
The Studies on Structure and Stability of CaBn Clusters

School of Chemistry and Chemical Engineering, Xuchang University of China, No. 88 of Bayi Road, Xuchang 461000, China
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(6), 1011; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061011
Submission received: 22 January 2019
/
Revised: 5 March 2019
/
Accepted: 5 March 2019
/
Published: 13 March 2019
(This article belongs to the Special Issue Boron Chemistry and Its Development in the 21st Century: In Memory of Professor Bohumil Štíbr (1940-2020))
Abstract
:Calcium-boron systems have excellent properties of hardness, strength, and chemical stability, and we studied a series of CaBn clusters to investigate their structures and relative stability. The results showed the most stable structures of CaBn clusters are not planar. The B atoms tend to get together and form the planar ring to stabilize the structure, and the Ca atoms are coordinated to the periphery of the formations. The average binding energy (Eb), fragmentation energy (EF), second-order energy difference (Δ2E), adiabatic detachment energy (ADE), and adiabatic electron affinity (AEA) of the CaBn clusters were calculated to investigate the relative stability and the ability of removing or obtaining an electron. As shown by the results, EF and Δ2E values had obvious odd-even alteration as n increased, which indicated that the formations CaB4, CaB6, and CaB8 were more stable. The ADE values for CaBn clusters with even values of n were higher than those with odd values of n, which indicated CaBn clusters with even values of n had difficultly removing an electron. The AEA values of CaB3 and CaB7 were larger than the others, which meant CaB3 and CaB7 easily obtained an electron. These results provide a useful reference for understanding the formation mechanism and stability of the alkaline earth metal boride as well as guidance for synthesizing the CaBn clusters.
1. Introduction
The boride compounds display excellent properties of hardness, strength, and chemical stability because of their structural characteristics. Due to the discovery of the superconductivity of MgB2 [1], the alkaline earth metal borides have been of great concern [1,2,3,4,5]. As an important member of the alkaline earth metal boride, the calcium boron systems are mentioned by many scientists [2,3,4,5,6]. Tian et al. [2] successfully synthesized CaB4 crystals under high-temperature high-pressure (HPHT) conditions and pointed out a potential synthesis method for the CaB2 crystal. Tian et al. [3] also prepared the CaB6 polycrystalline samples by solid phase sintering. At the same time, Zhang et al. [4,5] prepared the different composition CaB6 films by direct current (DC) magnetron sputtering. Due to the excellent properties of the alkaline earth metal boride systems, theoretical scientists have also shown interest in them [7,8,9,10,11]. Li et al. [7,8,9] studied the series of MB5+ (M = Be, Mg, Ca, Sr) and MB6 (M = Be, Mg, Ca, and Sr). Ju et al. [10] studied the geometric structures, stabilities, and electronic properties of (n = 1–7 and m = 0, 1). Last year, our group studied the structures and stabilities of BeBn+ (n = 1–8) clusters [11]. There have also been many theoretical works reported on the structures and stabilities of small-metal-atom-doped boron clusters [12,13,14,15,16,17,18,19,20]. However, until now, no systematic investigation for the CaBn cluster had been performed to see the effect of the Ca atom doping on boron clusters. In this context, we investigated the stable configurations of the small clusters CaBn (n = 1–8) formed by adding one Ca heteroatom into the corresponding Bn cluster. The stable configurations CaBn were analyzed and compared with MgBn clusters [10]. We also calculated the average binding energy (Eb), fragmentation energy (EF), second-order energy difference (Δ2E), adiabatic detachment energy (ADE), and adiabatic electron affinity (AEA) in order to evaluate the stability and ability of CaBn (n = 1–8) clusters to obtain and remove an electron.
2. Computational Details
In this paper, all calculations were performed with the Gaussian 03 [21] program package. The geometries of CaBn clusters were fully optimized by using the B3LYP [22,23] method with 6-311+G(d) basis set and were characterized as energy minima by frequency calculations at the same level. The zero-point energies (ZPE) were also obtained at this level. In order to get more reliable electronic energy, the single point energy calculations for all the local minima were obtained at the MP2 [24]/6-311+G(d) level based on the geometry optimized at the B3LYP/6-311+G(d) level. All the lower-lying isomer energies were obtained at the MP2/6-311+G(d) level with zero point energy correction from the B3LYP/6-311+G(d) level. In each group of isomers, the reference energy was taken as that of the lowest geometry. The Eb, EF, Δ2E, ADE, and AEA of the CaBn clusters were calculated to investigate the relative stability and the ability of removing or obtaining an electron at the same level of theory. The following formulas were used:
where E(B), E(CaBn), E(CaBn+1), and E(CaBn-1) are the energies of the most stable structures of B, CaBn, CaBn+1, and CaBn-1, respectively.
where E is the energy of the optimized structures of CaBn+, CaBn−, and CaBn, each in its vibrational ground state.
3. Results and Discussions
3.1. Stable Geometric Structures
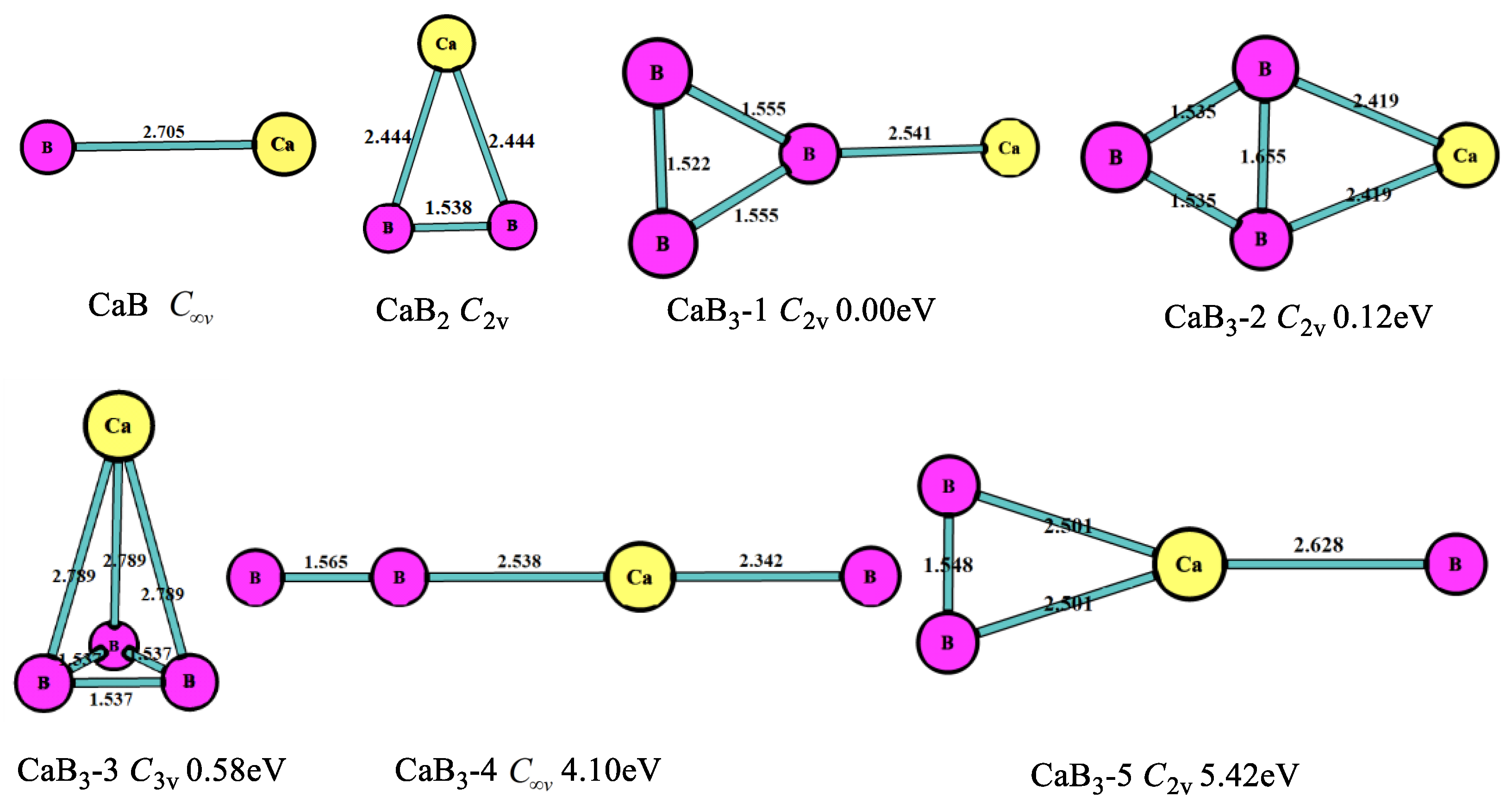
The optimized structures, symmetry point groups, and relative energies of the CaBn (n = 1–8) clusters are displayed in Figure 1, Figure 2, Figure 3, Figure 4, Figure 5 and Figure 6 and Table 1. These structures included the lowest-energy structures and their low-lying isomers, and they were ordered from the lowest to highest energy. All of them had no imaginary frequency, and the relative energies were given in eV based on the most stable ones. All of the structures of CaBn (n = 1–8) clusters were singlet state (where n is even number) or doublet state (where is odd number).
