Density Functional Theory Investigations on the Mechanism of Formation of Pa(V) Ion in Hydrous Solutions


Abstract
:1. Introduction
2. Results and Discussion
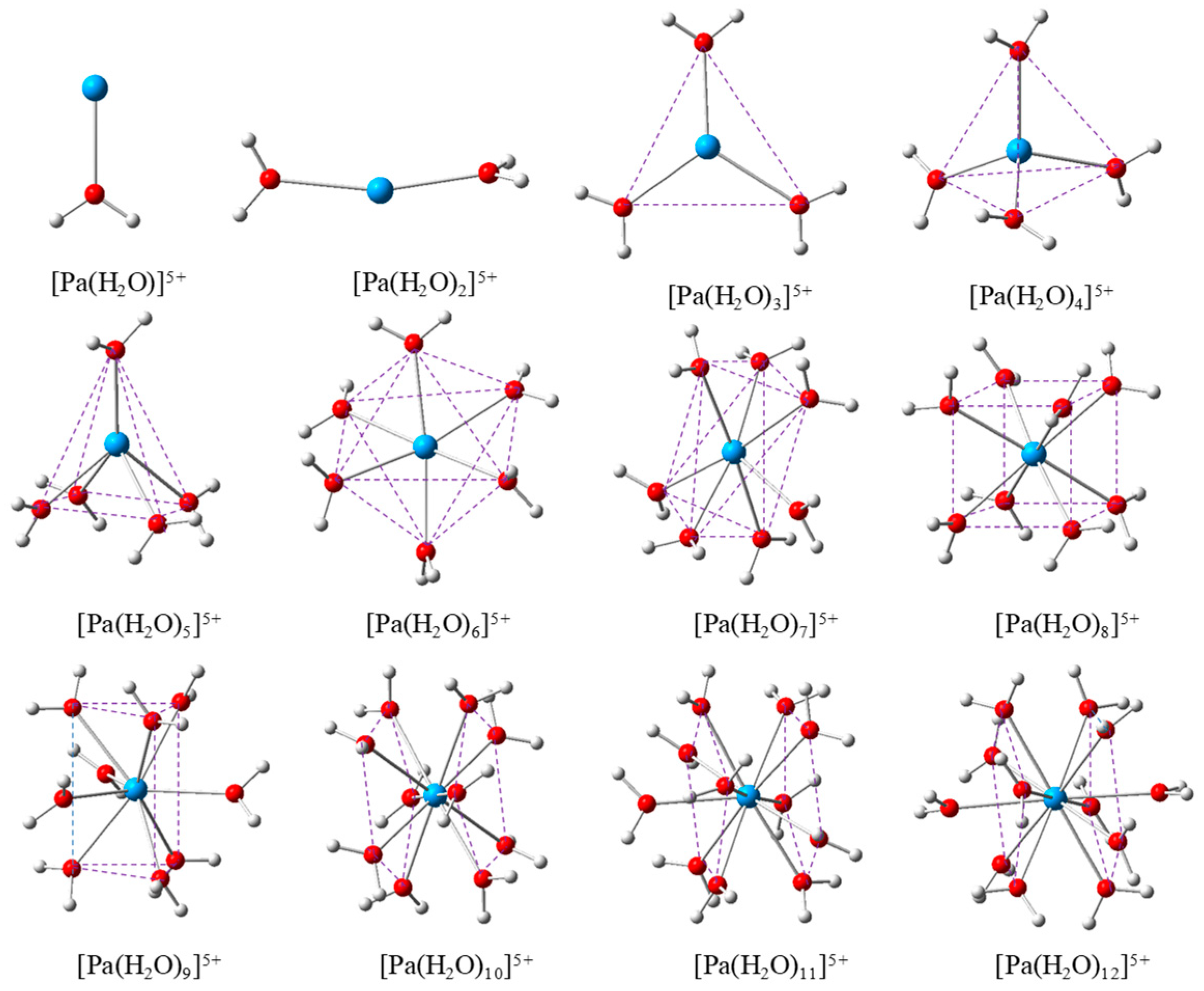
2.1. Structure of hydrated Pa(V) ions
2.2. Analysis on Pa-O Bond Distance
2.3. Thermodynamic analysis
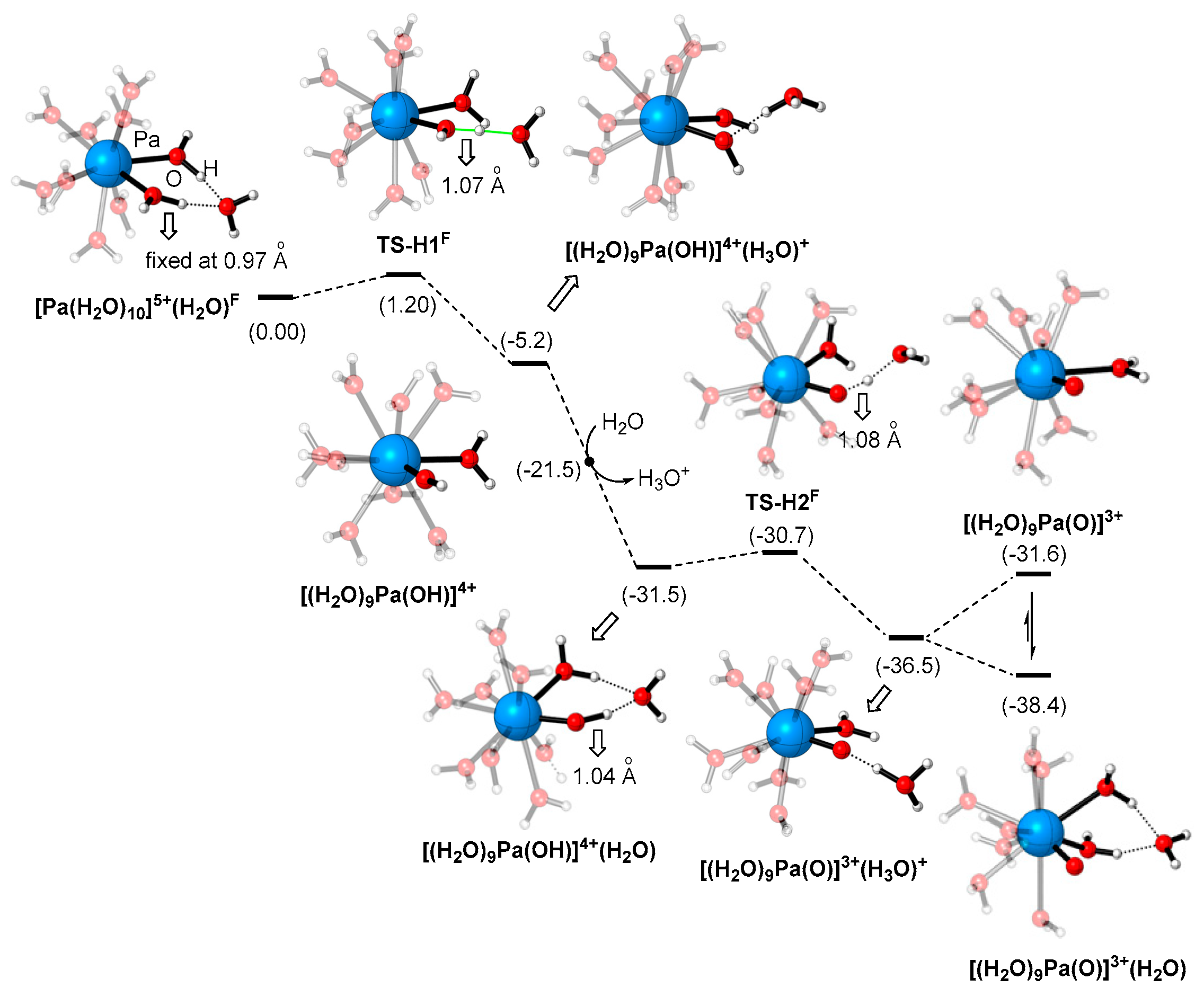
2.4. Formation of [Pa=O(H2O)x]3+ from [Pa(H2O)n]5+
3. Computation Methods
4. Conclusions
- (1)
- The maximum allowed coordination number of water ligands on the Pa(V) centre is 10.
- (2)
- [Pa(H2O)10]5+ is the initial species in aqueous solution, but is labile to the proton transfer with the water ligands on the second coordination sphere, to form the stable Pa=O complexes with higher thermodynamic stability. The transformations are kinetically highly feasible (with energy demand < 1.5 kcal/mol).
