
Clofibrate Treatment Decreases Inflammation and Reverses Myocardial Infarction-Induced Remodelation in a Rodent Experimental Model

 , ,
, ,  ,
,
Abstract
:1. Introduction
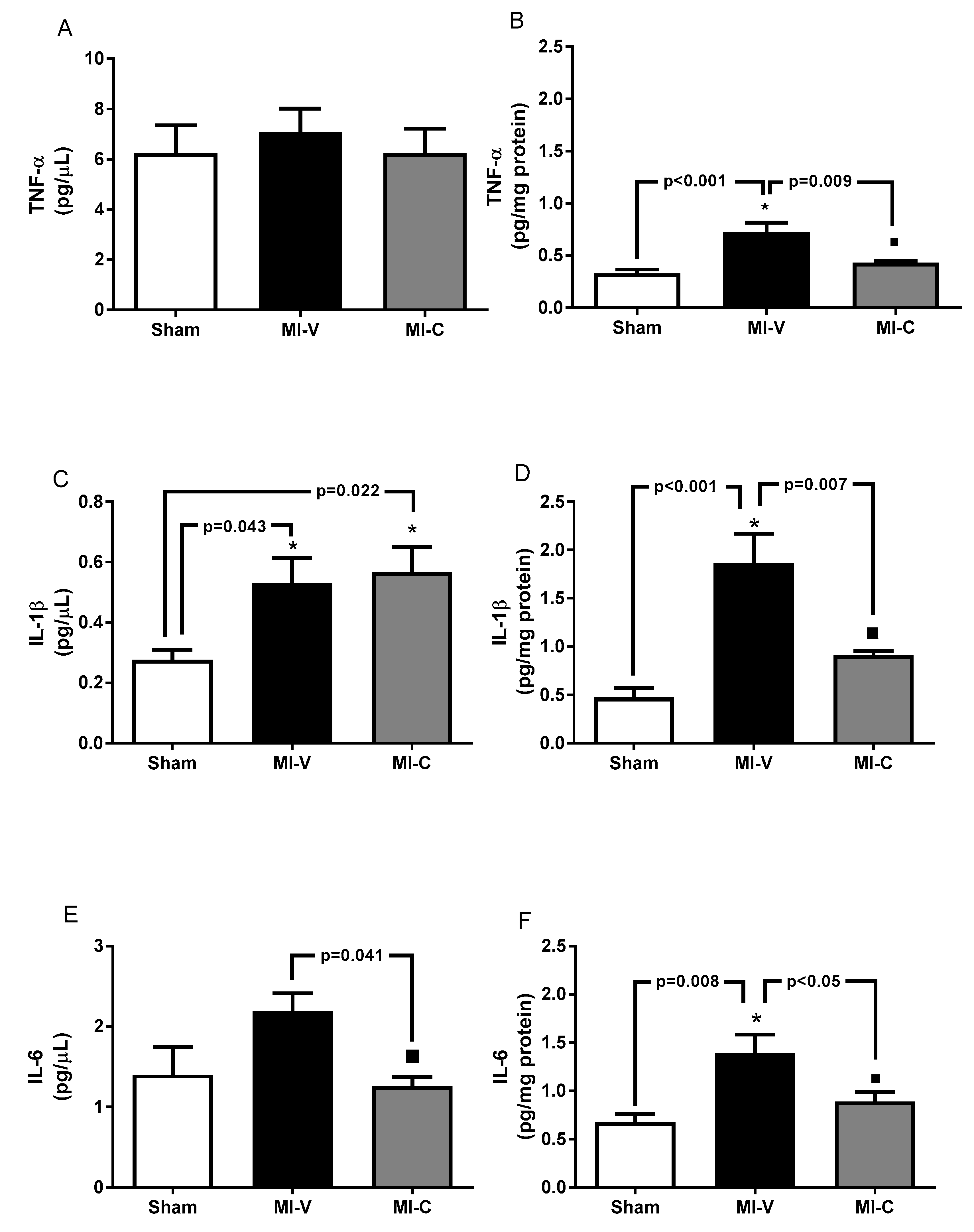
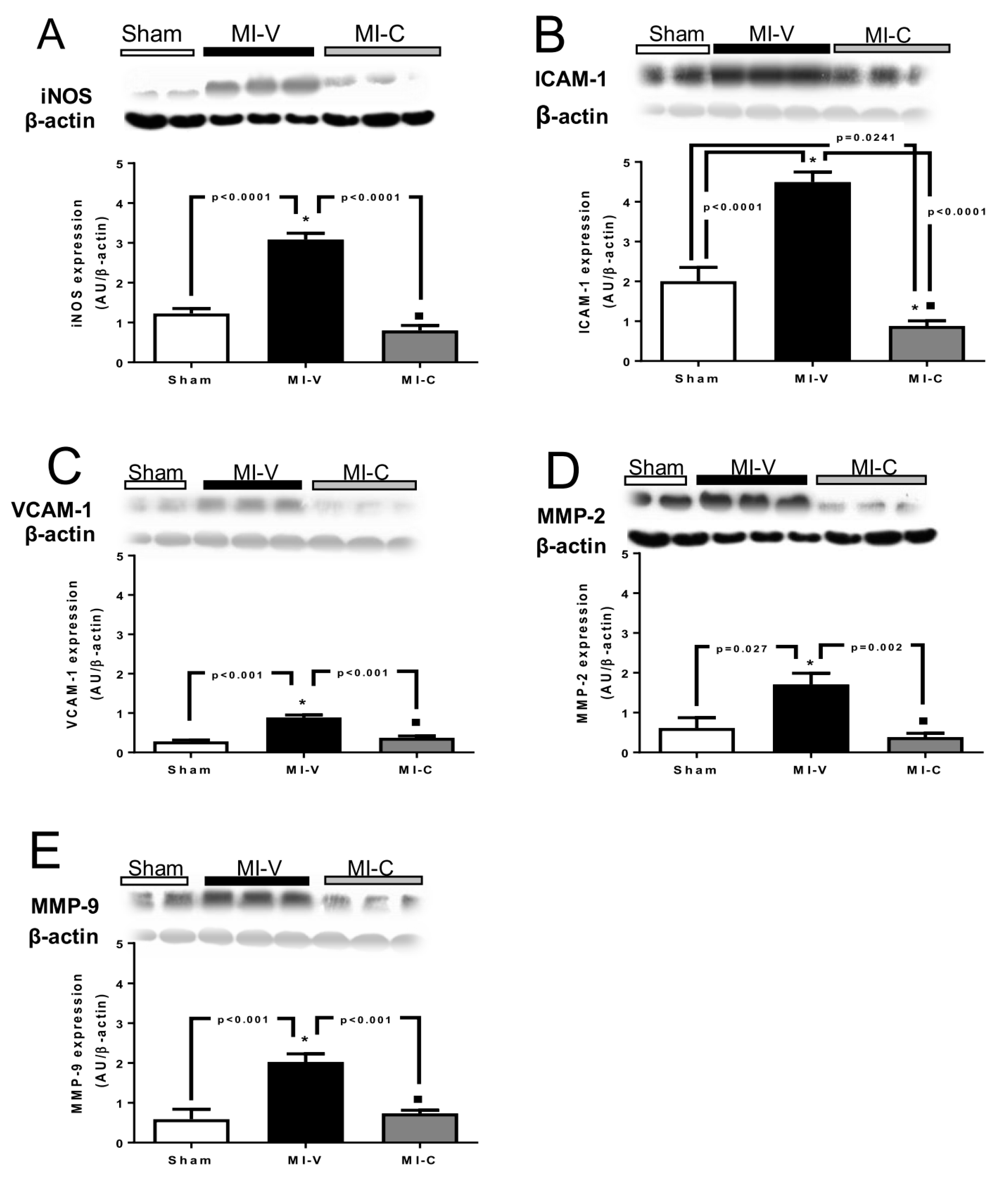
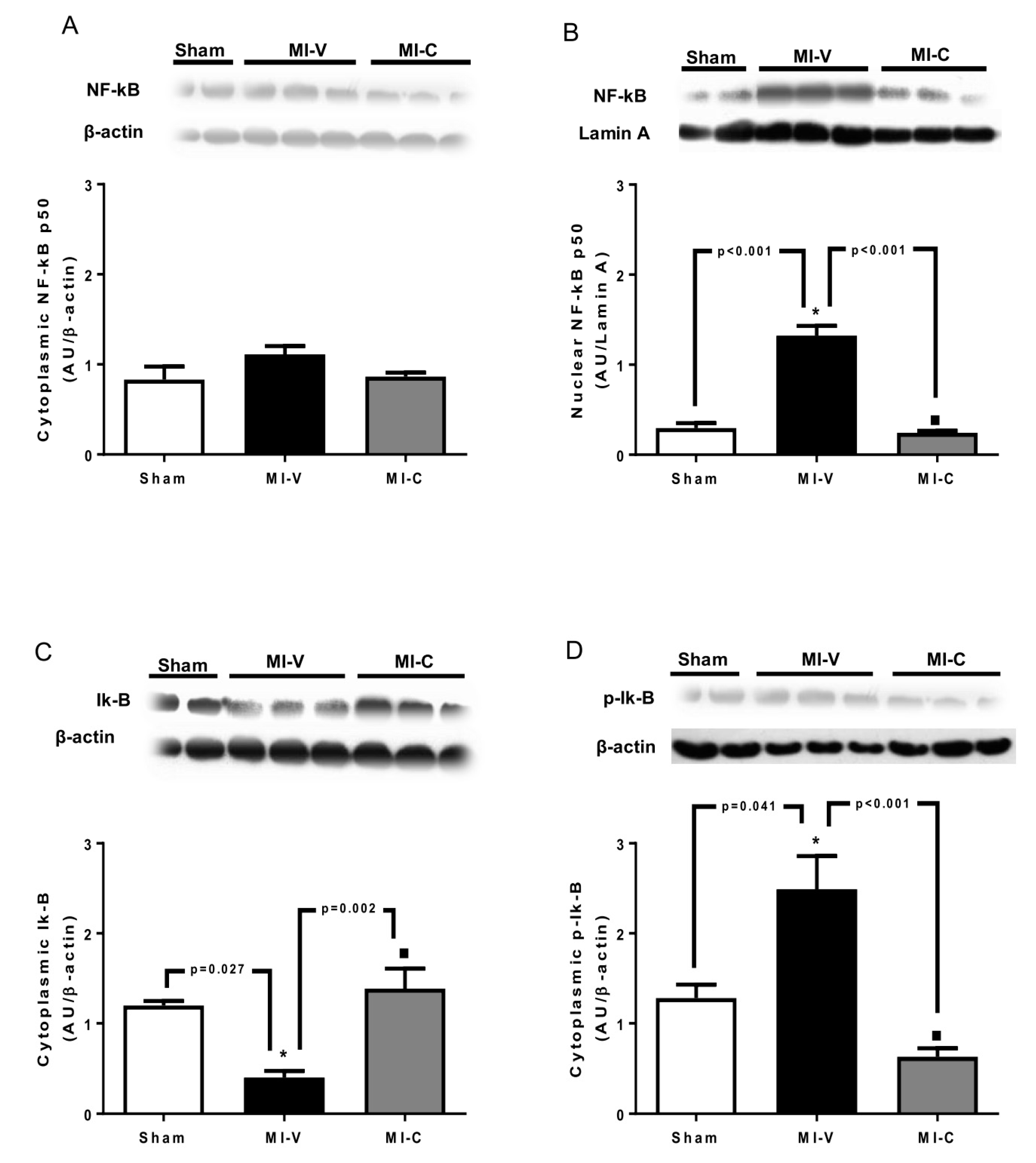
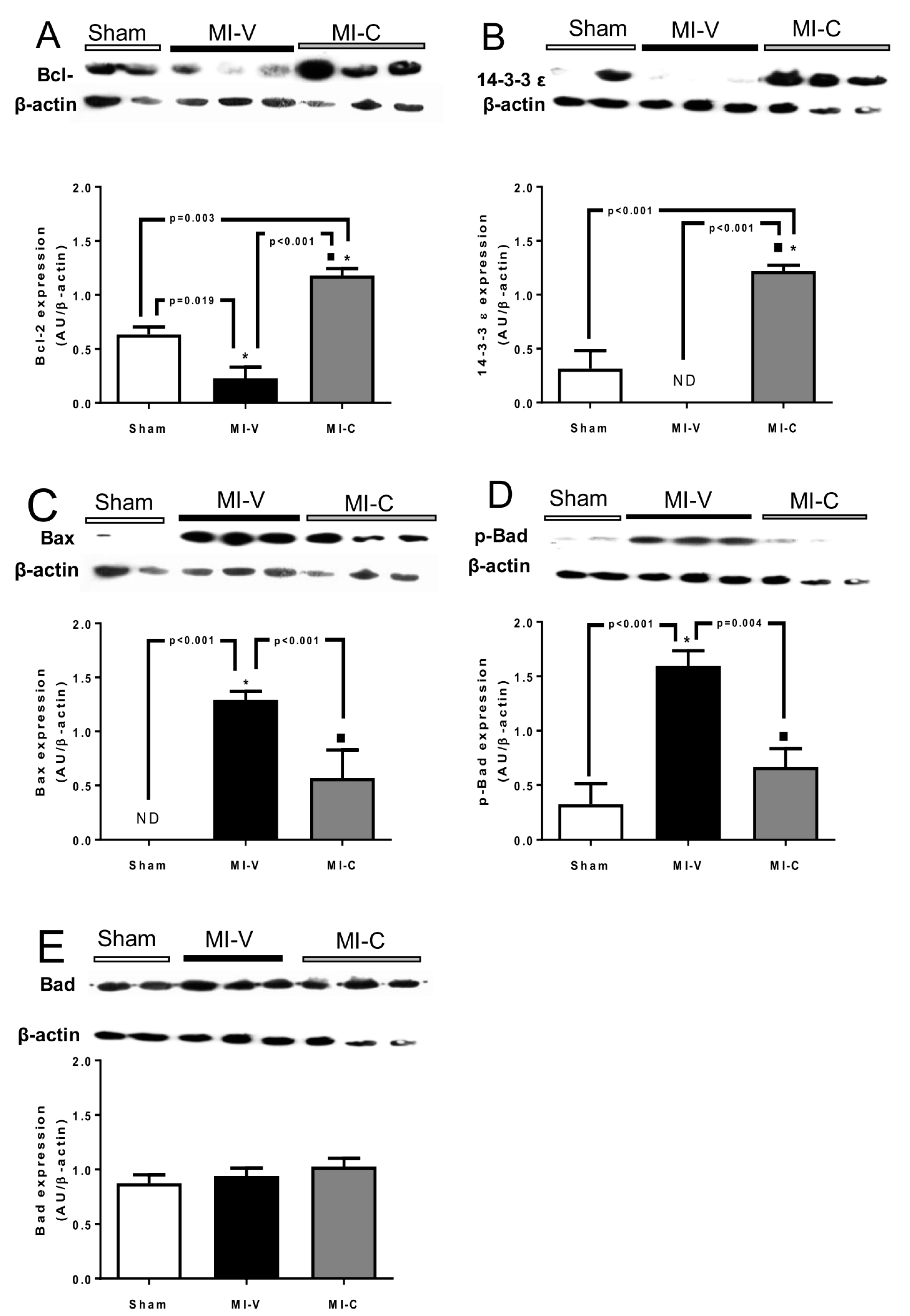
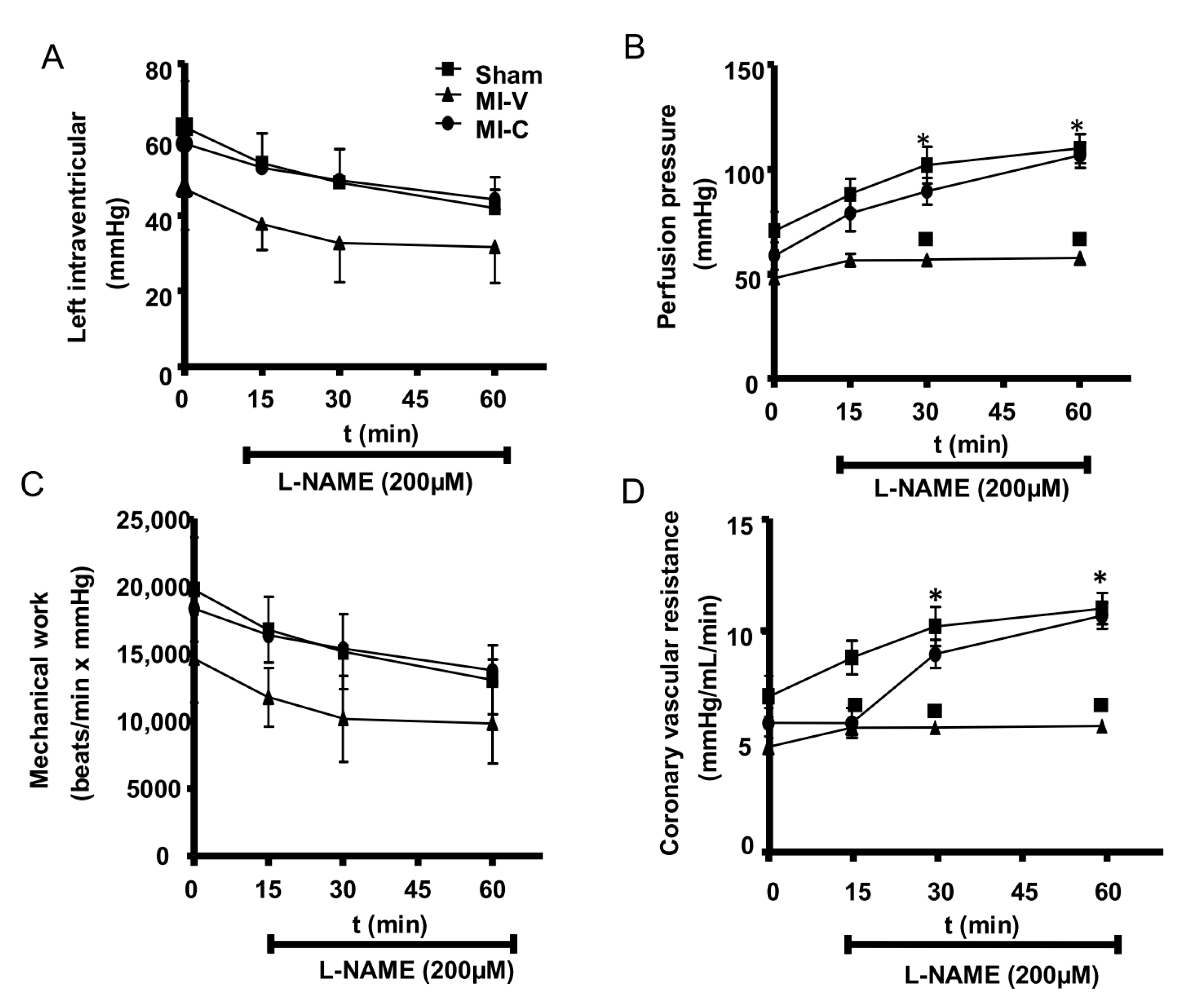
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Myocardial Ischemia
4.3. Cytokine Quantification
4.4. Western Blot
4.5. Nuclear and Cytoplasmic Protein Extraction
4.6. Histological Analysis by Electronic Microscopy
4.7. Echocardiogram
4.8. Isolated Perfused Peart
4.9. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kain, V.; Prabhu, S.D.; Halade, G.V. Inflammation revisited: Inflammation versus resolution of inflammation following myocardial infarction. Basic Res. Cardiol. 2014, 109, 444. [Google Scholar] [CrossRef] [PubMed]
- Seropian, I.M.; Toldo, S.; Van Tassell, B.W. Anti-inflammatory strategies for ventricular remodeling following st-segment elevation acute myocardial infarction. J. Am. Coll. Cardiol. 2014, 63, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.G.; Pfeffer, M.A.; Pasternak, R.C. Left ventricular remodeling after myocardial infarction: A corollary to infarct expansion. Circulation 1986, 74, 693–702. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef]
