Synthesis of Amino Acid–Naphthoquinones and In Vitro Studies on Cervical and Breast Cell Lines


 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Chemistry
2.2. Electrochemical Studies by Cyclic Voltammetry
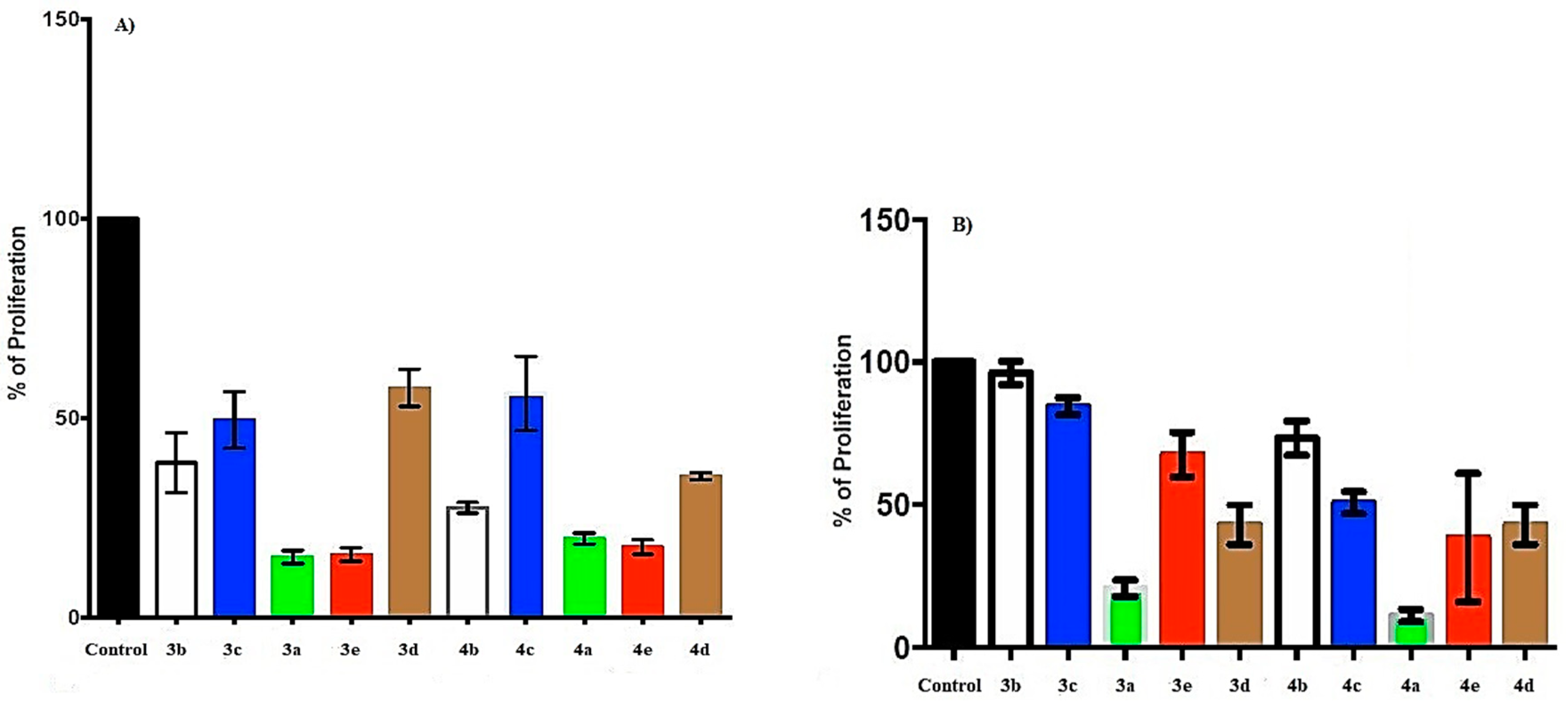
2.3. Effect of Naphthoquinone–Amino Acid Derivatives in Cell Proliferation of Cervical Cancer Cell Line SiHa and Breast Cancer Cell Line MCF-7
3. Discussion
4. Materials and Methods
4.1. General
4.2. General Procedure for the Optimization of the Reaction Conditions for the Synthesis of Compounds 3a–c
4.3. Optimization of the Reaction Conditions for the Synthesis of Compounds 3a–c Under Microwave Irradiation
4.4. Optimized Reaction Conditions Under Microwave Irradiation for the Synthesis of Compounds 3a–e and 4a–e
4.5. General Procedure for the Synthesis of Compounds 3a–e and 4a–6 Under Ultrasonic Irradiation
4.6. Spectroscopic Characterization of Amino Acid–1,4-naphthoquinone Derivatives
4.7. Spectroscopic Characterization of Amino Acid–2,3-dichloronaphthoquinone Derivatives
4.8. Cyclic Voltammetry
4.9. Cell Lines
4.10. Cell Proliferation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López, L.I.; Vaquera, J.J.; Nery, S.D.; Silva, S.Y. Naphthoquinones; biological properties and synthesis of lawsone and derivatives—A structured review. Vitae 2014, 21, 248–258. [Google Scholar]
- Leyva, E.; Loredo-Carrillo, S.E.; López, L.I.; Escobedo-Avellaneda, E.G.; Navarro-Tovar, G. Importancia química y biológica de naftoquinonas. Revisión bibliográfica. Afinidad 2017, 74, 36–50. [Google Scholar]
- Li, C.J.; Li, Y.Z.; Pinto, A.V.; Pardee, A.B. Potent inhibition of tumor survival in vivo by beta-lapachone plus taxol: Combining drugs imposes different artificial checkpoints. Proc. Natl. Acad. Sci. USA 1999, 96, 13369–13374. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Y.W.; Liu, G.; Afrasiabi, Z.; Sinn, E.; Padhye, S.; Ma, Y. The cytotoxicity and mechanisms of 1,2-naphthoquinone thiosemicarbazone and its metal derivatives against MCF-7 human breast cancer cells. Toxicol. Appl. Pharmacol. 2004, 197, 40–48. [Google Scholar] [CrossRef]
- Kongkathip, N.; Kongkathip, B.; Siripong, P.; Sangma, C.; Luangkamin, S.; Niyomdecha, M.; Pattanapa, S.; Piyaviriyagul, S.; Kongsaeree, P. Potent antitumor activity of synthetic 1,2-naphthoquinones and 1,4-naphthoquinones. Bioorg. Med. Chem. 2003, 11, 3179–3191. [Google Scholar] [CrossRef]
- Kongkathip, N.; Luangkamin, S.; Kongkathip, B.; Sangma, C.; Grigg, R.; Kongsaeree, P.; Prabpai, S.; Pradidphol, N.; Piyaviriyagul, S.; Siripong, P. Synthesis of novel rhinacanthins and related anticancer naphthoquinone esters. J. Med. Chem. 2004, 47, 4427–4438. [Google Scholar] [CrossRef]
