Direct Observation of Structure and Dynamics of Photogenerated Charge Carriers in Poly(3-hexylthiophene) Films by Femtosecond Time-Resolved Near-IR Inverse Raman Spectroscopy


Abstract
: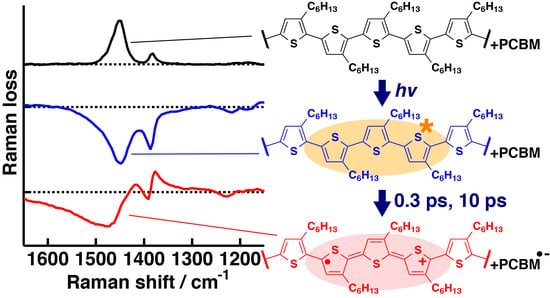
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
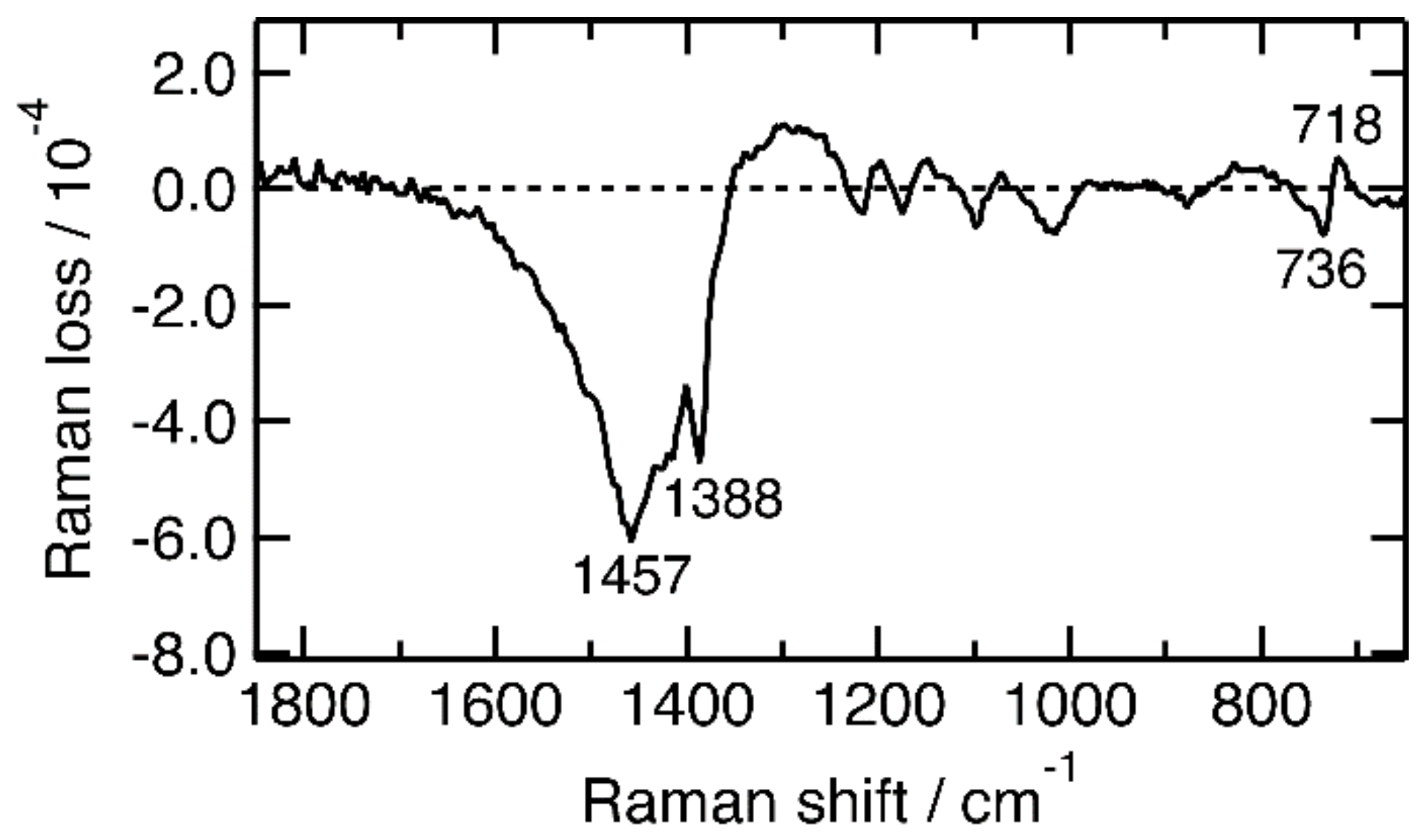
2.1. Steady-State Near-IR Inverse Raman Spectra of Pristine and FeCl3-Doped P3HT Films
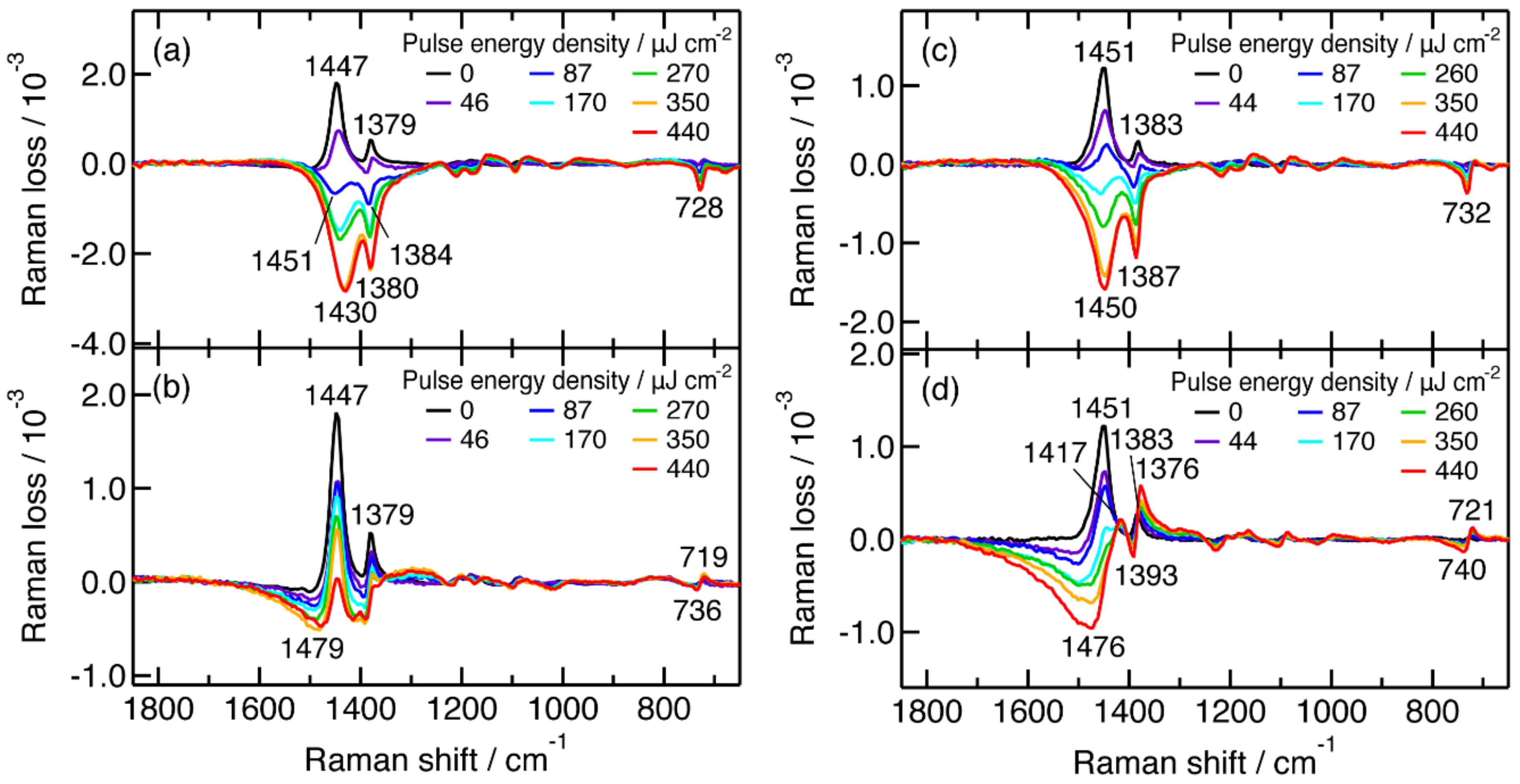
2.2. Femtosecond Time-Resolved Near-IR Inverse Raman Spectra of Pristine P3HT and P3HT:PCBM Blend Films
2.3. Actinic Pump Energy Density Dependence of Femtosecond Time-Resolved Near-IR Inverse Raman Spectra
3. Discussion
3.1. Structure of Photogenerated Singlet Excitons and Positive Polarons
3.2. Time Constants of Polaron Formation
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shirakawa, H.; Louis, E.J.; MacDiarmid, A.G.; Chang, C.K.; Heeger, A.J. Synthesis of Electrically Conducting Organic Polymers: Halogen Derivatives of Polyacetylene, (CH)x. J. Chem. Soc. Chem. Commun. 1977, 0, 578–580. [Google Scholar] [CrossRef]
- Etemad, S.; Mitani, T.; Ozaki, M.; Chung, T.C.; Heeger, A.J.; MacDiarmid, A.G. Photoconductivity in Polyacetylene. Solid State Commun. 1981, 40, 75–79. [Google Scholar] [CrossRef]
- Lauchlan, L.; Etemad, S.; Chung, T.-C.; Heeger, A.J.; MacDiarmid, A.G. Photoexcitations in Polyacetylene. Phys. Rev. B 1981, 24, 3701–3711. [Google Scholar] [CrossRef]
- Heeger, A.J.; Kivelson, S.; Schrieffer, J.R.; Su, W.-P. Solitons in Conducting Polymers. Rev. Mod. Phys. 1988, 60, 781–850. [Google Scholar] [CrossRef]
- Korovyanko, O.J.; Österbacka, R.; Jiang, X.M.; Vardeny, Z.V. Photoexcitation Dynamics in Regioregular and Regiorandom Polythiophene Films. Phys. Rev. B 2001, 64, 235122. [Google Scholar] [CrossRef]
- Jiang, X.M.; Österbacka, R.; An, C.P.; Vardeny, Z.V. Photoexcitations in Regio-Regular and Regio-Random Polythiophene Films. Synth. Met. 2003, 137, 1465–1468. [Google Scholar] [CrossRef]
- Grage, M.M.-L.; Zaushitsyn, Y.; Yartsev, A.; Chachisvilis, M.; Sundström, V.; Pullerits, T. Ultrafast Excitation Transfer and Trapping in a Thin Polymer Film. Phys. Rev. B 2003, 67, 205207. [Google Scholar] [CrossRef]
- Wells, N.P.; Boudouris, B.W.; Hillmyer, M.A.; Blank, D.A. Intramolecular Exciton Relaxation and Migration Dynamics in Poly(3-hexylthiophene). J. Phys. Chem. C 2007, 111, 15404–15414. [Google Scholar] [CrossRef]
