
Recent Strides in the Transition Metal-Free Cross-Coupling of Haloacetylenes with Electron-Rich Heterocycles in Solid Media

{kind=link}
{kind=link}
{kind=link}
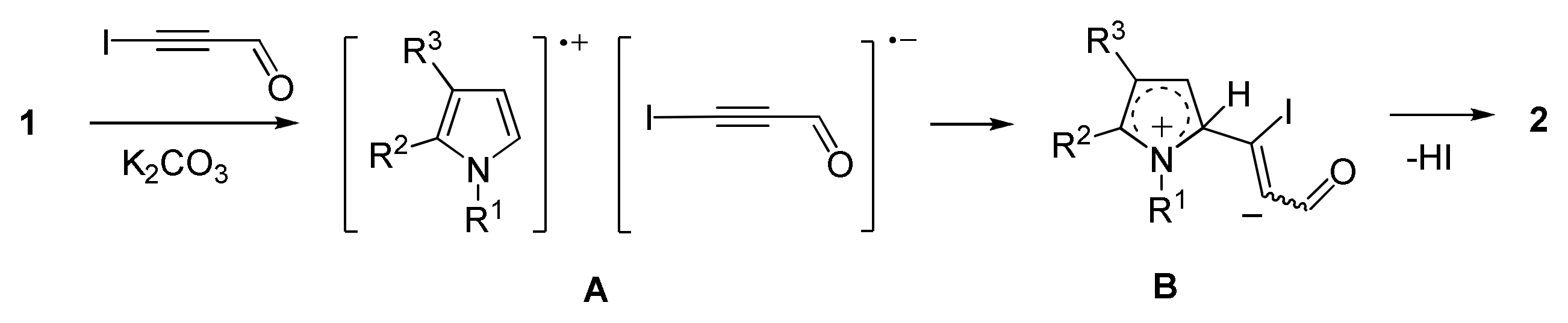{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
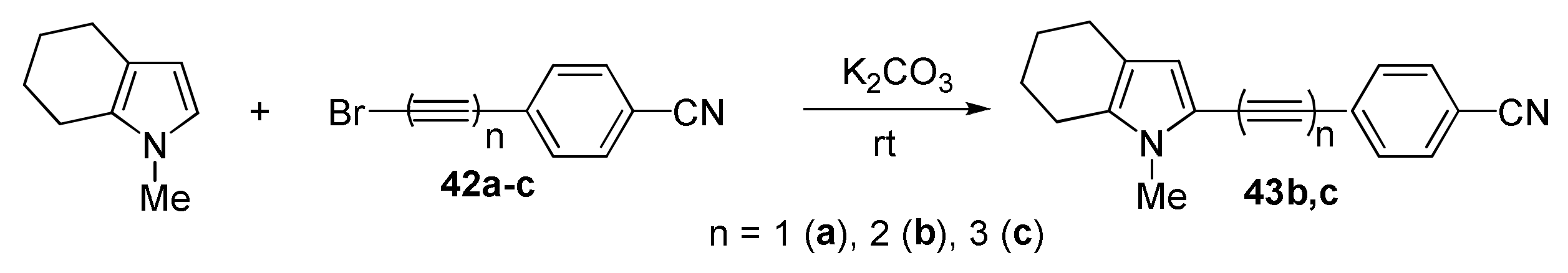{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cross-Coupling of Haloacetylenes with Electron-Rich Heterocycles