- Pedersen, J.A. On the application of electron paramagnetic resonance in the study of naturally occurring quinones and quinols. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2002, 58, 1257–1270. [Google Scholar] [CrossRef]
- Coates, C.S.; Ziegler, J.; Manz, K.; Good, J.; Kang, B.; Milikisiyants, S.; Chatterjee, R.; Hao, S.; Golbeck, J.H.; Lakshmi, K.V. The structure and function of quinones in biological solar energy transduction: A cyclic voltammetry, EPR, and hyperfine sub-level correlation (HYSCORE) spectroscopy study of model naphthoquinones. J. Phys. Chem. B 2013, 117, 7210–7220. [Google Scholar] [CrossRef]
- Maldonado, T.; Martínez-González, E.; Frontana, C. Intramolecular hydrogen bonding/selfprotonation processes modulated by the substituent effect in hydroxyl-substituted naphthoquinones. Electroanalysis 2016, 28, 2827–2833. [Google Scholar] [CrossRef]
- Smithson, C.S.; MacDonald, D.J.; Letvenuk, T.M.; Carello, C.E.; Jennings, M.; Lough, A.J.; Britten, J.; Decken, A.; Preuss, K.E. A 1,2,3-dithiazolyl-o-naphthoquinone: A neutral radical with isolable cation and anion oxidation states. Dalton Trans. 2016, 45, 9608–9620. [Google Scholar] [CrossRef]
- Tarábek, J.; Wen, J.; Dron, P.I.; Pospísil, L.; Michl, J. EPR Spectroscopy of radical ion of a 2,3-diamino-1,4-naphthoquinone derivative. J. Org. Chem. 2018, 83, 5474–5479. [Google Scholar] [CrossRef]
- Mondal, S.; Bera, S.; Ghosh, P. Redox cascades and making of a C-C bond: 1,2-benzodiazinyl radicals and a copper complex of a benzodiazine. J. Org. Chem. 2019, 84, 1871–1881. [Google Scholar] [CrossRef]
- Wardman, P. Electron transfer and oxidative stress as key factors in the design of drugs selectively active in hypoxia. Curr. Med. Chem. 2001, 8, 739–761. [Google Scholar] [CrossRef]
- Ham, S.W.; Choe, J.I.; Wang, M.F.; Peyregne, V.; Carr, B.I. Fluorinated quinoid inhibitor: Possible “pure” arylator predicted by the simple theoretical calculation. Bioorg. Med. Chem. Lett. 2004, 14, 4103–4105. [Google Scholar] [CrossRef]
- Kar, S.; Wang, M.; Ham, S.W.; Carr, B.I. Fluorinated Cpd 5, a pure arylating K-vitamin derivative, inhibits human hepatoma cell growth by inhibiting Cdc25 and activating MAPK. Biochem. Pharmacol. 2006, 72, 1217–1227. [Google Scholar] [CrossRef]
- Park, H.; Carr, B.I.; Li, M.; Ham, S.W. Fluorinated NSC as a Cdc25 inhibitor. Bioorg. Med. Chem. Lett. 2007, 17, 2351–2354. [Google Scholar] [CrossRef]
- Troshkova, N.M.; Goryunov, L.I.; Shteingarts, V.D.; Zakharova, O.D.; Ovchinnikova, L.P.; Nevinsky, G.A. Synthesis and cytotoxicity evaluation of polyfluorinated 1,4-naphthoquinones containing amino acid substituents. J. Fluorine Chem. 2014, 164, 18–26. [Google Scholar] [CrossRef]