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- eNian, M.; Lee, P.; Khaper, N. Inflammatory cytokines and postmyocardial infarction remodeling. Circ. Res. 2004, 94, 1543–1553. [Google Scholar] [CrossRef]
- eOno, K.; Matsumori, A.; Shioi, T. Cytokine gene expression after myocardial infarction in rat hearts: Possible implication in left ventricular remodeling. Circulation 1998, 98, 149–156. [Google Scholar] [CrossRef]
- Deten, A.; Volz, H.C.; Briest, W. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc. Res. 2002, 55, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Li, J. Inflammation and inflammatory cells in myocardial infarction and reperfusion injury: A double-edged sword. Clin. Med. Insights. Cardiol. 2016, 10, 79–84. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef] [Green Version]
- Marunouchi, T.; Tanonaka, K. Cell death in the cardiac myocyte. Biol. Pharm. Bull. 2015, 38, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.R. Role of neutrophils in myocardial ischemia and reperfusion. Circulation 1995, 91, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- eLaskin, D.L.; Pendino, K.J. Macrophages and inflammatory mediators in tissue injury. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Brower, G.L.; Gardner, J.D.; Forman, M.F. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur. J. Cardiothorac. Surg. 2006, 30, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T. Review: Real-world use of nonsteroidal antiinflammatory drugs is associated with acute myocardial infarction. Ann. Intern. Med. 2017, 167, JC30. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.; Wang, Y.; Berger, A.K. Nonsteroidal antiinflammatory drugs after acute myocardial infarction. Am. Heart J. 2002, 143, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, G.R.; Giugliano, R.P.; Gibson, C.M. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am. J. Cardiol. 2003, 91, 1055–1059. [Google Scholar] [CrossRef]
- Gibson, C.M.; Pride, Y.B.; Aylward, P.E. Association of non-steroidal anti-inflammatory drugs with outcomes in patients with st-segment elevation myocardial infarction treated with fibrinolytic therapy: An extract-timi 25 analysis. J. Thromb. Thrombolysis. 2009, 27, 11–17. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M. The PPARalpha-leukotriene b4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Li, A.C.; Palinski, W. Peroxisome proliferator-activated receptors: How their effects on macrophages can lead to the development of a new drug therapy against atherosclerosis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 1–39. [Google Scholar] [CrossRef]
- Ibarra-Lara, M.L.; Sánchez-Aguilar, M.; Soria, E. Peroxisome proliferator-activated receptors (PPAR) downregulate the expression of pro-inflammatory molecules in an experimental model of myocardial infarction. Can. J. Physiol. Pharmacol. 2016, 94, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, G.E.; Rhaleb, N.E.; D’Ambrosio, M.A. Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin ii-high salt-induced hypertension. J. Hypertens. 2015, 33, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Hasegawa, H.; Nagai, T. Implication of cardiac remodeling in heart failure: Mechanisms and therapeutic strategies. Intern. Med. 2003, 42, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Hilfiker, A.; Kaminski, K. Role of interleukin-6 for lv remodeling and survival after experimental myocardial infarction. FASEB J. 2003, 17, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yuan, Y.C.; Li, J.Y. Tumor necrosis factor-alpha and its role as a mediator in myocardial infarction: A brief review. Chronic. Dis. Transl. Med. 2015, 1, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Wallen, N.H.; Held, C.; Rehnqvist, N. Elevated serum intercellular adhesion molecule-1 and vascular adhesion molecule-1 among patients with stable angina pectoris who suffer cardiovascular death or non-fatal myocardial infarction. Eur. Heart J. 1999, 20, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessen, H.W.; Krijnen, P.A.; Visser, C.A. Intercellular adhesion molecule-1 in the heart. Ann. N. Y. Acad. Sci. 2002, 973, 573–585. [Google Scholar] [CrossRef]
- Sager, H.B.; Dutta, P.; Dahlman, J.E. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci. Transl. Med. 2016, 8, 342ra80. [Google Scholar] [CrossRef]