As seen in Figure 1, for n = 1, the most stable structure was the linear geometry with the Ca-B bond length of 2.705 Å and point group . For n = 2, the most stable structure was triangle (CaB2, C2v), and none of the chain isomers of CaB2 were stable—that is, they were different from the MgB2 cluster structures [10]. The Ca-B bond length of CaB2 structure was 2.444 Å, remarkably longer than the Mg-B bond length of isomer MgB2 (2.265 Å) [10]. In the case of n = 3, the most stable geometry (CaB3-1, C2v) was the triangle boron ring with Ca connecting to one B atom in the B3 clusters, which was similar to the MgB3 cluster. The second stable one (CaB3-2, C2v) was the planar four-member ring including a triangle boron ring. The B-B bond length (1.655 Å) in CaB3-2 was longer than the B-B bond (1.555 Å and 1.522 Å) in CaB3-1, thus the energy of CaB3-2 was higher (0.12 eV) than that of CaB3-1. The third stable one (CaB3-3, C3v) was the trigonal pyramid structure with the Ca-B bond length of 2.789 Å. There was only a linear structure (CaB3-4) with the Ca atom at the middle of the chain. Above, the most stable structures of CaBn (n = 1–3) were similar to MgBn (n = 1–3).
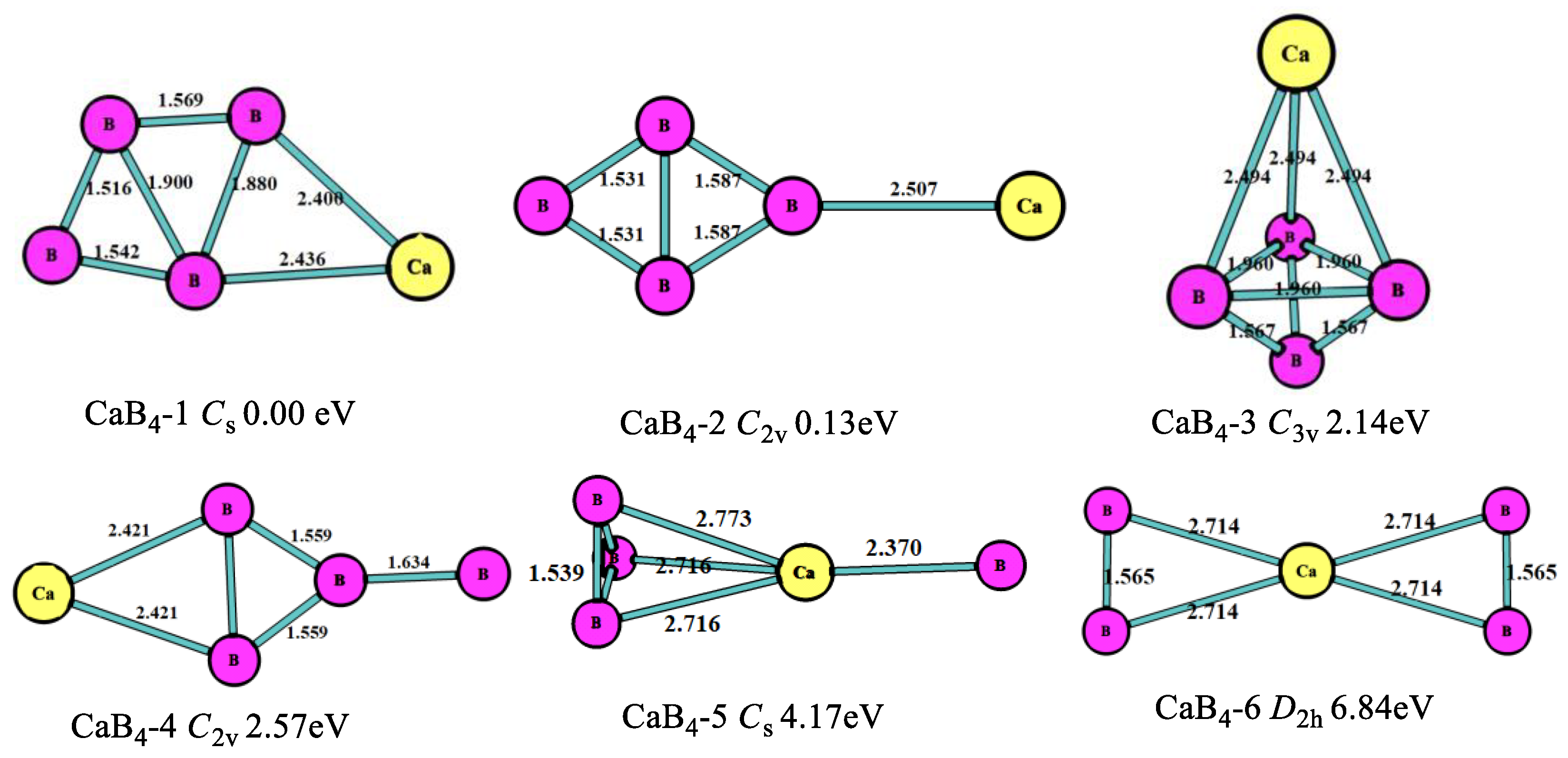
As shown in Figure 2, the lowest-energy structure (CaB4-1, Cs) was the planar five-member ring including a four-member boron ring, which was different from the MgB4 cluster. The second stable one (CaB4-2, C2v) was the four-member boron ring with the Ca connecting to one B atom in the B4 clusters, which had an energy higher (0.13 eV) than that of the most stable one. The third stable one (CaB4-3, C3v) was the tetrahedron, which was 2.14 eV higher than the most stable one. In this structure, the B-B bond lengths were 1.960 Å and 1.567 Å, and the Ca-B bond length was 2.494 Å. From the consideration of Figure 1 and Figure 2, we found B atoms tended to get together and make more stable structures.
