
A Suitable Functionalization of Nitroindazoles with Triazolyl and Pyrazolyl Moieties via Cycloaddition Reactions


 , ,
, ,  , , , , and
, , , , and
Abstract
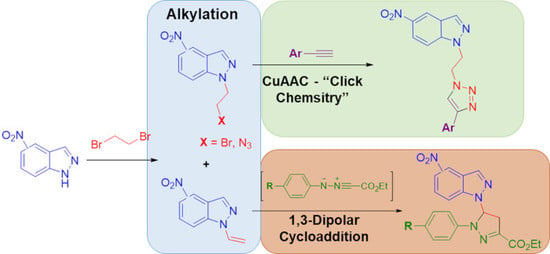
:
1. Introduction
2. Results
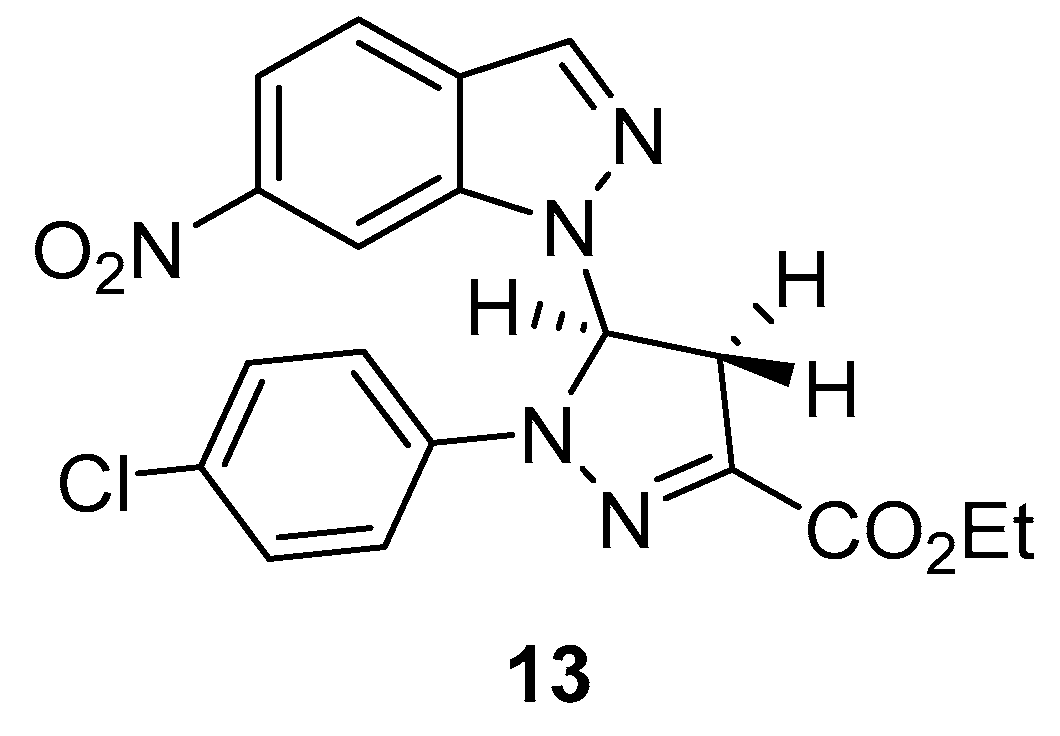
2.1. Reaction of Nitroindazoles 1a–1d with 1,2-Dibromoethane
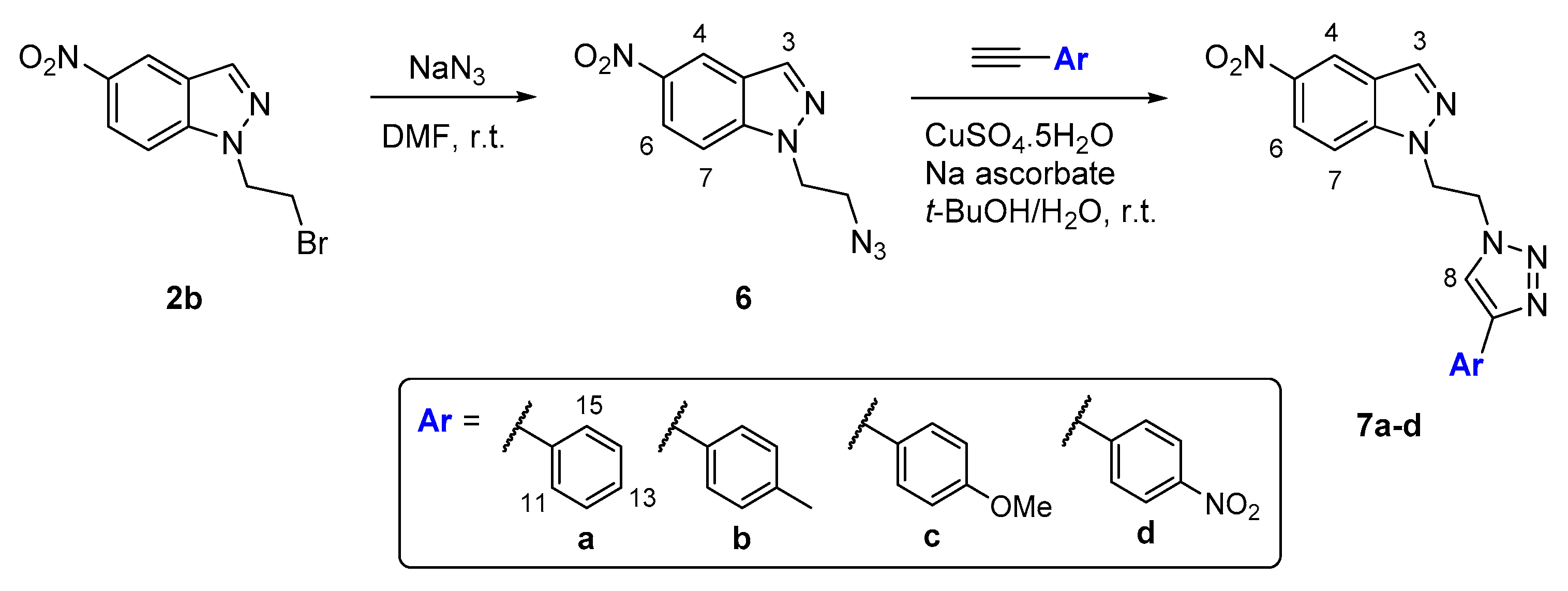
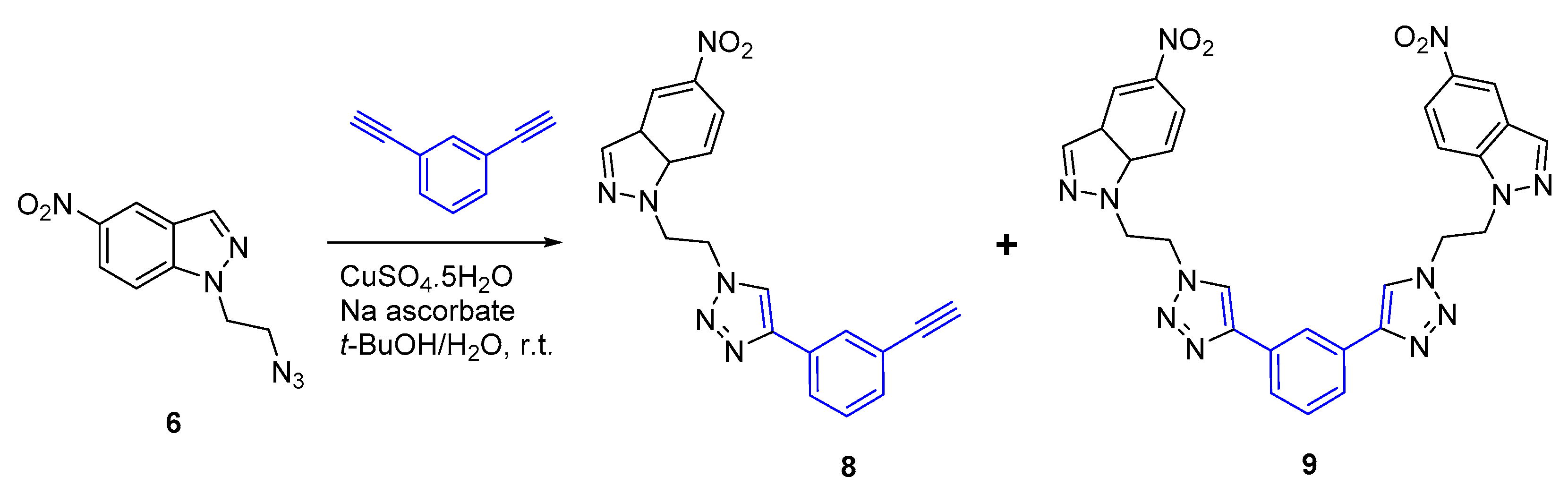
2.2. CuAAC of Azidoethyl-Nitroindazoles with Terminal Alkynes
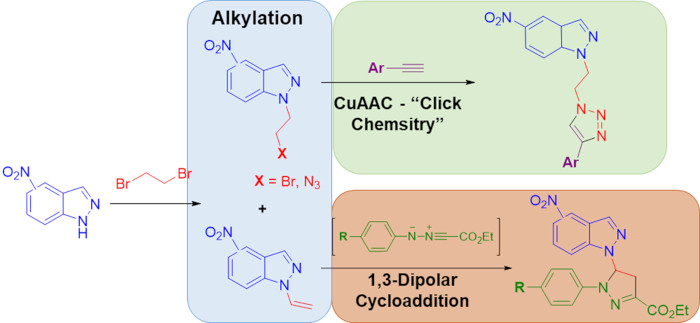
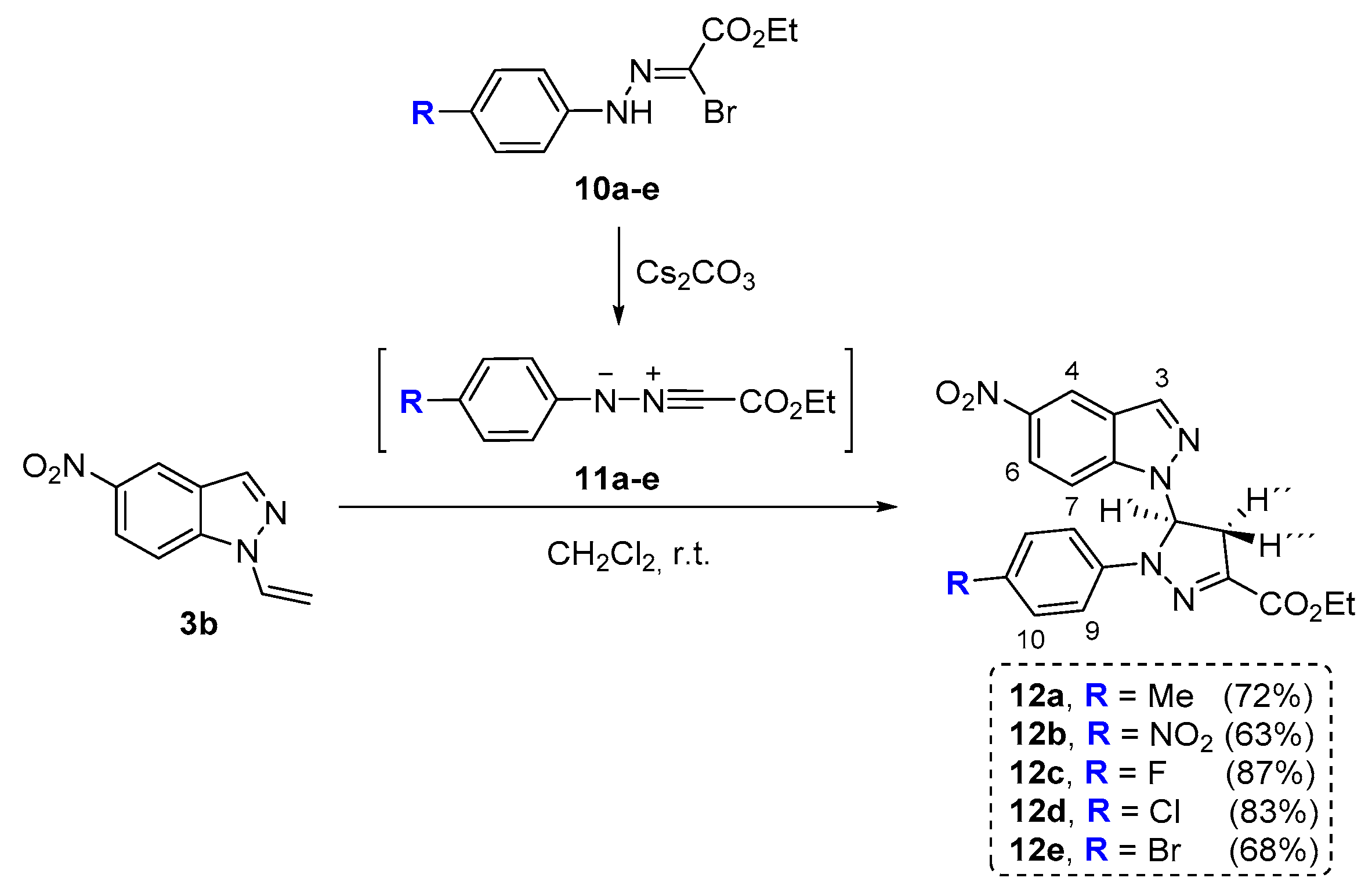
2.3. 1,3-Dipolar Cycloaddition Reactions of N-Vinyl-Nitroindazoles with Nitrile Imines
2.4. X-Ray Diffraction
3. Materials and Methods
3.1. General Remarks
3.2. N-alkylation of Nitroindazole Derivatives 1a-d with 1,2-Dibromoethane. General Procedure
3.3. Procedure for the Preparation of 1-(2-Azidoethyl)-5-nitro-1H-indazole Intermediate 6
3.4. General Procedure for 1,3-Dipolar Cycloaddition of Azides with Terminal Alkynes
3.5. General Procedure for 1,3-Dipolar Cycloaddition Reactions of N-Vinyl-Nitroindazoles with Nitrile Imines to Give Access to Compounds 12a–12e
3.6. Single-Crystal X-Ray Diffraction Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Martina, K.; Tagliapietra, S.; Veselov, V.V.; Cravotto, G. Green Protocols in Heterocycle Syntheses via 1,3-Dipolar Cycloadditions. Front. Chem. 2019, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Arrastia, I.; Arrieta, A.; Cossío, F.P. Application of 1,3-Dipolar Reactions between Azomethine Ylides and Alkenes to the Synthesis of Catalysts and Biologically Active Compounds. Eur. J. Org. Chem. 2018, 2018, 5889–5904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozorov, K.; Zhao, J.Y.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.W. Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2018; Volume 126. [Google Scholar]