- Paterniti, I.; Campolo, M.; Cordaro, M. PPAR-alpha modulates the anti-inflammatory effect of melatonin in the secondary events of spinal cord injury. Mol. Neurobiol. 2017, 54, 5973–5987. [Google Scholar] [CrossRef]
- Kingery, J.R.; Hamid, T.; Lewis, R.K. Leukocyte iNOS is required for inflammation and pathological remodeling in ischemic heart failure. Basic Res. Cardiol. 2017, 112, 19. [Google Scholar] [CrossRef]
- Chen, J.; Tung, C.H.; Allport, J.R. Near-infrared fluorescent imaging of matrix metalloproteinase activity after myocardial infarction. Circulation 2005, 111, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.; Moainie, S.L.; Baskin, J.M. Region- and type-specific induction of matrix metalloproteinases in post-myocardial infarction remodeling. Circulation 2003, 107, 2857–2863. [Google Scholar] [CrossRef] [PubMed]
- Radhiga, T.; Rajamanickam, C.; Sundaresan, A. Effect of ursolic acid treatment on apoptosis and DNA damage in isoproterenol-induced myocardial infarction. Biochimie 2012, 94, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Zeiher, A.M. Reactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factors. Regul. Pept. 2000, 90, 19–25. [Google Scholar] [CrossRef]
- Christia, P.; Frangogiannis, N.G. Targeting inflammatory pathways in myocardial infarction. Eur. J. Clin. Investig. 2013, 43, 986–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, A.; Lu, Y.; Zheng, Q. Qiliqiangxin attenuates cardiac remodeling via inhibition of tgf-beta1/smad3 and nf-kappab signaling pathways in a rat model of myocardial infarction. Cell. Physiol. Biochem. 