- (3)
- The most stable Pa=O structure in aqueous solution is [PaO(H2O)6]3+. This structure is formed by the proton transfer-water dissociation mechanism starting from [Pa(H2O)10]5+.
- (4)
- [PaO(H2O)6]3+ adopts a pentagonal bipyramidal geometry. The relatively weaker Pa-O bond trans- to the Pa=O bond (compared to the equatorial Pa-O bonds) results in an easier replacement in the ligand exchange and extraction reactions.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ramana, M.V. Nuclear power: Economic, safety, health, and environmental issues of near-term technologies. Annu. Rev. Environ. Resour. 2009, 34, 127–152. [Google Scholar] [CrossRef]
- Rodney, C.E. Nuclear fuel cycle: Environmental impact. MRS Bull. 2008, 33, 338–340. [Google Scholar]
- Hargraves, R.; Moir, R. Liquid Fluoride Thorium Reactors: An old idea in nuclear power gets reexamined. Am. Sci. 2010, 98, 304–313. [Google Scholar] [CrossRef]
- Ignatiev, V.V.; Feynberg, O.S.; Zagnitko, A.V.; Merzlyakov, A.V.; Panov, A.V.; Subbotin, V.G.; Afonichkin, V.K.; Khokhlov, V.A.; Kormilitsyn, M.V. Molten-salt reactors: New possibilities, problems and solutions. Russ. J. Gen. Chem. 2012, 112, 157–165. [Google Scholar]
- Clery, D. India’s homegrown thorium reactor. Science 2005, 309, 1174–1175. [Google Scholar]
- IAEA. Thorium Fuel Cycle-Potential Benefits and Challenges; IAEA-TECDOC-1450; IAEA: Vienna, Austria, 2005. [Google Scholar]
- Hladek, K.L.; Kusler, L.E. Production test IP-614-A: Irradiation of thorium target elements. HW-78789. 1963. [Google Scholar] [Green Version]
- Humphrey, U.; Khandaker, M. Viability of thorium-based nuclear fuel cycle for the next generation nuclear reactor: Issues and prospects. Renew. Sustain. Energ Rev. 2018, 97, 259–275. [Google Scholar] [CrossRef]
- Ashley, S.F.; Parks, G.T.; Nuttall, W.J.; Boxall, C.; Grimes, R.W. Thorium fuel has risks. Nature 2012, 492, 31–33. [Google Scholar] [CrossRef]
- Kimura, I. Review of cooperative research on thorium fuel cycle as a promising energy source in the next century. Prog. Nucl. Energy 1995, 29, 445–452. [Google Scholar] [CrossRef]
- Kumari, N.; Pathak, P.N.; Prabhu, D.R.; Manchanda, V.K. Solvent extraction studies of protactinium for its recovery from short-cooled spent fuel and high-level waste solutions in thorium fuel cycle using diisobutyl carbinol (DIBC) as extractant. Desalin. Water Treat. 2012, 38, 46–51. [Google Scholar] [CrossRef]
- Mendes, M.; Aupiais, J.; Jutier, C.; Pointurier, F. Determination of weight distribution ratios of Pa(V) with some extraction chromatography resins and the AG1-X8 resin. Anal. Chim. Acta 2013, 780, 110–116. [Google Scholar] [CrossRef]
- Ostapenko, V.; Sinenko, I.; Arefyeva, E.; Lapshina, E.; Ermolaev, S.; Zhuikov, B.; Kalmykov, S. Sorption of protactinium(V) on extraction chromatographic resins from nitric and hydrochloric solutions. J. Radioanal. Nucl. Chem. 2017, 311, 1545–1550. [Google Scholar] [CrossRef]
- Mendes, M.; Leguay, S.; Naour, C.; Hamadi, S.; Roques, J.; Moisy, P.; Guillaumont, D.; Topin, S.; Aupiais, J.; Auwer, C.; Hennig, C. Thermodynamic Study of the Complexation of Protactinium(V) with Diethylenetriaminepentaacetic Acid. Inorg. Chem. 2013, 52, 7497–7507. [Google Scholar] [CrossRef] [PubMed]