- Faisal, M.; Saeed, A.; Hussain, S.; Dar, P.; Larik, F.A. Recent developments in synthetic chemistry and biological activities of pyrazole derivatives. J. Chem. Sci. 2019, 131, 70. [Google Scholar] [CrossRef] [Green Version]
- Varghese, B.; Al-Busafi, S.N.; Suliman, F.O.; Al-Kindy, S.M.Z. Unveiling a versatile heterocycle: pyrazoline a review. RSC Adv. 2017, 7, 46999–47016. [Google Scholar] [CrossRef] [Green Version]
- Vinutha, V.S.; Badiadka, N.; Balladka, K.S.; Kullaiah, B. A Comprehensive Review on Recent Developments in the Field of Biological Applications of Potent Pyrazolines Derived from Chalcone Precursors. Lett. Drug Des. Discovery 2018, 15, 516–574. [Google Scholar]
- Pramod, S.; Jitendra, S.; Geeta, J.P.; Mohan, S.M.R. 2-Pyrazolines as Biologically Active and Fluorescent Agents, An Overview. Anti-Cancer Agents Med. Chem. 2018, 18, 1366–1385. [Google Scholar]
- Prasher, P.; Sharma, M. Tailored therapeutics based on 1,2,3-1H-triazoles: A mini review. MedChemComm 2019, 10, 1302–1328. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Kacprzak, K.; Skiera, I.; Piasecka, M.; Paryzek, Z. Alkaloids and Isoprenoids Modification by Copper(I)-Catalyzed Huisgen 1,3-Dipolar Cycloaddition (Click Chemistry): Toward New Functions and Molecular Architectures. Chem. Rev. 2016, 116, 5689–5743. [Google Scholar] [CrossRef]
- Dervaux, B.; Du Prez, F.E. Heterogeneous azide-alkyne click chemistry: towards metal-free end products. Chem. Sci. 2012, 3, 959–966. [Google Scholar] [CrossRef]
- Zheng, Z.J.; Wang, D.; Xu, Z.; Xu, L.W. Synthesis of bi- and bis-1,2,3-triazoles by copper-catalyzed Huisgen cycloaddition: A family of valuable products by click chemistry. Beilstein J. Org. Chem. 2015, 11, 2557–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struthers, H.; Mindt, T.L.; Schibli, R. Metal chelating systems synthesized using the copper(I) catalyzed azide-alkyne cycloaddition. Dalton Trans. 