2018, 45, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Linz, W.; Wohlfart, P.; Baader, M. The peroxisome proliferator-activated receptor-alpha (ppar-alpha) agonist, ave8134, attenuates the progression of heart failure and increases survival in rats. Acta Pharmacol. Sin. 2009, 30, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.P.; Bogaert, J.; Zalewski, J. Nitric oxide for inhalation in ST-elevation myocardial infarction (nomi): A multicentre, double-blind, randomized controlled trial. Eur. Heart J. 2018, 39, 2717–2725. [Google Scholar] [CrossRef]
- Pavon, N.; Martinez-Abundis, E.; Hernandez, L. Sexual hormones: Effects on cardiac and mitochondrial activity after ischemia-reperfusion in adult rats. Gender difference. J. Steroid Biochem. Mol. Biol. 2012, 132, 135–146. [Google Scholar] [CrossRef]
- Pavon, N.; Hernandez-Esquivel, L.; Buelna-Chontal, M. Antiarrhythmic effect of tamoxifen on the vulnerability induced by hyperthyroidism to heart ischemia/reperfusion damage. J. Steroid Biochem. Mol. Biol. 2014, 143, 416–423. [Google Scholar] [CrossRef]
- Ibarra-Lara, L.; Hong, E.; Soria-Castro, E. Clofibrate PPARalpha activation reduces oxidative stress and improves ultrastructure and ventricular hemodynamics in no-flow myocardial ischemia. J. Cardiovasc. Pharmacol. 