- Marastoni, M.; Trapella, C.; Scotti, A.; Fantinati, A.; Ferretti, V.; Marzola, E.; Eleonora, G.; Gavioli, R.; Preti, D. Naphthoquinone amino acid derivatives, synthesis and biological activity as proteasome inhibitors. J. Enzym. Inhib. Med. Ch. 2017, 32, 865–877. [Google Scholar] [CrossRef]
- Bittner, S.; Gorohovsky, S.; Paz-Tal, L.O.; Becker, J.Y. Synthesis, electrochemical and spectral properties of some ω-N-quinonyl amino acids. Amino Acids 2002, 22, 71–93. [Google Scholar] [CrossRef]
- Tandon, V.K.; Maurya, H.K. ‘On water’: Unprecedented nucleophilic substitution and addition reactions with 1,4-quinones in aqueous suspension. Tetrahedron Lett. 2009, 50, 5896–5902. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Huang, L.; Sakhuja, R. Efficient syntheses of naphthoquinone-dipeptides. Synthesis 2010, 12, 2011–2016. [Google Scholar] [CrossRef]
- Mital, A.; Sonawane, M.; Bindal, S.; Mahlavat, S.; Negi, V. Substituted 1,4-naphthoquinones as a new class of antimycobacterial agents. Der Pharm. Chem. 2010, 2, 63–73. [Google Scholar]
- de Moraes, T.A.; Filha, M.J.; Camara, C.A.; Silva, T.M.; Soares, B.M.; Bomfim, I.S.; Pessoa, C.; Ximenes, G.C.; Silva, V.A., Jr. Synthesis and cytotoxic evaluation of a series of 2-amino-naphthoquinones against human cancer cells. Molecules 2014, 19, 13188–13199. [Google Scholar] [CrossRef]
- Figurka, O.; Kochubei, V.; Khomyak, S.; Platonov, M.; Martynyuk, I.; Stadnichuk, O.; Gubriy, Z.; Kurka, M.; Novikov, V. Synthesis and characteristics of amino acid derivatives of 1,4-naphthoquinone. J. Chem. Pharm. Res. 2015, 7, 1289–1294. [Google Scholar]
- Janeczko, M.; Demchuk, O.M.; Strzelecka, D.; Kubinski, K.; Maslyk, M. New family of antimicrobial agents derived from 1,4-naphthoquinone. Eur. J. Med. Chem. 2016, 124, 1019–1025. [Google Scholar] [CrossRef]
- Fihurka, O.; Kochubei, V.; Khomyak, S.; Novikov, V. Thermal analysis of 2-amino-3-chloro-1,4-naphthoquinones. Int. J. ChemTech Res. 2017, 10, 985–993. [Google Scholar]
- Bittner, S.; Gorohovsky, S.; Lozinsky, E.; Shames, A.I. EPR study of anion radical of various N-quinonyl amino acids. Amino Acids 2000, 19, 439–449. [Google Scholar] [CrossRef]
- Rahimipour, S.; Weiner, L.; Fridkin, M.; Shresthe-Dawadi, P.B.; Bittner, S. Novel naphthoquinonyl derivatives: Potential structural components for the synthesis of cytotoxic peptides. Lett. Pept. Sci. 1996, 3, 263–274. [Google Scholar] [CrossRef]
- Gorohovsky, S.; Bittner, S. Novel N-quinonyl amino acids and their transformation to 3-substituted p-isoxazinones. Amino acids. 2001, 20, 134–144. [Google Scholar] [CrossRef]
- López, L.I.; Vaquera, J.J.; Sáenz, A.; Silva, S.Y. Ultrasonic and microwave assisted synthesis of nitrogen-containing derivatives of juglone as potential antibacterial agents. Lett. Org. Chem. 2014, 11, 573–582. [Google Scholar] [CrossRef]