2010, 39, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Simón-Fuentes, A.; Sanz-Cervera, J.F. Recent Advances in the Synthesis of Pyrazoles. A Review. Org. Prep. Proced. Int. 2009, 41, 253–290. [Google Scholar] [CrossRef]
- Karrouchi, K.R.S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibi, M.P.; Stanley, L.M.; Jasperse, C.P. An Entry to a Chiral Dihydropyrazole Scaffold: Enantioselective [3 + 2] Cycloaddition of Nitrile Imines. J. Am. Chem. Soc. 2005, 127, 8276–8277. [Google Scholar] [CrossRef]
- Mojahidi, S.; Sekkak, H.; Rakib, E.M.; Neves, M.G.P.M.S.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Zouihri, H. Alkylation and 1,3-Dipolar Cycloaddition of 6-Styryl-4,5-dihydro-2H-pyridazin-3-one: Synthesis of Novel N-Substituted Pyridazinones and Triazolo [4,3-b]pyridazinones. J. Chem. 2013, 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Moura, N.M.M.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Rakib, E.M.; Hannioui, A.; Mojahidi, S.; Hackbarth, S.; Röder, B.; Almeida Paz, F.A.; et al. Novel pyrazoline and pyrazole porphyrin derivatives: synthesis and photophysical properties. Tetrahedron 2012, 68, 8181–8193. [Google Scholar] [CrossRef]
- Moura, N.M.M.; Giuntini, F.; Faustino, M.A.F.; Neves, M.G.P.S.; Tomé, A.C.; Silva, A.M.S.; Rakib, E.M.; Hannioui, A.; Abouricha, S.; Röder, B.; et al. 1,3-Dipolar cycloaddition of nitrile imines to meso-tetraarylporphyrins. ARKIVOC 2010, 5, 24–33. [Google Scholar]
- Shaaban, M.R.; Mayhoub, A.S.; Farag, A.M. Recent advances in the therapeutic applications of pyrazolines. Expert Opin. Ther. Patents 2012, 22, 253–291. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Roman, O.; Lesyk, R. Synthetic approaches, structure activity relationship and biological applications for pharmacologically attractive pyrazole/pyrazoline–thiazolidine-based hybrids. Eur. J. Med. Chem. 2016, 113, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, J.; Sharma, S.; Jain, S.; Singh, A. The Synthetic and Biological Attributes of Pyrazole Derivatives: A Review. Mini-Rev. Med. Chem. 2018, 18, 918–947. [Google Scholar] [CrossRef] [PubMed]
- Larina, L.; Lopyrev, V. Nitroazoles: Synthesis, Structure and Applications; Springer: London, UK, 2009. [Google Scholar]
- Shrivastava, A.; Upmanyu, A.K.C.N.; Singh, A. Recent Progress in Chemistry and Biology of Indazole and its Derivatives: A Brief Review. Austin J. Anal. Pharm. Chem. 2016, 3, 1076. [Google Scholar]
- Claramunt, R.M.; Santa María, D.; Alkorta, I.; Elguero, J. The Structure of N-phenyl-pyrazoles and Indazoles: Mononitro, Dinitro, and Trinitro Derivatives. J. Heterocycl. Chem. 2018, 55, 44–64. [Google Scholar] [CrossRef]
- Denya, I.; Malan, S.F.; Joubert, J. Indazole derivatives and their therapeutic applications: A patent review (2013-2017). Expert Opin. Ther. Patents 2018, 28, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, D.D.; Chapolikar, A.D.; Devkate, C.G.; Warad, K.D.; Tayade, A.P.; Pawar, R.P.; Domb, A.J. Synthesis of indazole motifs and their medicinal importance: An overview. Eur. J. Med.Chem. 2015, 90, 707–731. [Google Scholar] [CrossRef]
- Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives. Molecules 2018, 23, 2783. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. Recent Advances in the Development of Indazole-based Anticancer Agents. ChemMedChem 2018, 13, 1490–1507. [Google Scholar] [CrossRef]
- Kang, Y.-S.; Yoon, T.; Mun, J.; Park, M.S.; Song, I.-Y.; Benayad, A.; Oh, S.M. Effective passivation of a high-voltage positive electrode by 5-hydroxy-1H-indazole additives. J. Mater. Chem. A 2014, 2, 14628–14633. [Google Scholar] [CrossRef]