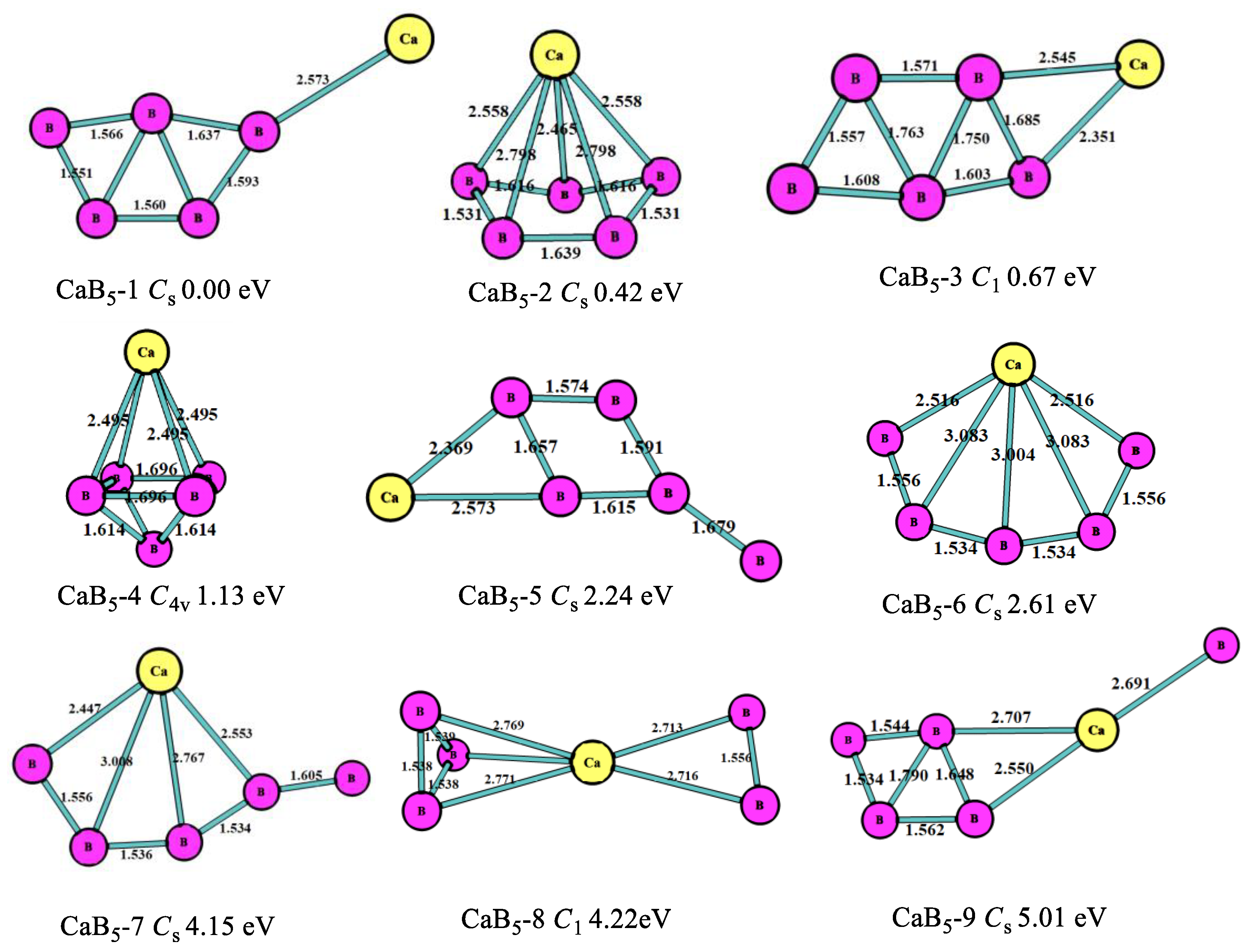
For n = 5, as shown in Figure 3, the most stable one (CaB5-1, Cs) was the five-member boron ring with the Ca atom connected to the B atom of B5 clusters, which was similar to the most stable one of the MgB5 cluster [10]. The second lowest-energy structure (CaB5-2, Cs) was the pentagonal pyramid including a five-member boron ring that was 0.42 eV higher in energy than CaB5-1 with Cs symmetry. The third most stable one (CaB5-3, C1) was the six-member ring including a five-member boron ring. The fourth lowest-energy structure was the quadrangular bipyramid with C4v symmetry, which was 1.13 eV larger in energy than CaB5-1. As depicted in Figure 3, the B atoms of all the structures tended to get together and form the planar or quasi-planar boron clusters that were the same characters as in MgBn and BeBn+ [10,11].
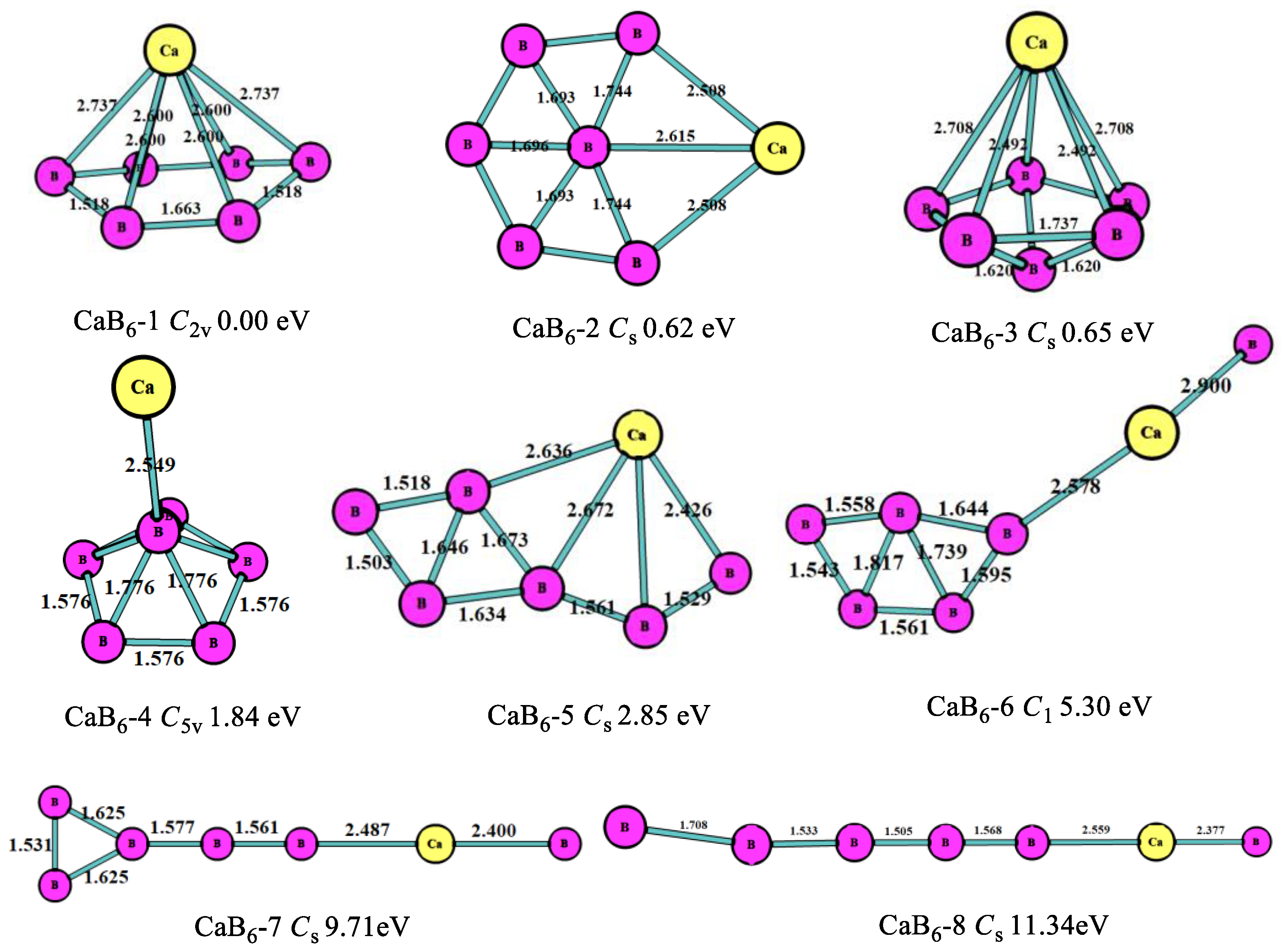
At n = 6, as shown in Figure 4, the lowest-energy structure (CaB6-1, C2v) for the CaB6 cluster was the hexagonal pyramid, which was different from MgB6 [10]. The second stable one (CaB6-2, Cs) was the planar six-member ring with a B atom in the middle, which was similar to the most stable MgB6 structure. The third stable one (CaB6-3, Cs) was the pentagonal bipyramid, which was 0.65 eV higher in energy than the most stable one. The fourth stable one (CaB6-4, C5v) was the pentagonal pyramid B6 with connected Ca atom. As is evident in Figure 4, the B atoms also tended to form planar or quasi-planar boron clusters and keep the structures more stable. This is because the planar or quasi-planar boron cluster stability is greatly defined by the aromaticity caused by the p- and d-delocalization [25], which requires a cyclic configuration consisting of at least three boron atoms. Thus, the clusters with cyclic boron configuration of CaBn clusters are more stable than others.