- Leyva, E.; López, L.I.; García de la Cruz, R.F.; Espinosa-González, C.G. Synthesis and studies of the antifungal activity of 2-anilino-/2,3-dianilino-/2-phenoxy- and 2,3-diphenoxy-1,4-naphthoquinones. Res. Chem. Intermed. 2017, 43, 1813–1827. [Google Scholar] [CrossRef]
- López, L.I.; Nery, S.D.; Sáenz, A.; de Loera, D. Facile synthesis of aminonaphthoquinone Mannich bases by noncatalytic multicomponent reaction. Synth. Commun. 2017, 47, 2247–2253. [Google Scholar] [CrossRef]
- Ge, Y.; Li, A.; Wu, J.; Feng, H.; Wang, L.; Liu, H.; Xu, Y.; Xu, Q.; Zhao, L.; Li, Y. Design, synthesis and biological evaluation of novel non-peptide boronic acid derivatives as proteasome inhibitors. Eur. J. Med. Chem. 2017, 128, 180–191. [Google Scholar] [CrossRef]
- Ruda, N.V.; Drachuk, O.P.; Stepanuyk, G.I. The acute toxicity study of new amino acid-containing derivatives of 1,4-naphthoquinone. Available online: http://nuph.edu.ua/wp-content/uploads/2015/04/Microsoft-Word-UBFG_3_2013_31.doc.pdf (accessed on 19 November 2019).
- Guin, P.S.; Das, S.; Mandal, P.C. Electrochemical reduction of quinones in different media: A review. Int. J. Electrochem. 2011, 2011, 1–22. [Google Scholar] [CrossRef]
- Song, Y.; Buettner, G.R. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic. Biol. Med. 2010, 49, 919–962. [Google Scholar] [CrossRef]
- Heinze, J. Cyclic voltammetry—“Electrochemical spectroscopy”. New analytical methods (25). Angew. Chem. Int. Edit. 1984, 23, 831–847. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons Inc.: New York, NY, USA, 2000. [Google Scholar]
- da Cruz, E.H.; Hussene, C.M.; Dias, G.G.; Diogo, E.B.; de Melo, I.M.; Rodrigues, B.L.; da Silva, M.G.; Valenca, W.O.; Camara, C.A.; de Oliveira, R.N.; et al. 1,2,3-Triazole-, arylamino- and thio-substituted 1,4-naphthoquinones: Potent antitumor activity, electrochemical aspects, and bioisosteric replacement of C-ring-modified lapachones. Bioorg. Med. Chem. 2014, 22, 1608–1619. [Google Scholar] [CrossRef] [Green Version]
- Francisco, A.I.; Casellato, A.; Neves, A.P.; Carneiro, J.W.; Vargas, M.D.; Visentin, L.do.C.; Magalhaes, A.; Camara, C.A.; Pessoa, C.; Costa-Lotufo, L.V.; et al. Novel 2-(R-phenyl)amino-3-(2-methylpropenyl)-[1,4]-naphthoquinones: Synthesis, characterization, electrochemical behavior and antitumor activity. J. Braz. Chem. Soc. 2010, 21, 169–178. [Google Scholar] [CrossRef]
- Leyva, E.; López, L.I.; Loredo-Carrillo, S.E.; Rodríguez-Kessler, M.; Montes-Rojas, A. Synthesis, spectral and electrochemical characterization of novel 2-(fluoroanilino)-1,4-naphthoquinones. J. Fluor. Chem. 2011, 132, 94–101. [Google Scholar] [CrossRef]