- Ahmed, Z.; Iftikhar, K. Efficient Layers of Emitting Ternary Lanthanide Complexes for Fabricating Red, Green, and Yellow OLEDs. Inorg. Chem. 2015, 54, 11209–11225. [Google Scholar] [CrossRef]
- Fan, K.W.; Roberts, J.J.; Martens, P.J.; Stenzel, M.H.; Granville, A.M. Copolymerization of an indazole ligand into the self-polymerization of dopamine for enhanced binding with metal ions. J. Mater. Chem. B 2015, 3, 7457–7465. [Google Scholar] [CrossRef]
- Qiang, Y.; Zhang, S.; Xu, S.; Li, W. Experimental and theoretical studies on the corrosion inhibition of copper by two indazole derivatives in 3.0% NaCl solution. J. Colloid Interface Sci. 2016, 472, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kouakou, A.; Chicha, H.; Rakib, E.M.; Gamouh, A.; Hannioui, A.; Chigr, M.; Viale, M. SnCl2/RSH: a versatile catalytic system for the synthesis of 4-alkylsulfanyl-indazole derivatives. J. Sulfur Chem. 2015, 36, 86–95. [Google Scholar] [CrossRef]
- Bassou, O.; Chicha, H.; Allam, A.; Monticone, M.; Gangemi, R.; Maric, I.; Viale, M.; Rakib, E.M. Synthesis and Anti-proliferative Activity of Novel Polysubstitued Indazole Derivatives. J. Heterocycl. Chem. 2019, 56, 343–348. [Google Scholar] [CrossRef] [Green Version]
- López, P.; Seipelt, C.G.; Merkling, P.; Sturz, L.; Álvarez, J.; Dölle, A.; Zeidler, M.D.; Cerdán, S.; Ballesteros, P. N-2-(Azol-1(2)-yl)ethyliminodiacetic acids: a novel series of Gd(III) chelators as T2 relaxation agents for magnetic resonance imaging. Bioorg. Med. Chem. 1999, 7, 517–527. [Google Scholar] [CrossRef]
- Micheletti, G.; Kouakou, A.; Boga, C.; Franchi, P.; Calvaresi, M.; Guadagnini, L.; Lucarini, M.; Rakib, E.M.; Spinelli, D.; Tonelli, D.; et al. Comparative spectroscopic and electrochemical study of N-1 or N-2-alkylated 4-nitro and 7-nitroindazoles. Arabian J. Chem. 2017, 10, 823–836. [Google Scholar] [CrossRef] [Green Version]