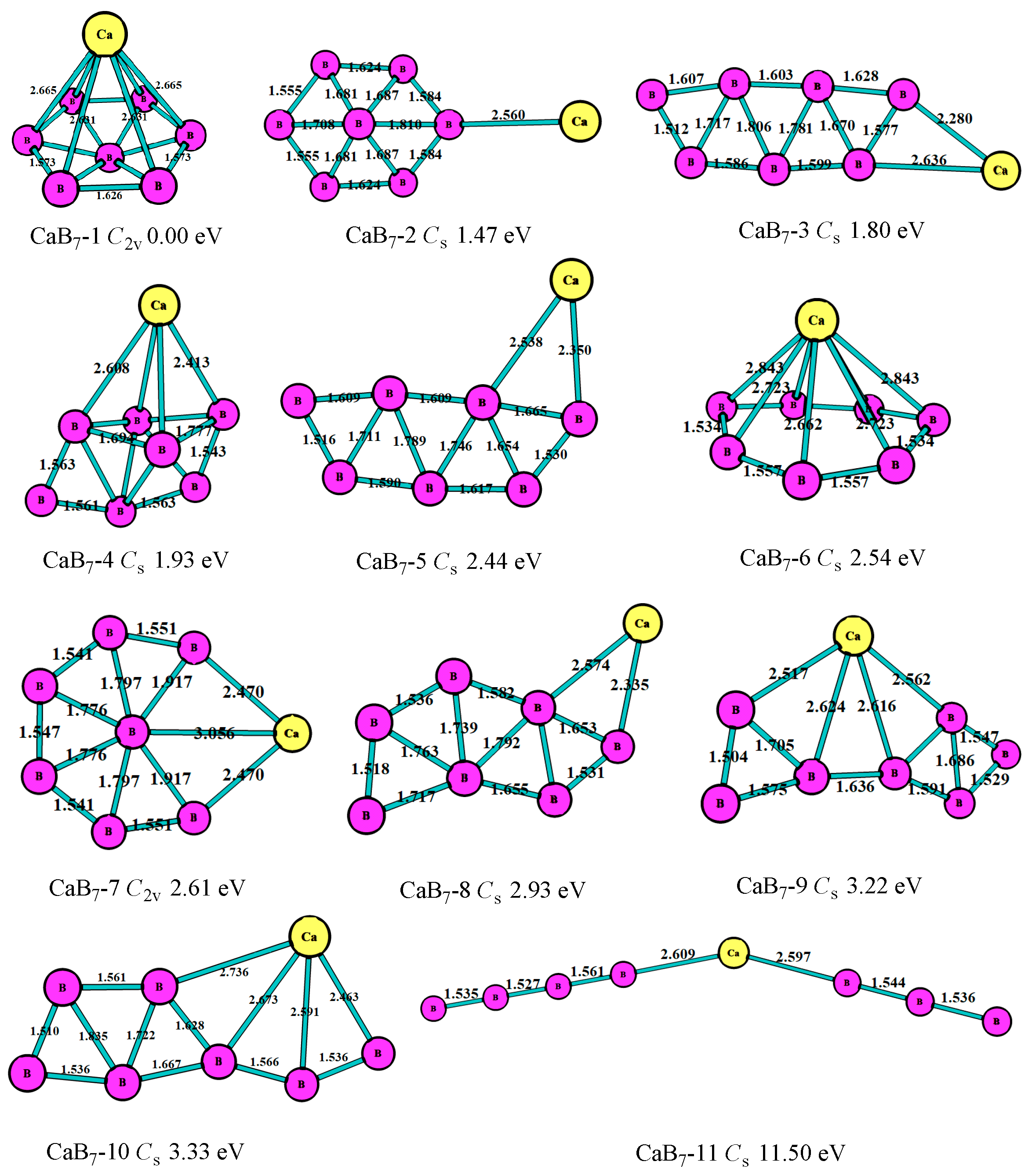
In the case of n = 7, as shown in Figure 5, the most stable (CaB7-1, C2v) of the CaB7 cluster was the hexagonal bipyramid geometry. The second lowest-energy structure (CaB7-2, Cs) was the hexagonal pyramid of B7 with a connected Ca atom, which was similar to the most stable structure of MgB7. The third lowest-energy structure (CaB7-3, Cs) was the planar seven-member ring, which was 1.80 eV higher than the most stable one. Figure 5 displays that most of the CaB7 clusters were not planar, which was different from that of the MgB7 clusters. As with the bond characteristics, the B atoms tended to get together and form the triangle boron ring, and the Ca atom was at the periphery.