- Diogo, E.B.; Dias, G.G.; Rodrigues, B.L.; Guimarães, T.T.; Valença, W.O.; Camara, C.A.; de Oliveira, R.N.; da Silva, M.G.; Ferreira, V.F.; de Paiva, Y.G.; et al. Synthesis and anti-trypanosoma cruzi activity of naphthoquinone-containing triazoles: Electrochemical studies on the effects of the quinoidal moiety. Bioorg. Med. Chem. 2013, 21, 6337–6348. [Google Scholar] [CrossRef] [Green Version]
- Emadi, A.; Le, A.; Harwood, C.J.; Stagliano, K.W.; Kamangar, F.; Ross, A.E.; Cooper, C.R.; Dang, C.V.; Karp, J.E.; Vulca-Ross, M. Metabolic and electrochemical mechanisms of dimeric naphthoquinones cytotoxicity in breast cancer cells. Bioorg. Med. Chem. 2011, 19, 7057–7062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez, M.; González, F.J.; González, I. Effect of host and guest structures on hydrogen bonding association: Influence on stoichiometry and equilibrium constants. J. Electrochem. Soc. 2003, 150, 527–534. [Google Scholar] [CrossRef]
- Gupta, N.; Linschitz, H. Hydrogen-bonding and protonation effects in electrochemistry of quinones in aprotic solvents. J. Am. Chem. Soc. 1997, 119, 6384–6391. [Google Scholar] [CrossRef]
- Shi, R.R.; Tessensohn, M.E.; Lauw, S.J.; Foo, N.A.; Webster, R.D. Tuning the reduction potential of quinones by controlling the effects of hydrogen bonding, protonation and proton-coupled electron transfer reactions. Chem. Commun. 2019, 55, 2277–2280. [Google Scholar] [CrossRef] [PubMed]
- Gómez, M.; González, F.J.; González, I. A Model for characterization of successive hydrogen bonding interactions with electrochemically generated charged species. The quinone electroreduction in the presence of donor protons. Electroanalysis 2003, 15, 635–645. [Google Scholar] [CrossRef]
- Goulart, M.O.; Zani, C.L.; Tonholo, J.; Freitas, L.R.; de Abreu, F.C.; Oliveira, A.B.; Raslan, D.D.; Starling, S.; Chiari, E. Trypanocidal activity and redox potential of heterocyclic- and 2-hydroxy-naphthoquinones. Bioorg. Med. Chem. Lett. 1997, 7, 2043–2048. [Google Scholar] [CrossRef]
- Ashnagar, A.; Bruce, J.M.; Dutton, P.L.; Prince, R.C. One- and two-electron reduction of hydroxy-1,4-naphthoquinones and hydroxy-9,10-anthraquinones: The role of internal hydrogen bonding and its bearing on the redox chemistry of the anthracycline antitumour quinones. Biochim. Biophys. Acta 1984, 801, 351–359. [Google Scholar] [CrossRef]
- Aguilar-Martínez, M.; Cuevas, G.; Jiménez-Estrada, M.; González, I.; Lotina-Hennsen, B.; Macías-Ruvalcaba, N. An experimental and theoretical study of the substituent effects on the redox properties of 2-[(R-phenyl)amine]-1,4-naphthalenediones in acetonitrile. J. Org. Chem. 1999, 64, 3684–3694. [Google Scholar] [CrossRef]
- Hillard, E.A.; de Abreu, F.C.; Ferreira, D.C.; Jaouen, G.; Goulart, M.O.; Amatore, C. Electrochemical parameters and techniques in drug development, with an emphasis on quinones and related compounds. Chem. Commun. 2008, 23, 2612–2628. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3a–e and 4a–e are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Method | Compound Yield (%) | |||