- Cook, S.; Furube, A.; Katoh, R. Analysis of the Excited States of Regioregular Polythiophene P3HT. Energy Environ. Sci. 2008, 1, 294–299. [Google Scholar] [CrossRef]
- Guo, J.; Ohkita, H.; Benten, H.; Ito, S. Near-IR Femtosecond Transient Absorption Spectroscopy of Ultrafast Polaron and Triplet Exciton Formation in Polythiophene Films with Different Regioregularities. J. Am. Chem. Soc. 2009, 131, 16869–16880. [Google Scholar] [CrossRef]
- Lee, Y.H.; Yabushita, A.; Hsu, C.S.; Yang, S.H.; Iwakura, I.; Luo, C.W.; Wu, K.H.; Kobayashi, T. Ultrafast Relaxation Dynamics of Photoexcitations in Poly(3-hexylthiophene) for the Determination of the Defect Concentration. Chem. Phys. Lett. 2010, 498, 71–76. [Google Scholar] [CrossRef]
- Banerji, N.; Cowan, S.; Vauthey, E.; Heeger, A.J. Ultrafast Relaxation of the Poly(3-hexylthiophene) Emission Spectrum. J. Phys. Chem. C 2011, 115, 9726–9739. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.-W.; Moses, D.; Heeger, A.J. Photoinduced Carrier Generation in P3HT/PCBM Bulk Heterojunction Materials. J. Phys. Chem. C 2008, 112, 4350–4354. [Google Scholar] [CrossRef]
- Ohkita, H.; Cook, S.; Astuti, Y.; Duffy, W.; Tierney, S.; Zhang, W.; Heeney, M.; McCulloch, I.; Nelson, J.; Bradley, D.D.C.; et al. Charge Carrier Formation in Polythiophene/Fullerene Blend Films Studied by Transient Absorption Spectroscopy. J. Am. Chem. Soc. 2008, 130, 3030–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, S.; Katoh, R.; Furube, A. Ultrafast Studies of Charge Generation in PCBM:P3HT Blend Films following Excitation of the Fullerene PCBM. J. Phys. Chem. C 2009, 113, 2547–2552. [Google Scholar] [CrossRef]
- Piris, J.; Dykstra, T.E.; Bakulin, A.A.; van Loosdrecht, P.H.M.; Knulst, W.; Trinh, M.T.; Schins, J.M.; Siebbeles, D.A. Photogeneration and Ultrafast Dynamics of Excitons and Charges in P3HT/PCBM Blends. J. Phys. Chem. C 2009, 113, 14500–14506. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Ohkita, H.; Benten, H.; Ito, S. Charge Generation and Recombination Dynamics in Poly(3-hexylthiophene)/Fullerene Blend Films with Different Regioregularities and Morphologies. J. Am. Chem. Soc. 2010, 132, 6154–6164. [Google Scholar] [CrossRef]