2012, 60, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Resendiz, S.; Roldan, F.J.; Correa, F. Postconditioning protects against reperfusion injury in hypertensive dilated cardiomyopathy by activating MEK/ERK1/2 signaling. J. Card. Fail. 2013, 19, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Silva-Palacios, A.; Ostolga-Chavarria, M.; Buelna-Chontal, M. 3-np-induced huntington’s-like disease impairs nrf2 activation without loss of cardiac function in aged rats. Exp. Gerontol. 2017, 96, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Yang, X.P.; Nass, O. Chronic heart failure induced by coronary artery ligation in lewis inbred rats. Am. J. Physiol. 1997, 272, H722–H727. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.B.; Tiwari, S.; Thomas, P. Effects of anesthesia on echocardiographic assessment of left ventricular structure and function in rats. Basic Res Cardiol. 2007, 102, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Lara, L.; Del Valle-Mondragon, L.; Soria-Castro, E. Peroxisome proliferator-activated receptor-alpha stimulation by clofibrate favors an antioxidant and vasodilator environment in a stressed left ventricle. Pharmacol. Rep. 2016, 68, 692–702. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | MI-V | MI-C | |
|---|---|---|---|
| Heart rate (bpm) | 276.6 ± 17.33 | 267.9 ± 21.50 | 243.3 ± 6.83 |
| LVEF (%) | 74.86 ± 2.74 | 64.71 ± 2.89 | 66.14 ± 3.03 |
| Posterior wall thickness (mm) | 18.57 ± 0.42 | 19.43 ± 0.57 | 19.29 ± 0.28 |
| Septum thickness (mm) | 18.86 ± 0.76 | 15.00 ± 1.26 * | 14.17 ± 1.07 * |