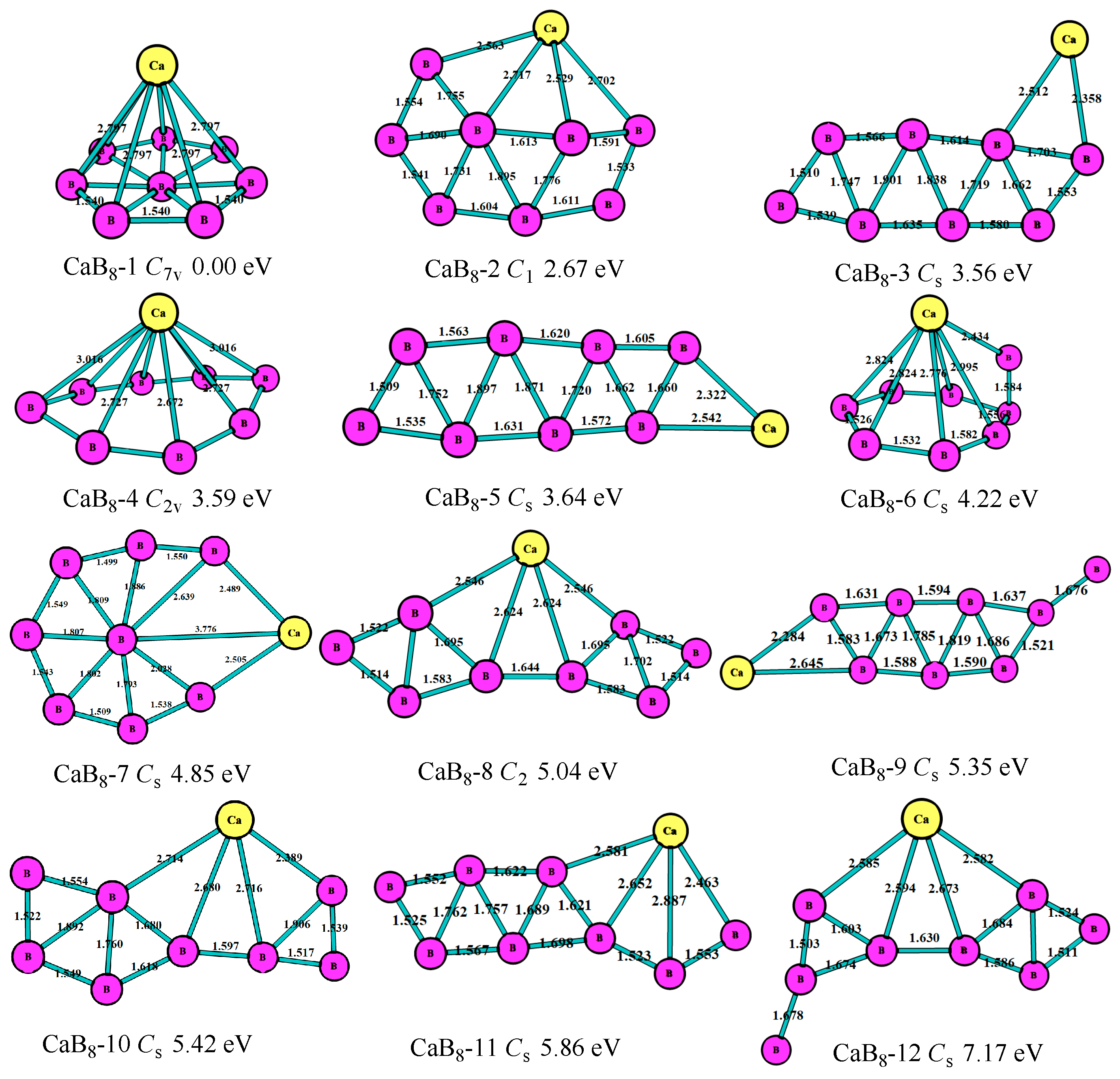
For n = 8, as shown in Figure 6, the lowest-energy structure (CaB8-1, Cs) was the six-member ring of B7 with the Ca atom above the boron ring, which was the same with the most stable of the BeB8+ cluster [11]. The second lowest-energy structure (CaB8-2, C2v) was the seven-member ring with two B atoms above the ring, which had an energy 2.67 eV higher than that of the most stable one. The third lowest-energy structure (CaB8-3, Cs) was the eight-member boron ring with the Ca atom connected, which was 3.56 eV higher than the most stable one. The fourth stable one (CaB8-4, C2v) was the eight-member boron ring with the Ca atom above the ring. The fifth stable one (CaB8-5, Cs) was the nine-member boron ring with a Ca atom at the periphery. Figure 6 shows the B atoms also tended to get together to form the triangle boron ring to keep the structure more stable for the CaB8 clusters.
3.2. Relative Stability
To evaluate the relative stability of CaBn clusters, Table 1 shows the Eb, EF, and Δ2E at the MP2/6-311+G(d) level. These parameters were obtained by the following formula and plotted as the function of the cluster size n in Figure 7, Figure 8 and Figure 9.
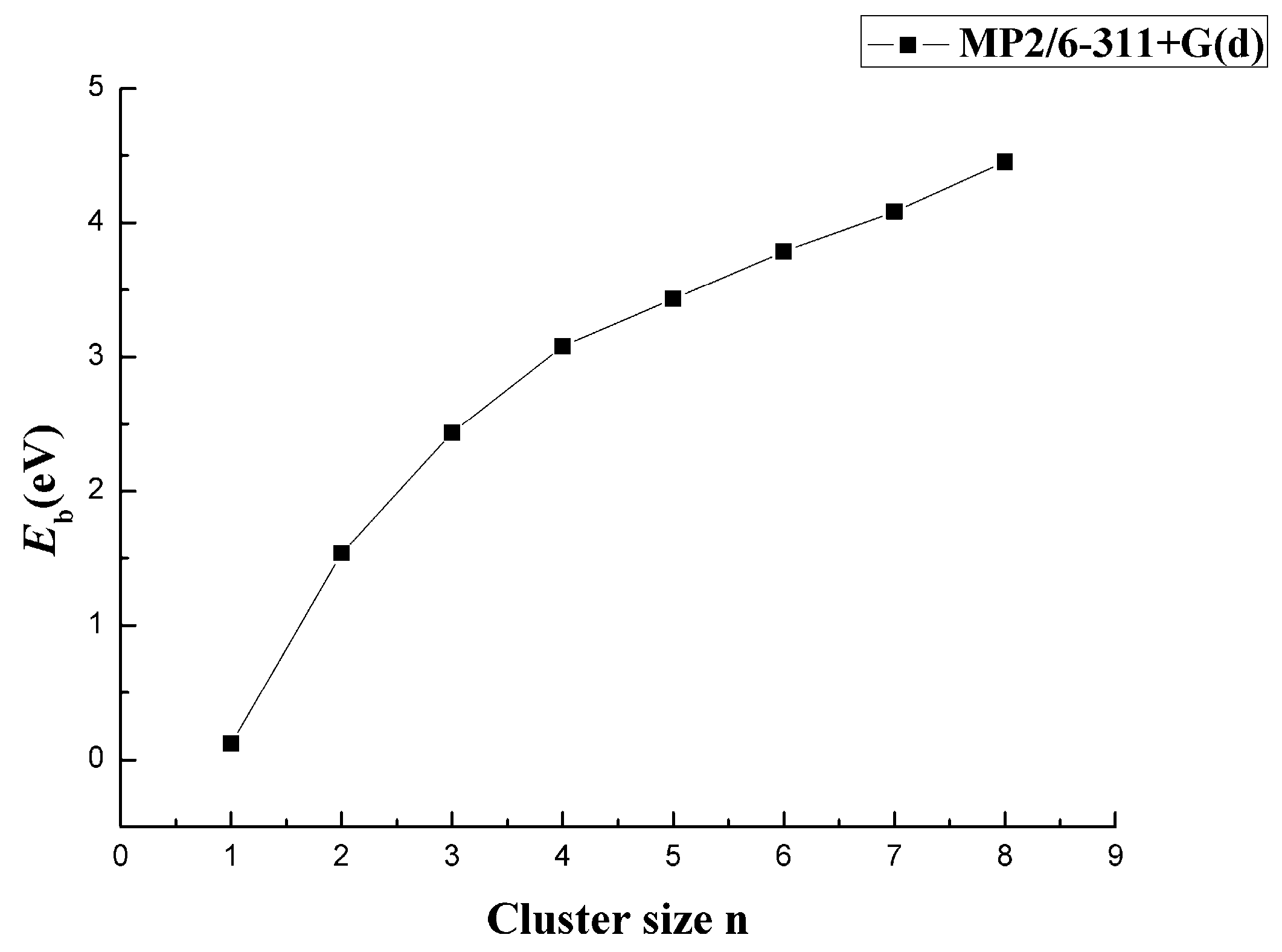
In Figure 7, the energy Eb versus size n at the MP2/6-311+G(d) level is shown. All average binding energies Eb of the lowest-energy clusters increased as the size n increased, but after n = 3, the increase became smaller and stable. This was due to the bond tendency to saturate with the increase in the number of atoms. Eb increased as the number of B atoms increased. If the number of Ca atoms did not change, the system could form a stable, large-sized CaBn cluster.