- Abbassi, N.; Rakib, E.M.; Hannioui, A. Alkylation and Reduction of N-Alkyl-4-nitroindazoles with Anhydrous SnCl2 in Ethanol: Synthesis of Novel 7-Ethoxy-N-alkylindazole Derivatives. Heterocycles 2011, 83, 891–900. [Google Scholar] [CrossRef]
- Abbassi, N.; Rakib, E.M.; Chicha, H.; Bouissane, L.; Hannioui, A.; Aiello, C.; Gangemi, R.; Castagnola, P.; Rosano, C.; Viale, M. Synthesis and Antitumor Activity of Some Substituted Indazole Derivatives. Arch. Pharm. 2014, 347, 423–431. [Google Scholar] [CrossRef]
- Noelting, E. Ueber Bildung von Indazolen aus nitrirten orthomethylirten Aminen. Ber. Dtsch. Chem. Ges. 1904, 37, 2556–2597. [Google Scholar] [CrossRef]
- Cooper, K.A.; Hughes, E.D.; Ingold, C.K.; MacNulty, B.J. 418. Mechanism of elimination reactions. Part VIII. Temperature effects on rates and product-proportions in uni- and bi-molecular substitution and elimination reactions of alkyl halides and sulphonium salts in hydroxylic solvents. J. Chem. Soc. 1948, 2049–2054. [Google Scholar] [CrossRef]
- Catalan, J.; del Valle, J.C.; Claramunt, R.M.; Boyer, G.; Laynez, J.; Gomez, J.; Jimenez, P.; Tomas, F.; Elguero, J. Acidity and Basicity of Indazole and its N-Methyl Derivatives in the Ground and in the Excited State. J. Phys. Chem. 1994, 98, 10606–10612. [Google Scholar] [CrossRef]
- Schmidt, A.; Beutler, A.; Snovydovych, B. Recent Advances in the Chemistry of Indazoles. Eur. J. Org. Chem. 2008, 2008, 4073–4095. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Hao, X.; Jing, L.L.; Wu, G.C.; Kang, D.W.; Liu, X.Y.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert. Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Arslan, M.; Acik, G.; Tasdelen, M.A. The emerging applications of click chemistry reactions in the modification of industrial polymers. Polym. Chem. 2019, 10, 3806–3821. [Google Scholar] [CrossRef]