- De Sio, S.M.; Wilson, R.E. EXAFS study of the speciation of protactinium(V) in aqueous hydrofluoric acid solutions. Inorg. Chem. 2014, 53, 12643–12649. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.; Hamadi, S.; Naour, C.; Roques, J.; Jeanson, A.; Auwer, C.; Moisy, P.; Topin, S.; Aupiais, J.; Hennig, C.; Giandomenico, M. Thermodynamical and Structural Study of Protactinium(V) Oxalate Complexes in Solution. Inorg. Chem. 2010, 49, 9962–9971. [Google Scholar] [CrossRef]
- Giandomenico, M.V.; Naour, C. Complex formation between protactinium(V) and sulfate ions at 10 and 60 °C. Inorg. Chim. Acta 2009, 362, 3253–3258. [Google Scholar] [CrossRef]
- Mitsuji, T.; Suzuki, S. The chemistry of protactinium. III. A Study of the sulfate complex of protactinium(V) by the solvent extraction technique using TTA as the chelating agent. Bull. Chem. Soc. Jpn. 1967, 40, 821–826. [Google Scholar] [CrossRef]
- Luchini, C.; Leguay, S.; Aupiais, J.; Cannes, C.; Le Naour, C. Complexation of protactinium(V) with nitrilotriacetic acid: a study at the tracer scale. New J. Chem. 2018, 42, 7789–7795. [Google Scholar] [CrossRef]
- Myasoedov, B.F.; Kirby, H.W.; Tananaev, I.G. Protactinium. In The Chemistry of the Actinide and Transactinide Elements; Moss, L.R., Edelstein, N.M., Fuger, J., Eds.; Springer: Berlin, Germany, 2006; p. 161. [Google Scholar]
- Dau, P.D.; Wilson, R.E.; Gibson, J.K. Elucidating protactinium hydrolysis: The relative stabilities of PaO2(H2O)+ and PaO(OH)2+. Inorg. Chem. 2015, 54, 7474–7480. [Google Scholar] [CrossRef] [PubMed]
- Naour, C.; Trubert, D.; Jaussaud, C. Hydrolysis of protactinium(V). II. Equilibrium constants at 40 and 60℃: A solvent extraction study with TTA in the aqueous system Pa(V)/H2O/H+/Na+/ClO4−. J. Solu. Chem. 2003, 32, 489–504. [Google Scholar] [CrossRef]
- Toraishi, T.; Tsuneda, T.; Tanaka, S. Theoretical study on molecular property of protactinium(V) and uranium(VI) oxocations: Why does protactinium(V) form monooxo cations in aqueous solution? J. Phys. Chem. A 2006, 110, 13303–13309. [Google Scholar] [CrossRef]
- Naour, C.L.; Trubert, D.; Di Giandomenico, M.V.; Fillaux, C.; Den Auwer, C.; Moisy, P.; Hennig, C. First structural characterization of a protactinium (V) single oxo bond in aqueous media. Inorg. Chem. 2005, 44, 9542–9546. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.W.; Eitrheim, E.S.; Nelson, A.W.; Nelson, S.; Schultz, M.K. A simple-rapid method to separate uranium, thorium, and protactinium for U-series age-dating of materials. J. Environ. Radioactiv. 2014, 134, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, A.W.; Nelson, A.W.; Eitrheim, E.S.; Forbes, T.Z.; Schultz, M.K. A chromatographic separation of neptunium and protactinium using 1-octanol impregnated onto a solid phase support. J. Radioanal. Nucl. Chem. 2016, 307, 59–67. [Google Scholar] [CrossRef]
- Knight, A.W.; Eitrheim, E.S.; Nelson, A.W.; Peterson, M.; McAlister, D.; Forbes, T.Z.; Schultz, M.K. Trace-level extraction behavior of actinide elements by aliphatic alcohol extractants in mineral acids: Insights into the trace solution chemistry of protactinium. Solvent Extr. Ion Exc. 2016, 34, 509–521. [Google Scholar] [CrossRef]
- Tsushima, S.; Suzuki, A. Hydration numbers of pentavalent and hexavalent uranyl, neptunyl, and plutonyl. J. Mol. Struct. 2000, 529, 21–25. [Google Scholar] [CrossRef]
- Druchok, M.; Bryk, T.; Holovko, M. A molecular dynamics study of uranyl hydration. J. Mol. Liq. 2005, 120, 11–14. [Google Scholar] [CrossRef]