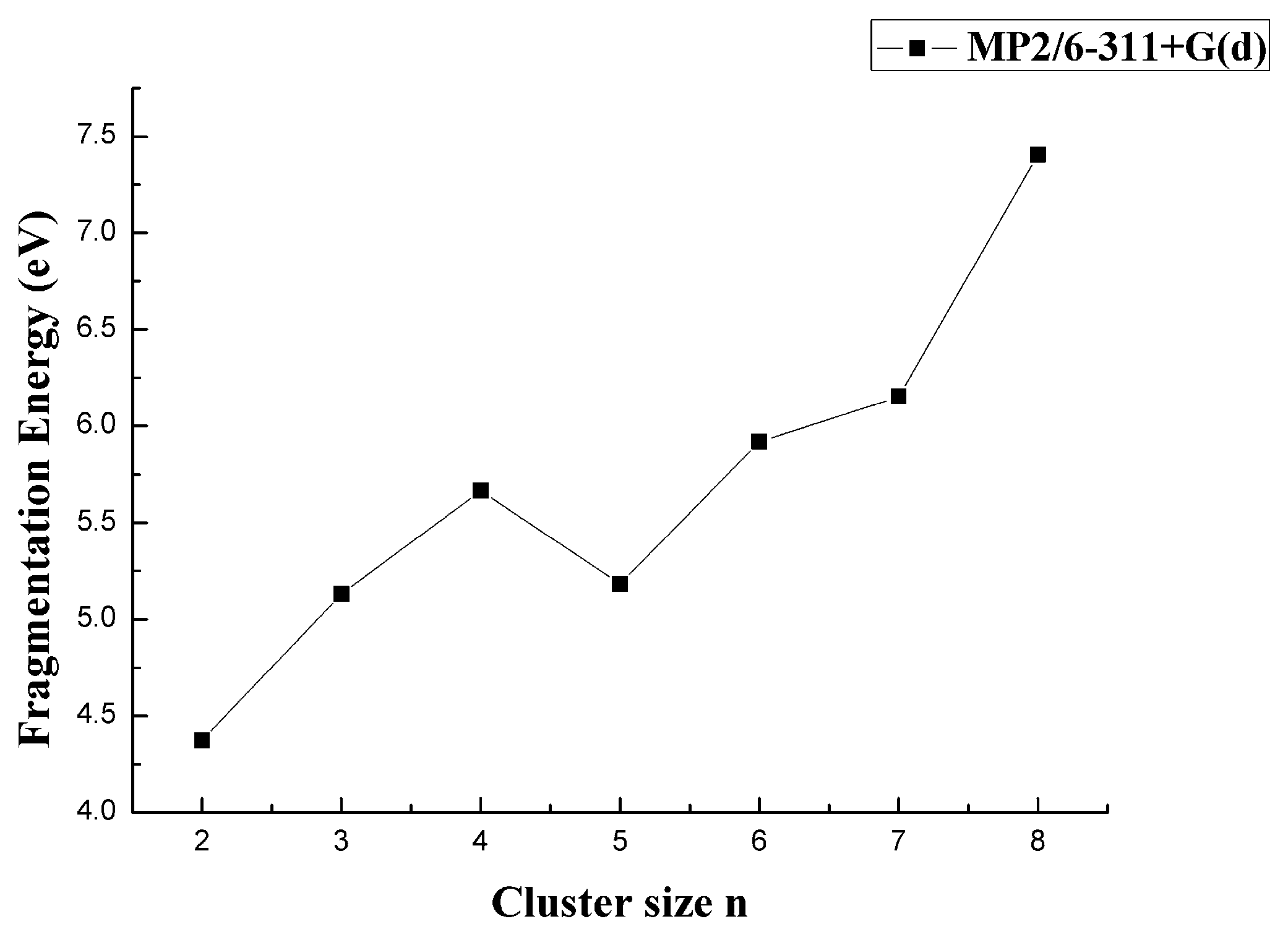
As shown in Figure 8 by the MP2 result for all the clusters, the global minimum of fragmentation energy appeared at n = 2. The energy followed obvious odd-even alterations as the size n increased from n = 3. However, it was noted that the fragmentation energy had its local-maximum when n was even, which indicated the CaB4, CaB6, and CaB8 were more stable. These results opposed those obtained for MgBn and BeBn+. Among all the CaBn (n = 1–8) clusters, the CaB8 cluster was the most stable.
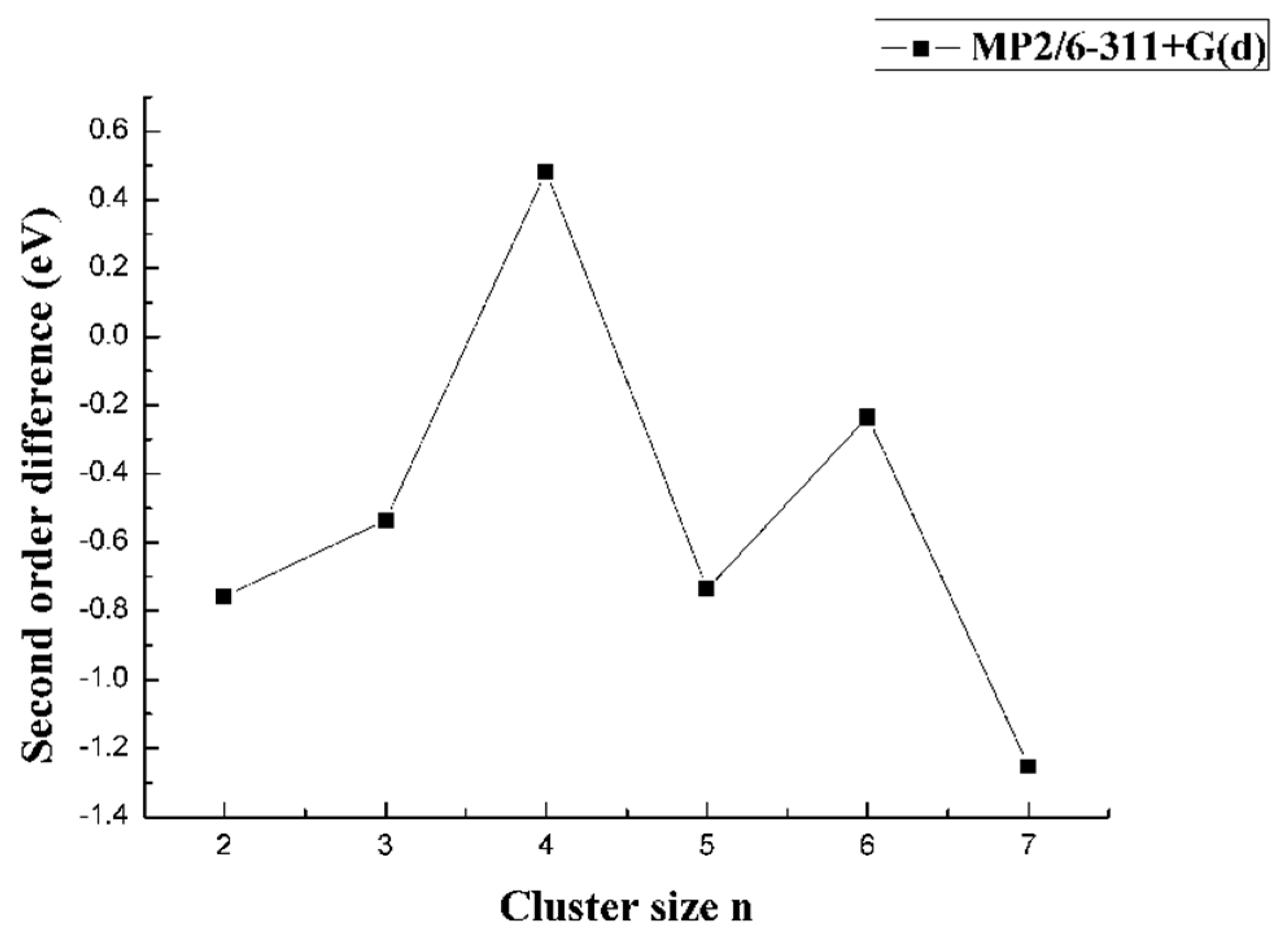
The second-order difference in total energy is considered a very useful quantity that can reflect the relative cluster stability in the field of cluster physics [26]. From the MP2 result, the second-order difference energy followed a clear “odd-even oscillation” phenomenon for n = 3–7, as shown in Figure 9. When n was even, the value was at the peak, which indicated the higher stability of these clusters. The results were consistent with the information revealed in Figure 8. When n = 4, the second-order difference energy was the largest, which meant the CaB4 was the most stable among CaBn (n = 3–7).