- Kirkpatrick, J.; Keivanidis, P.E.; Bruno, A.; Ma, F.; Haque, S.A.; Yartsev, A.; Sundstrom, V.; Nelson, J. Ultrafast Transient Optical Studies of Charge Pair Generation and Recombination in Poly-3-Hexylthiophene(P3ht):[6,6]Phenyl C61 Butyric Methyl Acid Ester (PCBM) Blend Films. J. Phys. Chem. B 2011, 115, 15174–15180. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, R.; Li, D.; Huo, M.-M.; Ai, X.-C.; Zhang, J.-P. Primary Dynamics of Exciton and Charge Photogeneration in Solvent Vapor Annealed P3HT/PCBM Films. J. Phys. Chem. C 2012, 116, 4298–4310. [Google Scholar] [CrossRef]
- Kaunisto, K.M.; Vivo, P.; Dubey, R.K.; Chukharev, V.I.; Efimov, A.; Tkachenko, N.V.; Lemmetyinen, H.J. Charge-Transfer Dynamics in Poly(3-hexylthiophene):Perylenediimide-C60 Blend Films Studied by Ultrafast Transient Absorption. J. Phys. Chem. C 2014, 118, 10625–10630. [Google Scholar] [CrossRef]
- Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C. Plastic Solar Cells. Adv. Funct. Mater. 2001, 11, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Reyes, M.; Kim, K.; Carroll, D.L. High-Efficiency Photovoltaic Devices Based on Annealed Poly(3-hexylthiophene) and 1-(3-Methoxycarbonyl)-propyl-1-phenyl-(6,6)C61 Blends. Appl. Phys. Lett. 2005, 87, 83506. [Google Scholar] [CrossRef]
- Ma, W.; Yang, C.; Gong, X.; Lee, K.; Heeger, A.J. Thermally Stable, Efficient Polymer Solar Cells with Nanoscale Control of the Interpenetrating Network Morphology. Adv. Funct. Mater. 2005, 15, 1617–1622. [Google Scholar] [CrossRef]
- Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. High-Efficiency Solution Processable Polymer Photovoltaic Cells by Self-Organization of Polymer Blends. Nat. Mater. 2005, 4, 864–868. [Google Scholar] [CrossRef]
- Kim, Y.; Cook, S.; Tuladhar, S.M.; Choulis, S.A.; Nelson, J.; Durrant, J.R.; Bradley, D.D.C.; Giles, M.; McCulloch, I.; Ha, C.-S.; et al. A Strong Regioregularity Effect in Self-Organizing Conjugated Polymer Films and High-Efficiency Polythiophene:Fullerene Solar Cells. Nat. Mater. 2006, 5, 197–203. [Google Scholar] [CrossRef]
- Österbacka, R.; An, C.P.; Jiang, X.M.; Verdeny, Z.V. Two-Dimensional Electronic Excitations in Self-Assembled Conjugated Polymer Nanocrystals. Science 2000, 287, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Österbacka, R.; Jiang, X.M.; An, C.P.; Horovitz, B.; Verdeny, Z.V. Photoinduced Quantum Interference Antiresonances in π-Conjugated Polymers. Phys. Rev. Lett. 2002, 88, 226401. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Takao, H.; Yamamoto, J.; Furukawa, S. Infrared Absorption Induced by Field-Effect Doping from Poly(3-alkylthiophene)s. Synth. Met. 2003, 135–136, 341–342. [Google Scholar] [CrossRef]