- Siboulet, B.; Marsden, C.J.; Vitorge, P. A theoretical study of uranyl solvation: Explicit modelling of the second hydration sphere by quantum mechanical methods. Chem. Phys. 2006, 326, 289–296. [Google Scholar] [CrossRef]
- Johansson, G.; Magini, M.; Ohtaki, H. Coordination around thorium(IV) in aqueous perchlorate, chloride and nitrate solutions. J. Solution Chem. 1991, 20, 775–792. [Google Scholar] [CrossRef]
- Moll, H.; Denecke, M.A.; Jalilehvand, F.; Sandström, M.; Grenthe, I. Structure of the aqua ions and fluoride complexes of uranium(IV) and thorium(IV) in aqueous solution an EXAFS study. Inorg. Chem. 1999, 38, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.E.; Skanthakumar, S.; Burns, P.C.; Soderholm, L. Structure of the homoleptic thorium(IV) aqua ion [Th(H2O)10]Br4. Angew. Chem. Int. Ed. 2007, 46, 8043–8045. [Google Scholar] [CrossRef] [PubMed]
- Torapava, N.; Persson, I.; Eriksson, L.; Lundberg, D. Hydration and hydrolysis of thorium(IV) in aqueous solution and the structures of two crystalline thorium(IV) hydrates. Inorg. Chem. 2009, 48, 11712–11723. [Google Scholar] [CrossRef] [PubMed]
- Atta-Fynn, R.; Bylaska, E.J.; Jong, W.A. Strengthening of the coordination shell by counter ions in aqueous Th4+ solutions. J. Phys. Chem. A 2016, 120, 10216–10222. [Google Scholar] [CrossRef]
- Giaquinta, D.M.; Soderholm, L.; Yuchs, S.E.; Wasserman, S.R. Hydrolysis of uranium and thorium in surface-modified bentonite under hydrothermal conditions. J. Alloys Compd. 1997, 249, 142–145. [Google Scholar] [CrossRef]
- Rothe, J.; Denecke, M.A.; Neck, V.; Müller, R.; Kim, J.I. XAFS investigation of the structure of aqueous thorium(IV) species, colloids, and solid thorium(IV) oxide/hydroxide. Inorg. Chem. 2002, 41, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Amador, D.H.T.; Sambrano, J.R.; Ricardo, G.; Macedo, L. Computational study of Th4+ and Np4+ hydration and hydrolysis of Th4+ from first principles. J. Mol. Model. 2017, 23, 69–75. [Google Scholar] [CrossRef]
- Rèal, F.; Trumm, M.; Vallet, V.; Schimmelpfennig, B.; Masella, M.; Flament, J. Quantum chemical and molecular dynamics study of the coordination of Th(IV) in aqueous solvent. J. Phys. Chem. B 2010, 114, 15913–15924. [Google Scholar]
- Hay, P.J.; Martin, R.L.; Schreckenbach, G. Theoretical studies of the properties and solution chemistry of AnO22+ and AnO2+ aquo complexes for An = U, Np, and Pu. J. Phys. Chem. A 2000, 104, 6259–6270. [Google Scholar] [CrossRef]
- Ikeda-Ohno, A.; Hennig, C.; Tsushima, S.; Scheinost, A.C.; Bernhard, G.; Yaita, T. Speciation and structural study of U(IV) and -(VI) in perchloric and nitric acid solutions. Inorg. Chem. 2009, 48, 7201–7210. [Google Scholar] [CrossRef]
- Ikeda, A.; Hennig, C.; Rossberg, A.; Tsushima, S.; Scheinost, A.C.; Bernhard, G. Structural determination of individual chemical species in a mixed system by iterative transformation factor analysis-based X-ray absorption spectroscopy combined with UV-visible absorption and quantum chemical calculation. Anal. Chem. 2008, 80, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.G.; Bucher, J.J.; Shuh, D.K.; Edelstein, N.M.; Reich, T. Investigation of aquo and chloro complexes of UO22+, NpO2+, Np4+, and Pu3+ by X-ray absorption fine structure spectroscopy. Inorg. Chem. 1997, 36, 4676–4683. [Google Scholar] [CrossRef]