- Yanez-Sedeno, P.; Gonzalez-Cortes, A.; Campuzano, S.; Pingarron, J.M. Copper(I)-Catalyzed Click Chemistry as a Tool for the Functionalization of Nanomaterials and the Preparation of Electrochemical (Bio)Sensors. Sensors 2019, 19, 29. [Google Scholar] [CrossRef] [Green Version]
- Hong, T.T.; Liu, W.F.; Li, M.; Chen, C.P. Click chemistry at the microscale. Analyst 2019, 144, 1492–1512. [Google Scholar] [CrossRef]
- Li, P.Z.; Wang, X.J.; Zhao, Y.L. Click chemistry as a versatile reaction for construction and modification of metal-organic frameworks. Coord. Chem. Rev. 2019, 380, 484–518. [Google Scholar] [CrossRef]
- Takayama, Y.; Kusamori, K.; Nishikawa, M. Click Chemistry as a Tool for Cell Engineering and Drug Delivery. Molecules 2019, 24, 20. [Google Scholar] [CrossRef] [Green Version]
- Sharp, D.B.; Hamilton, C.S. Derivatives of 1,2,4-Triazole and of Pyrazole. J. Am. Chem. Soc. 1946, 68, 588–591. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Oxford, UK, 2013. [Google Scholar]
- Kottke, T.; Stalke, D.J. Crystal handling at low temperatures. Appl. Crystallogr. 1993, 26, 615–619. [Google Scholar] [CrossRef] [Green Version]
- APEX3. Data Collection Software Version 2016.9-0; Bruker AXS: Delft, The Netherlands, 2005–2016. [Google Scholar]
- Cryopad. Remote monitoring and control, Version 1.451; Oxford Cryosystems: Oxford, UK, 2006. [Google Scholar]
- SAINT+. Data Integration Engine v. 8.37a©; Bruker AXS: Madison, WI, USA, 1997–2015. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittric, B. ShelXle: a Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, K. DIAMOND, Version 3.2f; Crystal Impact GbR: Bonn, Germany, 1997–2010. [Google Scholar]
Sample Availability: Samples of the compounds 1–5, 6, 7a–7d, 8, 9, 10a–10e, 12a–12e and 13 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Base | Time (h) | Yield (%) | |||