- Horovitz, B. Infrared Activity of Peiers Systems and Application to Polyacetylene. Solid State Commun. 1982, 41, 729–734. [Google Scholar] [CrossRef]
- Torii, H.; Tasumi, M. Charge Fluxes and Changes in Electronic Structures as the Origin of Infrared Intensities in the Ground and Excited Electronic States. J. Phys. Chem. B 1997, 101, 466–471. [Google Scholar] [CrossRef]
- Meskers, S.C.J.; van Hal, P.A.; Spiering, A.J.H.; Hummelen, J.C.; van der Meer, A.F.G.; Janssen, R.A.J. Time-Resolved Infrared-Absorption Study of Photoinduced Charge Transfer in a Polythiophene-Methanofullerene Composite Film. Phys. Rev. B 2000, 61, 9917–9920. [Google Scholar] [CrossRef]
- Sakamoto, A.; Takezawa, M. Picosecond Time-Resolved Infrared Absorption Study on Photoexcited Dynamics of Regioregular Poly(3-hexylthiophene). Synth. Met. 2009, 159, 809–812. [Google Scholar] [CrossRef]
- Louarn, G.; Trznadel, M.; Buisson, J.P.; Laska, J.; Pron, A.; Lapkowski, M.; Lefrant, S. Raman Spectroscopic Studies of Regioregular Poly(3-alkylthiophenes). J. Phys. Chem. 1996, 100, 12532–12539. [Google Scholar] [CrossRef]
- Shoute, L.C.T.; Pekas, N.; Wu, Y.; McCreery, R.L. Redox Driven Conductance Changes for Resistive Memory. Appl. Phys. A 2011, 102, 841–850. [Google Scholar] [CrossRef]
- Kumar, R.; Pillai, R.G.; Pekas, N.; Wu, Y.; McCreery, R.L. Spatially Resolved Raman Spectroelectrochemistry of Solid-State Polythiophene/Viologen Memory Devices. J. Am. Chem. Soc. 2012, 134, 14869–14876. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Furukawa, Y. Electronic and Vibrational Spectra of Positive Polarons and Bipolarons in Regioregular Poly(3-hexylthiophene) Doped with Ferric Chloride. J. Phys. Chem. B 2015, 119, 4788–4794. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Furukawa, Y. Raman Characterization and Electrical Properties of Poly(3-hexylthiophene) Doped Electrochemically in an Ionic-Liquid-Gated Transistor Geometry. Org. Electron. 2016, 28, 82–87. [Google Scholar] [CrossRef]
- Yamamoto, J.; Furukawa, Y. Raman study of the Interaction between Regioregular Poly(3-hexylthiophene) (P3HT) and Transition-Metal Oxides MoO3, V2O5, and WO3 in Polymer Solar Cells. Chem. Phys. Lett. 2016, 644, 267–270. [Google Scholar] [CrossRef]