| Systolic diameter (mm) | 30.14 ± 2.58 | 46.71 ± 1.47 * | 41.00 ± 2.28 * |
| Diastolic diameter (mm) | 59.14 ± 2.81 | 78.57 ± 1.99 * | 70.14 ± 1.94 *■ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibarra-Lara, L.; Sánchez-Aguilar, M.; Soria-Castro, E.; Vargas-Barrón, J.; Roldán, F.J.; Pavón, N.; Torres-Narváez, J.C.; Cervantes-Pérez, L.G.; Pastelín-Hernández, G.; Sánchez-Mendoza, A. Clofibrate Treatment Decreases Inflammation and Reverses Myocardial Infarction-Induced Remodelation in a Rodent Experimental Model. Molecules 2019, 24, 270. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24020270
Ibarra-Lara L, Sánchez-Aguilar M, Soria-Castro E, Vargas-Barrón J, Roldán FJ, Pavón N, Torres-Narváez JC, Cervantes-Pérez LG, Pastelín-Hernández G, Sánchez-Mendoza A. Clofibrate Treatment Decreases Inflammation and Reverses Myocardial Infarction-Induced Remodelation in a Rodent Experimental Model. Molecules. 2019; 24(2):270. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24020270
Chicago/Turabian StyleIbarra-Lara, Luz, María Sánchez-Aguilar, Elizabeth Soria-Castro, Jesús Vargas-Barrón, Francisco J. Roldán, Natalia Pavón, Juan C. Torres-Narváez, Luz G. Cervantes-Pérez, Gustavo Pastelín-Hernández, and Alicia Sánchez-Mendoza. 2019. "Clofibrate Treatment Decreases Inflammation and Reverses Myocardial Infarction-Induced Remodelation in a Rodent Experimental Model" Molecules 24, no. 2: 270. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24020270