|---|---|---|---|---|
| Base | 3a | 3b | 3c | |
| RTS | None | Nr | Nr | Nr |
| TEA | Tp | Tp | Tp | |
| K2CO3 | Tp | Tp | Tp | |
| RS | None | Tp | Tp | Tp |
| TEA | 32 | 50 | 30 | |
| K2CO3 | 23 | 50 | 20 | |
| MAS | None | 30 | 30 | 28 |
| TEA | 74 | 73 | 71 | |
| K2CO3 | 65 | 64 | 62 | |
| AcONa | 59 | 56 | 52 | |
| KOH | 74 | 73 | 72 | |
| Compound | Nq–aa | MAS a (%) | UAS b (%) | ||
|---|---|---|---|---|---|
| TEA | KOH | TEA | KOH | ||
| 3a | 1:2.0 | 80 | 86 | 68 | 78 |
| 3b | 1:1.2 | 85 | 91 | 75 | 81 |
| 3c | 1:1.5 | 82 | 87 | 71 | 65 |
| 3d | 1:1.5 | 81 | 85 | 67 | 78 |
| 3e | 1:1.5 | 79 | 80 | 77 | 82 |
| 4a | 1:2.0 | 91 | 86 | 86 | 80 |
| 4b | 1:1.2 | 95 | 87 | 85 | 70 |
| 4c | 1:1.5 | 78 | 89 | 80 | 68 |
| 4d | 1:1.5 | 91 | 82 | 75 | 85 |
| 4e | 1:1.5 | 78 | 85 | 75 | 85 |
| Compound | Epa | Epc | ΔEp b | E1/2 c | ipa | ipc | |ipa/ipc| |
|---|---|---|---|---|---|---|---|
| (V) | (V) | (V) | (V) | (mA cm−2) | |||
| 3a | - | - | - | - | - | - | - |
| 3b | −1.18 | −1.30 | 0.12 | −1.24 | 0.28 | −0.29 | 0.95 |
| 3c | −1.15 | −1.30 | 0.15 | −1.23 | 0.16 | −0.17 | 0.93 |
| 3d | −1.17 | −1.31 | 0.13 | −1.24 | 0.28 | −0.29 | 0.94 |
| 3e | −1.16 | −1.32 | 0.16 | −1.24 | 0.05 | −0.06 | 0.91 |
| 4a | −1.04 | −1.18 | 0.14 | −1.11 | 0.36 | −0.38 | 0.97 |
| 4b | −1.07 | −1.19 | 0.12 | −1.13 | 0.37 | −0.41 | 0.90 |
| 4c | −1.04 | −1.16 | 0.12 | −1.10 | 0.27 | −0.31 | 0.89 |
| 4d | −1.07 | −1.20 | 0.13 | −1.13 | 0.28 | −0.26 | 1.06 |
| 4e | −1.05 | −1.15 | 0.11 | −1.10 | 0.26 | −0.26 | 0.98 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Ávalos, E.; de Loera, D.; Araujo-Huitrado, J.G.; Escalante-García, I.L.; Muñoz-Sánchez, M.A.; Hernández, H.; López, J.A.; López, L. Synthesis of Amino Acid–Naphthoquinones and In Vitro Studies on Cervical and Breast Cell Lines. Molecules 2019, 24, 4285. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24234285
Rivera-Ávalos E, de Loera D, Araujo-Huitrado JG, Escalante-García IL, Muñoz-Sánchez MA, Hernández H, López JA, López L. Synthesis of Amino Acid–Naphthoquinones and In Vitro Studies on Cervical and Breast Cell Lines. Molecules. 2019; 24(23):4285. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24234285
Chicago/Turabian StyleRivera-Ávalos, Ernesto, Denisse de Loera, Jorge Gustavo Araujo-Huitrado, Ismailia Leilani Escalante-García, Miguel Antonio Muñoz-Sánchez, Hiram Hernández, Jesús Adrián López, and Lluvia López. 2019. "Synthesis of Amino Acid–Naphthoquinones and In Vitro Studies on Cervical and Breast Cell Lines" Molecules 24, no. 23: 4285. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24234285