- Yu, W.; Zhou, J.; Bragg, A.E. Exciton Conformational Dynamics of Poly(3-hexylthiophene) (P3HT) in Solution from Time-Resolved Resonant-Raman Spectroscopy. J. Phys. Chem. Lett. 2012, 3, 1321–1328. [Google Scholar] [CrossRef]
- Magnanelli, T.J.; Bragg, A.E. Time-Resolved Raman Spectroscopy of Polaron Pair Formation in Poly(3-hexylthiophene) Aggregates. J. Phys. Chem. Lett. 2015, 6, 438–445. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Kurosawa, M. Femtosecond Time-Resolved Raman Spectroscopy Using Stimulated Raman Scattering. Phys. Rev. A 2000, 61, 13808. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond Stimulated Raman Spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef]
- Provencher, F.; Bérubé, N.; Parker, A.W.; Greetham, G.M.; Towrie, M.; Hellmann, C.; Côté, M.; Stingelin, N.; Silva, C.; Hayes, S.C. Direct Observation of Ultrafast Long-Range Charge Separation at Polymer–Fullerene Heterojunctions. Nat. Commum. 2014, 5, 4288. [Google Scholar] [CrossRef]
- Umapathy, S.; Lakshmanna, A.; Mallick, B. Ultrafast Raman Loss Spectroscopy. J. Raman Spectrosc. 2009, 40, 235–237. [Google Scholar] [CrossRef]
- Mallick, B.; Lakshmanna, A.; Umapathy, S. Ultrafast Raman Loss Spectroscopy (URLS): Instrumentation and Principle. J. Raman Spectrosc. 2011, 42, 1883–1890. [Google Scholar] [CrossRef]
- Umapathy, S.; Mallick, B.; Lakshmanna, A. Mode-Dependent Dispersion in Raman Line Shapes: Observation and Implications from Ultrafast Raman Loss Spectroscopy. J. Chem. Phys. 2010, 133, 24505. [Google Scholar] [CrossRef]
- Ai, X.; Beard, M.C.; Knutsen, K.P.; Shaheen, S.E.; Rumbles, G.; Ellingson, R.J. Photoinduced Charge Carrier Generation in a Poly(3-hexylthiophene) and Methanofullerene Bulk Heterojunction Investigated by Time-Resolved Terahertz Spectroscopy. J. Phys. Chem. B 2006, 110, 25462–25471. [Google Scholar] [CrossRef]
- Saeki, A.; Seki, S.; Koizumi, Y.; Tagawa, S. Dynamics of Photogenerated Charge Carrier and Morphology Dependence in Polythiophene Films Studied by in situ Time-Resolved Microwave Conductivity and Transient Absorption Spectroscopy. J. Photochem. Photobiol. A 2007, 186, 158–165. [Google Scholar] [CrossRef]