- Skanthakumar, S.; Antonio, M.R.; Wilson, R.E.; Soderholm, L. The curium aqua ion. Inorg. Chem. 2007, 46, 3485–3491. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Bursten, B.E. Speciation of the curium(III) ion in aqueous solution: A combined study by quantum chemistry and molecular dynamics simulation. Inorg. Chem. 2006, 45, 5291–5301. [Google Scholar] [CrossRef] [PubMed]
- Skanthakumar, S.; Antonio, M.R.; Soderholm, L. A comparison of neptunyl(V) and neptunyl(VI) solution coordination: The stability of cation-cation interactions. Inorg. Chem. 2008, 47, 4591–4595. [Google Scholar] [CrossRef] [PubMed]
- Di Giandomenico, M.V.; Le Naour, C.; Simoni, E.; Guillaumont, D.; Moisy, P.; Hennig, C.; Conradson, S.D.; Den Auwer, C. Structure of early actinides (V) in acidic solutions. Radiochim. Acta 2009, 97, 347–353. [Google Scholar] [CrossRef]
- Hagberg, D.; Bednarz, E.; Edelstein, N.M.; Gagliardi, L. A quantum chemical and molecular dynamics study of teh coordination of Cm(III) in water. J. Am. Chem. Soc. 2007, 129, 14136–14137. [Google Scholar] [CrossRef] [PubMed]
- Farkas, I.; Grenthe, I. The rates and mechanisms of water exchange of actinide aqua ions: A variable temperature 17O NMR study of U(H2O)104+, UF(H2O)93+, and Th(H2O)104+. J. Phys. Chem. A 2000, 104, 1201–1206. [Google Scholar] [CrossRef]
- Soderholm, L.; Skanthakumar, S.; Neuefeind, J. Determination of actinide speciation in solution using high-energy X-ray scattering. Anal. Bioanal. Chem. 2005, 383, 48–55. [Google Scholar] [CrossRef] [PubMed]
- El-Sweify, F.H.; Abdel Fattah, A.A.; Ali, S.M. Comparative studies on the extraction of protactinium using different kinds of organic extractants. Sep. Sci. Technol. 2009, 44, 753–772. [Google Scholar] [CrossRef]
- Pathak, P.N.; Prabhu, D.R.; Kanekar, A.S.; Manchanda, V.K. Distribution studies on Th (IV), U (VI) and Pu (IV) using tri-n-butylphosphate and N, N-dialkyl amides. Radiochim. Acta 2006, 94, 193–198. [Google Scholar] [CrossRef]
- Pathak, P.N.; Prabhu, D.R.; Manchanda, V.K. Distribution behaviour of U (VI), Th (IV) and Pa (V) from nitric acid medium using linear and branched chain extractants. Solv. Extr. Ion Exch. 2000, 18, 821–840. [Google Scholar] [CrossRef]
- Allen, P.G.; Bucher, J.J.; Shuh, D.K.; Edelstein, N.M.; Craig, I. Coordination Chemistry of Trivalent LanTEMPthanide and Actinide Ions in Dilute and Concentrated Chloride Solutions. Inorg. Chem. 2000, 39, 595–601. [Google Scholar] [CrossRef]
- Atwood, J.L.; Orr, G.W.; Robinson, K.D. First structural autantication of third-sphere coordination: [p-sulfonatocalix[4 ]arene]5- as a third-sphere ligand for Eu3+. Supramol. Chem. 1994, 3, 89–91. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comp. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Pascual-Ahuir, J.L.; Silla, E.; Tuñon, I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for teh computation of a solvent-excluding surface. J. Comp. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Miertuš, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J. Mol. Struct. 1988, 169, 41–62. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Carpenter, J.E. The Structure of Small Molecules and Ions; Naaman, R., Vager, Z., Eds.; Springer: New York, NY, USA, 1988; pp. 227–236. [Google Scholar]