3.3. The Ability of Obtaining or Removing an Electron
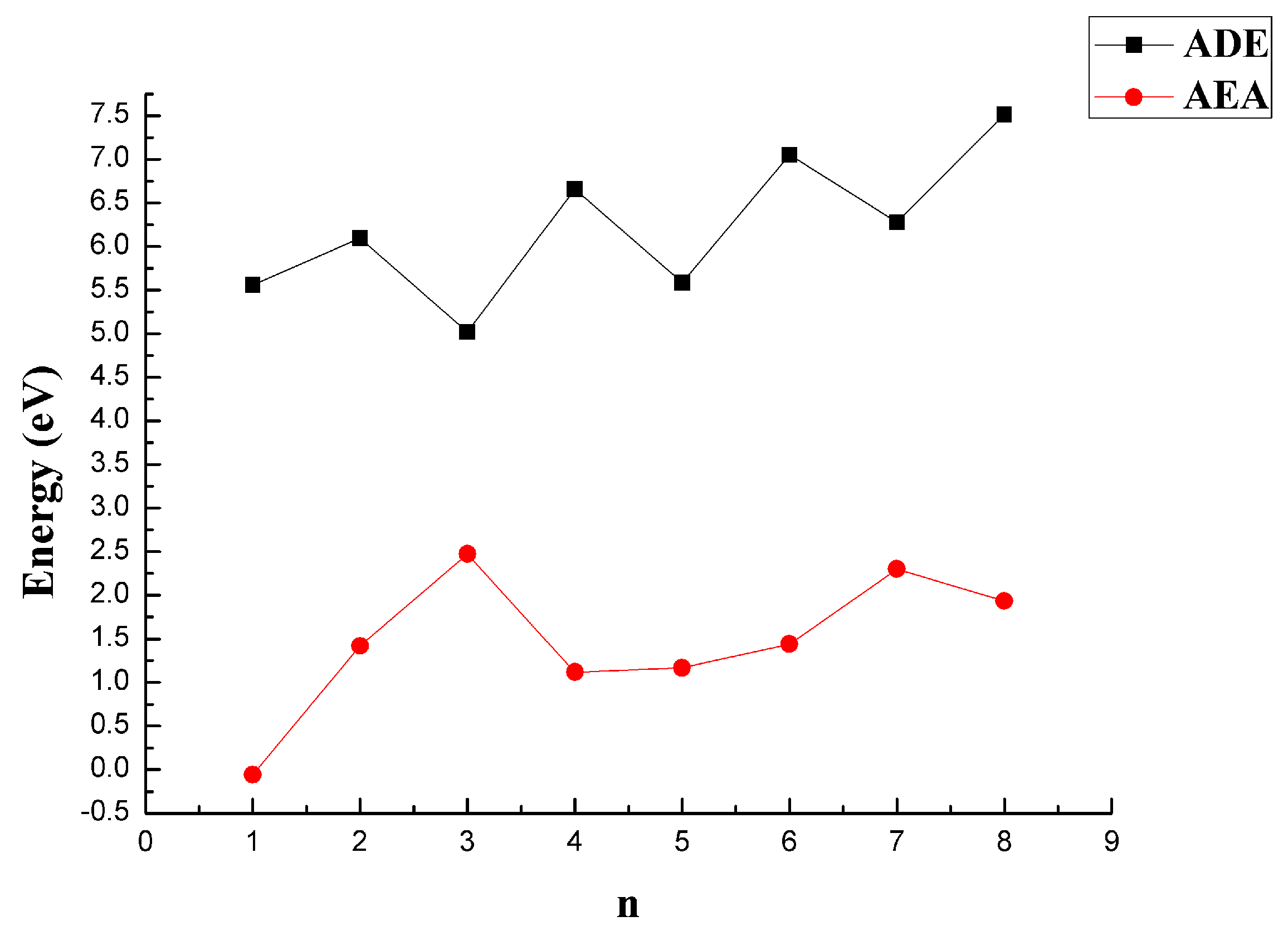
The ADE and AEA of the CaBn clusters were calculated to investigate the ability of the most stable CaBn clusters to remove or obtain an electron. A larger value of ADE meant that it was difficult for the cluster to remove an electron, and a larger value of AEA meant that it was easy to gain an electron. The results are shown in Figure 10 and Table 1. The ADE values for CaBn clusters with even values of n were higher than those with odd values of n. This indicated that CaBn clusters with even values of n had more difficulty removing an electron than those with odd values of n. The AEA values first increased and then decreased over the range of n = 1–4, and they subsequently increased and then decreased for n = 4–8. The larger AEA values for n = 3 and n = 7 revealed that obtaining an electron was easy.
4. Conclusions
The geometries, stabilities, and electronic properties of CaBn clusters up to n = 8 were systematically investigated using the B3LYP and MP2 method. It was found that the most stable structures of CaBn clusters as n increased were not the planar configurations. The B atoms tended to form the planar ring to keep the structure more stable, and the Ca atoms were coordinated to the structure periphery. For the most stable structures, the average binding energy, the fragmentation energy, and second-order difference of total energies were widely used to evaluate the relative stability of clusters. The results showed they had obvious odd-even alterations as the size n increased. When n was even, it had its local-maximum, which indicated the CaB4, CaB6, and CaB8 were more stable. The ADE and AEA of the CaBn clusters were calculated to investigate the ability of removing or obtaining an electron. The results showed the ADE values for CaBn clusters with even values of n were higher than those with odd values of n, which indicated CaBn clusters with even values of n had difficultly removing an electron, and the AEA values of CaB3 and CaB7 were larger than the others, which meant CaB3 and CaB7 easily obtained an electron.
Author Contributions
Data curation, F.C. and B.Q.; Formal analysis, C.L.; Funding acquisition, C.L.; Investigation, P.H.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 21703195, 31601447; the Foundation of Henan Educational Committee of China, grant number 17B150013; and Henan Scientific Committee of China, grant number 172102210466.
Conflicts of Interest
The authors declare no competing financial interest.
