. Analyst 2019, 144, 5558–5570. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.M.; Camargo, N.S.; Ganassin, R.; Rocha, M.C.O.; Merker, C.; Böttner, J.; Estrela-Lopis, I.; Py-Daniel, K.R.; Jardim, K.V.; Sousa, M.H.; et al. Oily core/amphiphilic polymer shell nanocapsules change the intracellular fate of doxorubicin in breast cancer cells. J. Mater. Chem. B 2019. [Google Scholar] [CrossRef]
- Ganassin, R.; Merker, C.; Rodrigues, M.C.; Guimaraes, N.F.; Sodre, C.S.C.; Ferreira, Q.D.S.; da Silva, S.W.; Ombredane, A.S.; Joanitti, G.A.; Py-Daniel, K.R.; et al. Nanocapsules for the co-delivery of selol and doxorubicin to breast adenocarcinoma 4T1 cells in vitro. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2002–2012. [Google Scholar] [CrossRef]
- Arnida; Janát-Amsbury, M.M.; Ray, A.; Peterson, C.M.; Ghandehari, H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur. J. Pharm. Biopharm. 2011, 77, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, R.; Sat-Klopsch, Y.N.; Burger, A.M.; Bibby, M.C.; Fiebig, H.H.; Sausville, E.A. Validation of tumour models for use in anticancer nanomedicine evaluation: The EPR effect and cathepsin B-mediated drug release rate. Cancer Chemother. Pharmacol. 2013, 72, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Yanagie, H.; Maruyama, K.; Takizawa, T.; Ishida, O.; Ogura, K.; Matsumoto, T.; Sakurai, Y.; Kobayashi, T.; Shinohara, A.; Rant, J.; et al. Application of boron-entrapped stealth liposomes to inhibition of growth of tumour cells in the In Vivo boron neutron-capture therapy model. Biomed. Pharmacother. 2006, 60, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Koganei, H.; Ueno, M.; Tachikawa, S.; Tasaki, L.; Ban, H.S.; Suzuki, M.; Shiraishi, K.; Kawano, K.; Yokoyama, M.; Maitani, Y.; et al. Development of high boron content liposomes and their promising antitumor effect for neutron capture therapy of cancers. Bioconjugate Chem. 2013, 24, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Yinghuai, Z.; Peng, A.T.; Carpenter, K.; Maguire, J.A.; Hosmane, N.S.; Takagaki, M. Substituted carborane-appended water-soluble single-wall carbon nanotubes: New approach to boron neutron capture therapy drug delivery. J. Am. Chem. Soc. 2005, 127, 9875–9880. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lin, Y.; Zhu, Y.Z.; Lu, J.; Maguire, J.A.; Hosmane, N.S. Boron drug delivery via encapsulated magnetic nanocomposites: A new approach for BNCT in cancer treatment. J. Nanomater. 2010, 2010. [Google Scholar] [CrossRef]
- Gao, S.; Zhu, Y.; Hosmane, N. Nanostructured Boron Compounds for Boron Neutron Capture Therapy (BNCT) in Cancer Treatment. In Boron-Based Compounds: Potential and Emerging Applications in Medicine; Hey-Hawkins, E., Viñas, C., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2018; pp. 371–388. [Google Scholar]
- Mulware, S.J.; Baxley, J.D.; Rout, B.; Reinert, T. Efficiency calibration of an HPGe X-ray detector for quantitative PIXE analysis. Nucl. Instr. Methods Phys. Res. Sect. B-Beam Interact. Mater. Atoms 2014, 332, 95–98. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulagam, K.R.; Gona, K.B.; Gómez-Vallejo, V.; Meijer, J.; Zilberfain, C.; Estrela-Lopis, I.; Baz, Z.; Cossío, U.; Llop, J. Gold Nanoparticles as Boron Carriers for Boron Neutron Capture Therapy: Synthesis, Radiolabelling and In Vivo Evaluation. Molecules 2019, 24, 3609. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24193609
Pulagam KR, Gona KB, Gómez-Vallejo V, Meijer J, Zilberfain C, Estrela-Lopis I, Baz Z, Cossío U, Llop J. Gold Nanoparticles as Boron Carriers for Boron Neutron Capture Therapy: Synthesis, Radiolabelling and In Vivo Evaluation. Molecules. 2019; 24(19):3609. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24193609
Chicago/Turabian StylePulagam, Krishna R., Kiran B. Gona, Vanessa Gómez-Vallejo, Jan Meijer, Carolin Zilberfain, Irina Estrela-Lopis, Zuriñe Baz, Unai Cossío, and Jordi Llop. 2019. "Gold Nanoparticles as Boron Carriers for Boron Neutron Capture Therapy: Synthesis, Radiolabelling and In Vivo Evaluation" Molecules 24, no. 19: 3609. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24193609