- Spezia, R.; Jeanvoine, Y.; Beuchat, C.; Gagliardid, L.; Vuilleumier, R. Hydration properties of Cm (III) and Th (IV) combining coordination free energy profiles wif electronic structure analysis. Phys. Chem. Chem. Phys. 2014, 16, 5824–5832. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method too the cyclopropylcatbinyl and cyclobutyl cation and too bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Pa(H2O)]5+ | [Pa(H2O)2]5+ | [Pa(H2O)3]5+ | [Pa(H2O)4]5+ | [Pa(H2O)5]5+ | [Pa(H2O)6]5+ | ||
| Bond Distances | Pa-O | 2.246 | 2.320 | 2.317–2.420 | 2.329–2.344 | 2.301–2.391 | 2.344–2.401 |
| Pa-OAVG | 2.246 | 2.320 | 2.353 | 2.338 | 2.350 | 2.375 | |
| Effective coordination number (ECN) | 0.93 | 1.83 | 2.73 | 3.64 | 4.54 | 5.43 | |
| NBO Charges on | Pa | 4.687 | 4.462 | 4.239 | 3.944 | 3.702 | 3.365 |
| O | −0.927 | −0.928 | −0.910 to −0.928 | −0.908 to −0.910 | −0.881 to −0.915 | −0.872 to −0.893 | |
| OAVG | −0.927 | −0.928 | −0.922 | −0.909 | −0.906 | −0.882 | |
| [Pa(H2O)7]5+ | [Pa(H2O)8]5+ | [Pa(H2O)9]5+ | [Pa(H2O)10]5+ | [Pa(H2O)11]5+ | [Pa(H2O)12]5+ | ||
| Bond distances | Pa-O | 2.342–2.431 | 2.347–2.377 | 2.345–2.450 | 2.423–2.506 | 2.433–2.544 | 2.480–2.517 |
| Pa-OAVG | 2.389 | 2.362 | 2.399 | 2.455 | 2.475 | 2.496 | |
| Effective coordination number (ECN) | 6.32 | 7.26 | 8.10 | 8.90 | 9.74 | 10.59 | |
| NBO Charges on | Pa | 3.004 | 2.288 | 1.933 | 1.815 | 1.371 | 0.931 |
| O | −0.851 to −0.870 | −0.818 to −0.822 | −0.802 to −0.813 | −0.802 to −0.808 | −0.783 to −0.790 | −0.769 to −0.773 | |
| OAVG | −0.861 | −0.820 | −0.807 | −0.804 | −0.787 | −0.771 | |
| n | Reaction (1) | Reaction (2) | |||||
|---|---|---|---|---|---|---|---|
| ΔHn (kcal/mol) | ΔGn (kcal/mol) | TΔSn (kcal/mol) | K | ΔHnref (kcal/mol) | ΔGnref (kcal/mol) | K’ | |
| 2 | −23.2 | −12.3 | −10.9 | 1.1 × 109 | −23.2 | −12.3 | 1.1 × 109 |
| 3 | −14.1 | −6.7 | −7.3 | 8.7 × 104 | −37.3 | −19.0 | 8.7 × 1013 |
| 4 | −12.9 | −1.9 | −11.0 | 2.5 × 101 | −50.1 | −20.9 | 2.1 × 1015 |
| 5 | −28.6 | −15.1 | −13.5 | 1.2 × 1011 | −78.7 | −36.0 | 2.6 × 1026 |
| 6 | −16.1 | −5.1 | −11.0 | 5.5 × 103 | −94.8 | −41.1 | 1.4 × 1030 |
| 7 | −24.3 | −12.5 | −11.8 | 1.5 × 109 | −119.1 | −53.6 | 2.1 × 1039 |
| 8 | −13.4 | −1.6 | −11.8 | 1.5 × 101 | −132.5 | −55.3 | 3.7 × 1040 |
| 9 | −21.8 | −10.9 | −10.9 | 9.9 × 107 | −154.3 | −66.2 | 3.7 × 1048 |
| 10 | −10.8 | −1.3 | −9.4 | 9.0 × 100 | −165.0 | −67.5 | 3.3 × 1049 |
| 11 | −14.3 | −3.3 | −11.0 | 2.6 × 102 | −179.4 | −70.8 | 8.6 × 1051 |
| 12 | −12.2 | −3.0 | −9.2 | 1.6 × 102 | −191.6 | −73.8 | 1.4 × 1054 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Yang, C.; Han, J.; Yu, J.; Hu, S.; Yu, H.; Long, X. Density Functional Theory Investigations on the Mechanism of Formation of Pa(V) Ion in Hydrous Solutions. Molecules 2019, 24, 1169. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061169
Ma J, Yang C, Han J, Yu J, Hu S, Yu H, Long X. Density Functional Theory Investigations on the Mechanism of Formation of Pa(V) Ion in Hydrous Solutions. Molecules. 2019; 24(6):1169. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061169
Chicago/Turabian StyleMa, Jun, Chuting Yang, Jun Han, Jie Yu, Sheng Hu, Haizhu Yu, and Xinggui Long. 2019. "Density Functional Theory Investigations on the Mechanism of Formation of Pa(V) Ion in Hydrous Solutions" Molecules 24, no. 6: 1169. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061169