References
- Buzea, C.; Yamashita, T. Review of the superconducting properties of MgB2. Supercond. Sci. Technol. 2001, 14, R115–R146. [Google Scholar] [CrossRef]
- Liu, Z.; Han, X.; Yu, D.; Sun, Y.; Xu, B.; Zhou, X.; He, J.; Wang, H.; Tian, Y. Formation, structure, and electric property of CaB4 single crystal synthesized under high pressure. Appl. Phys. Lett. 2010, 96, 031903. [Google Scholar] [CrossRef]
- Xin, S.; Han, X.; Liu, S.; Liu, Z.; Bo, X.; Tian, Y.; Yu, D. CaB6 single crystals grown under high pressure and hightemperature. J. Cryst. Growth. 2010, 313, 47–50. [Google Scholar] [CrossRef]
- Zhang, L.; Min, G.; Yu, H. Sintering process and high temperature stability investigation for nano-scale CaB6 materials. Ceram. Int. 2010, 36, 2253–2257. [Google Scholar] [CrossRef]
- Zhang, L.; Min, G.; Yu, H. Reaction mechanism and size control of CaB6 micron powder synthesized by the boroncarbide method. Ceram. Int. 2009, 35, 3533–3536. [Google Scholar]
- Chen, F.; Ju, M.; Kuang, X.Y.; Yeung, Y. Insights into the Microstructure and Transition Mechanism for Nd3+ Doped Bi4Si3O12: A Promising Near-Infrared Laser Material. Inorg. Chem. 2018, 57, 4563–4570. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Jin, Q. Aromaticity of Planar B5− Anion in the MB5 (M = Li, Na, K, Rb, and Cs) and MB5+ (M = Be, Mg, Ca, and Sr) Clusters. J. Phys. Chem. A 2004, 108, 855–860. [Google Scholar] [CrossRef]
- Li, Q.S.; Jin, Q. Theoretical Study on the Aromaticity of the Pyramidal MB6 (M = Be, Mg, Ca, and Sr) Clusters. J. Phys. Chem. A 2003, 107, 7869–7873. [Google Scholar] [CrossRef]
- Gong, L.F.; Guo, W.L.; Wu, X.M.; Li, Q.S. B7− as a novel ligand: Theoretical investigations on structures and chemical bonding of LiB7 and BeB7+. Chem. Phys. Lett. 2006, 429, 326–334. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Zhao, F.Q.; Ju, X.H. DFT study on structure and stability of MgBn±m clusters. Comput. Theor. Chem. 2014, 1027, 151–159. [Google Scholar] [CrossRef]
- Liu, C.; Si, H.; Han, P.; Tang, M. Density functional theory study on structure and stability of BeBn+ clusters. Rapid Commun. Mass Spectrom. 2017, 31, 1437–1444. [Google Scholar] [CrossRef]
- Lei, X.L.; Zhu, H.J.; Ge, G.X.; Wang, X.M.; Luo, Y.H. Structures and magnetism of BnNi (n = 6–12) clusters from density-functional theory. Acta Phys.Sin. 2008, 57, 5491–5499. [Google Scholar]
- Yang, Z.; Yan, Y.L.; Zhao, W.J.; Lei, X.L.; Ge, G.X.; Luo, Y.H. Structures and magnetism of FeBN (N ≤ 6) clusters. Acta. Phys. Sin. 2007, 56, 2590–2595. [Google Scholar]
- Liu, X.; Zhao, G.F.; Guo, L.J.; Jing, Q.; Luo, Y.H. Structural, electronic, and magnetic properties of MBn (M = Cr, Mn, Fe, Co, Ni, n ≤ 7) clusters. Phys. Rev. A 2007, 75, 063201. [Google Scholar] [CrossRef]
- Böyükata, M.; Güvenc, Z.B. Density functional study of AlBn clusters for n = 1–14. J. Alloys Compd. 2011, 509, 4214–4234. [Google Scholar] [CrossRef]
- Truong, B.T.; Nguyen, M.T. Thermochemical properties, electronic structure and bonding of mixed lithium boron clusters (BnLi, n = 1–8) and their anions. Chem. Phys. 2010, 375, 35–45. [Google Scholar]
- Yao, J.G.; Wang, X.W.; Wang, Y.X. A theoretical study on structural and electronic properties of Zr-doped B clusters: ZrBn (n = 1–12). Chem. Phys. 2008, 351, 1–6. [Google Scholar] [CrossRef]
- Feng, X.J.; Luo, Y.H. Structure and Stability of Al-Doped Boron Clusters by the Density-Functional Theory. J. Phys. Chem. A 2007, 111, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.F.; Ma, L.J.; Wang, J.F. Structures and stabilities of ScBn (n = 1–12) clusters: An ab initio investigation. J. Mol. Model. 2013, 19, 3255–3261. [Google Scholar] [CrossRef]
- Jia, J.F.; Li, X.R.; Li, Y.N.; Ma, L.J.; Wu, H.S. Density functional theory investigation on the structure and stability of Sc2Bn (n = 1–10) clusters. Comput. Theor. Chem. 2014, 1027, 128–134. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A. Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Aihara, J.; Ishida, T. Aromaticity of planar boron clusters confirmed. J. Am. Chem. Soc. 2005, 127, 13324–13330. [Google Scholar] [CrossRef]
- Wang, J.L.; Wang, G.H.; Zhao, J. Structure and electronic properties of Gen (n = 2–25) clusters from density-functional theory. Phys. Rev. B. 2001, 64, 205411. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
Figure 1.
Geometry of the CaBn (n = 1–3) clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.
Figure 1.
Geometry of the CaBn (n = 1–3) clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.

Figure 2.
The geometry structures of the CaB4 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.
Figure 2.
The geometry structures of the CaB4 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.

Figure 3.
The geometry structures of the CaB5 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.
Figure 3.
The geometry structures of the CaB5 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.

Figure 4.
The geometry structures of the CaB6 clusters obtained MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.
Figure 4.
The geometry structures of the CaB6 clusters obtained MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.

Figure 5.
The geometry structures of the CaB7 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.
Figure 5.
The geometry structures of the CaB7 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.

Figure 6.
The geometry structures of the CaB8 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.
Figure 6.
The geometry structures of the CaB8 clusters obtained at MP2/6-311+G(d)//B3LYP/6-311+G(d). The bond length unit is Å.

Figure 7.
Size dependence of the average binding energy of CaBn (n = 1–8) clusters.

Figure 8.
Size dependence of the fragmentation energy of CaBn (n = 1–8) clusters.

Figure 9.
Size dependence of the second-order difference energy of CaBn (n = 1–8) clusters.

Figure 10.
ADE and AEA of the most stable structures of CaBn (n = 1–8) clusters at the MP2/6-311+G(d)//B3LYP/6-311+G(d) level.
Figure 10.
ADE and AEA of the most stable structures of CaBn (n = 1–8) clusters at the MP2/6-311+G(d)//B3LYP/6-311+G(d) level.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The energies, the average binding energy (Eb), the fragmentation energy (EF), second-order difference of total energy (Δ2E), adiabatic detachment energy (ADE), and adiabatic electron affinity (AEA) of the CaBn clusters of the most stable structures of CaBn at the MP2/6-311+G(d)//B3LYP/6-311+G(d) level.
Table 1.
The energies, the average binding energy (Eb), the fragmentation energy (EF), second-order difference of total energy (Δ2E), adiabatic detachment energy (ADE), and adiabatic electron affinity (AEA) of the CaBn clusters of the most stable structures of CaBn at the MP2/6-311+G(d)//B3LYP/6-311+G(d) level.
| CaBn | Energies/a.u. | Eb/eV | EF/eV | Δ2E/eV | ADE/eV | AEA/eV |
|---|---|---|---|---|---|---|
| CaB | −701.5209 | 0.1196 | — | — | 5.5593 | −0.0595 |
| CaB2 | −726.2515 | 1.5378 | 4.3741 | −0.7567 | 6.0984 | 1.4198 |
| CaB3 | −751.0098 | 2.4361 | 5.1308 | −0.5352 | 5.0175 | 2.4738 |
| CaB4 | −775.7879 | 3.0820 | 5.6660 | 0.4833 | 6.6577 | 1.1190 |
| CaB5 | −800.5481 | 3.4321 | 5.1826 | −0.7357 | 5.5863 | 1.1684 |
| CaB6 | −825.3354 | 3.7873 | 5.9184 | −0.2340 | 7.0507 | 1.4423 |
| CaB7 | −850.1314 | 4.0829 | 6.1523 | −1.2530 | 6.2788 | 2.3017 |
| CaB8 | −874.9733 | 4.4521 | 7.4053 | — | 7.5131 | 1.9359 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, P.; Chai, F.; Qiao, B.; Liu, C. The Studies on Structure and Stability of CaBn Clusters. Molecules 2019, 24, 1011. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061011
AMA Style
Han P, Chai F, Qiao B, Liu C. The Studies on Structure and Stability of CaBn Clusters. Molecules. 2019; 24(6):1011. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061011
Chicago/Turabian StyleHan, Peilin, Fengli Chai, Bolin Qiao, and Chunhui Liu. 2019. "The Studies on Structure and Stability of CaBn Clusters" Molecules 24, no. 6: 1011. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061011