|---|---|---|---|---|---|---|---|
| 2b | 3b | 4b | 5b | ||||
| 1 | Acetone | KOH | 48 | 39 | 9 | 12 | 25 |
| 2 | Acetone | KOH a | 4 | 29 | 25 | Traces | 17 |
| 3 | Acetone | KOH b | 2 | 27 | 21 | Traces | 19 |
| 4 | THF | KOH b | 72 | 30 | 11 | 14 | 10 |
| 5 | MeOH | KOH b | 120 c | 15 | Traces | 7 | Traces |
| 6 | Acetone | Cs2CO3 | 1 | 35 | 6 | 12 | 15 |
| 7 | Acetone | K2CO3 | 68 | 47 | 10 | 20 | 8 |
| Entry | Product | Ar | Time (h) | Yield (%) |
|---|---|---|---|---|
| 1 | 7a | Ph | 12 | 74 |
| 2 | 7b | p-Me-Ph | 12 | 82 |
| 3 | 7c | p-OMe-Ph | 14 | 87 |
| 4 | 7d | p-NO2-Ph | 16 | 71 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eddahmi, M.; Moura, N.M.M.; Bouissane, L.; Amiri, O.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Mendes, R.F.; Paz, F.A.A.; Neves, M.G.P.M.S.; Rakib, E.M. A Suitable Functionalization of Nitroindazoles with Triazolyl and Pyrazolyl Moieties via Cycloaddition Reactions. Molecules 2020, 25, 126. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25010126
Eddahmi M, Moura NMM, Bouissane L, Amiri O, Faustino MAF, Cavaleiro JAS, Mendes RF, Paz FAA, Neves MGPMS, Rakib EM. A Suitable Functionalization of Nitroindazoles with Triazolyl and Pyrazolyl Moieties via Cycloaddition Reactions. Molecules. 2020; 25(1):126. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25010126
Chicago/Turabian StyleEddahmi, Mohammed, Nuno M. M. Moura, Latifa Bouissane, Ouafa Amiri, M. Amparo F. Faustino, José A. S. Cavaleiro, Ricardo F. Mendes, Filipe A. A. Paz, Maria G. P. M. S. Neves, and El Mostapha Rakib. 2020. "A Suitable Functionalization of Nitroindazoles with Triazolyl and Pyrazolyl Moieties via Cycloaddition Reactions" Molecules 25, no. 1: 126. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25010126