- Xie, Y.; Li, Y.; Xiao, L.; Qiao, Q.; Dhakal, R.; Zhang, Z.; Gong, Q.; Galipeau, D.; Yan, X. Femtosecond Time-Resolved Fluorescence Study of P3HT/PCBM Blend Films. J. Phys. Chem. C 2010, 114, 14590–14600. [Google Scholar] [CrossRef]
- Furukawa, Y. Reexamination of the Assignments of Electronic Absorption Bands of Polarons and Bipolarons in Conducting Polymers. Synth Met. 1995, 69, 629–632. [Google Scholar] [CrossRef]
- Takaya, T.; Iwata, K. Relaxation Mechanism of β-carotene from S2 (1Bu+) State to S1 (2Ag–) State: Femtosecond Time-Resolved Near-IR Absorption and Stimulated Resonance Raman Studies in 900–1550 nm Region. J. Phys. Chem. A 2014, 118, 4071–4078. [Google Scholar] [CrossRef] [PubMed]
- Takaya, T.; Iwata, K. Development of a Femtosecond Time-Resolved Near-IR Multiplex Stimulated Raman Spectrometer in Resonance with Transitions in the 900–1550 nm Region. Analyst 2016, 141, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Takaya, T.; Anan, M.; Iwata, K. Vibrational Relaxation Dynamics of β-Carotene and Its Derivatives with Substituents on Terminal Rings in Electronically Excited States as Studied by Femtosecond Time-Resolved Stimulated Raman Spectroscopy in the Near-IR Region. Phys. Chem. Chem. Phys. 2018, 20, 3320–3327. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds, P3HT, PCBM, FeCl3, chlorobenzene and acetonitrile, and film samples made from these compounds are not available from the authors. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takaya, T.; Enokida, I.; Furukawa, Y.; Iwata, K. Direct Observation of Structure and Dynamics of Photogenerated Charge Carriers in Poly(3-hexylthiophene) Films by Femtosecond Time-Resolved Near-IR Inverse Raman Spectroscopy. Molecules 2019, 24, 431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24030431
Takaya T, Enokida I, Furukawa Y, Iwata K. Direct Observation of Structure and Dynamics of Photogenerated Charge Carriers in Poly(3-hexylthiophene) Films by Femtosecond Time-Resolved Near-IR Inverse Raman Spectroscopy. Molecules. 2019; 24(3):431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24030431
Chicago/Turabian StyleTakaya, Tomohisa, Ippei Enokida, Yukio Furukawa, and Koichi Iwata. 2019. "Direct Observation of Structure and Dynamics of Photogenerated Charge Carriers in Poly(3-hexylthiophene) Films by Femtosecond Time-Resolved Near-IR Inverse Raman Spectroscopy" Molecules 24, no. 3: 431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24030431