2.1. Cross-Coupling of Haloacetylenes with Pyrroles
2.1.1. Cross-Coupling of Bromo- and Iodopropiolaldehydes with Pyrroles
2.1.2. Cross-Coupling of Acylbromoacetylenes with Pyrroles
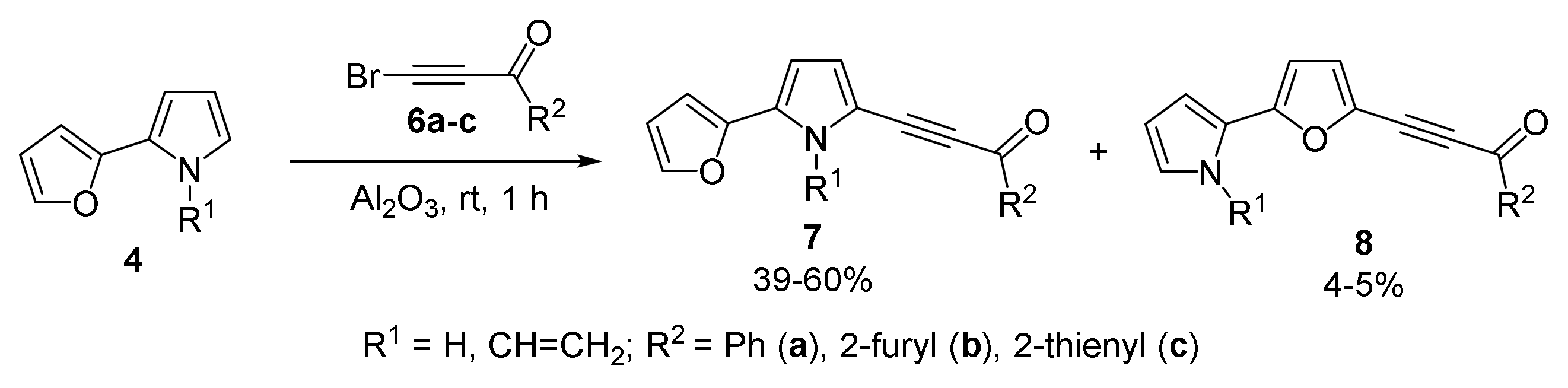
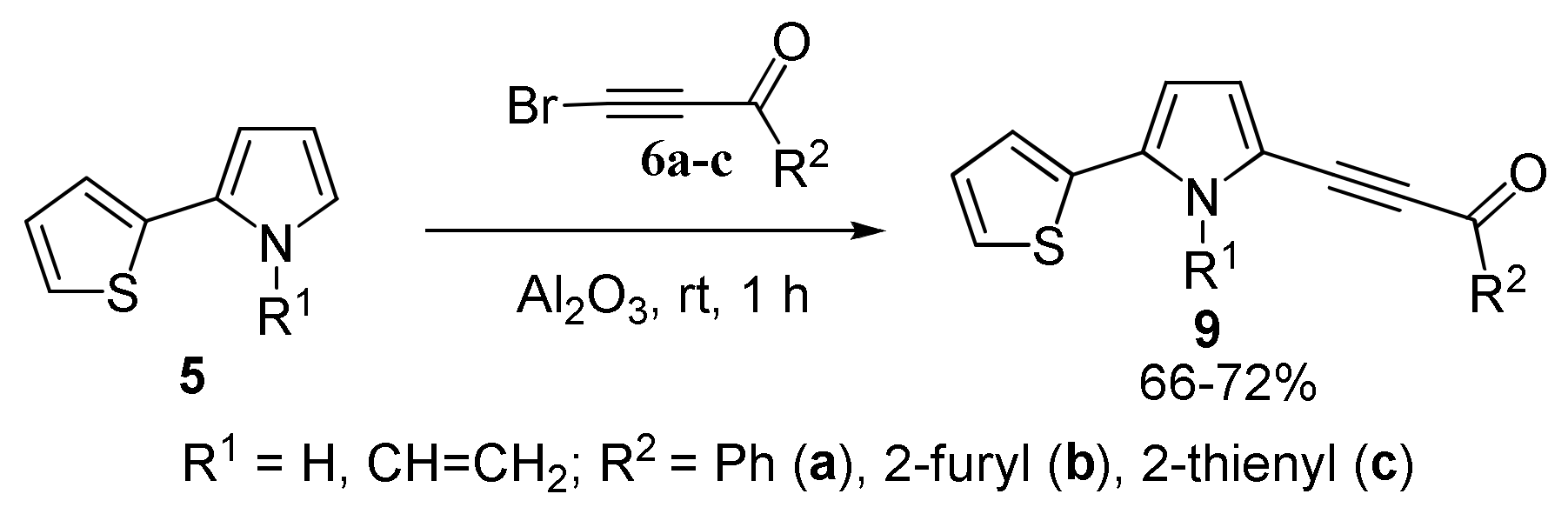
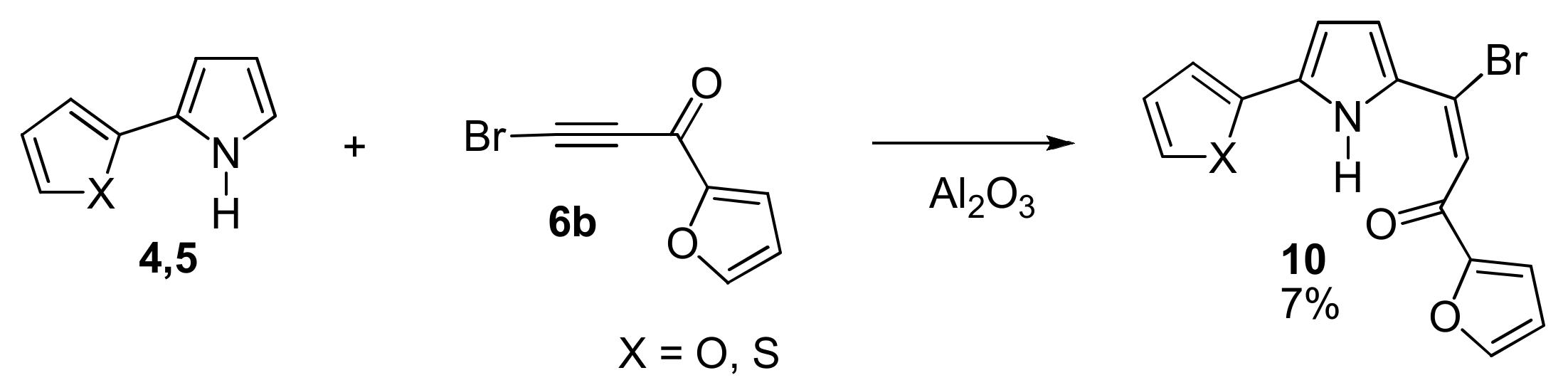
With 2-(Furan-2-yl)- and 2-(2-Thiophen-2-yl)pyrroles
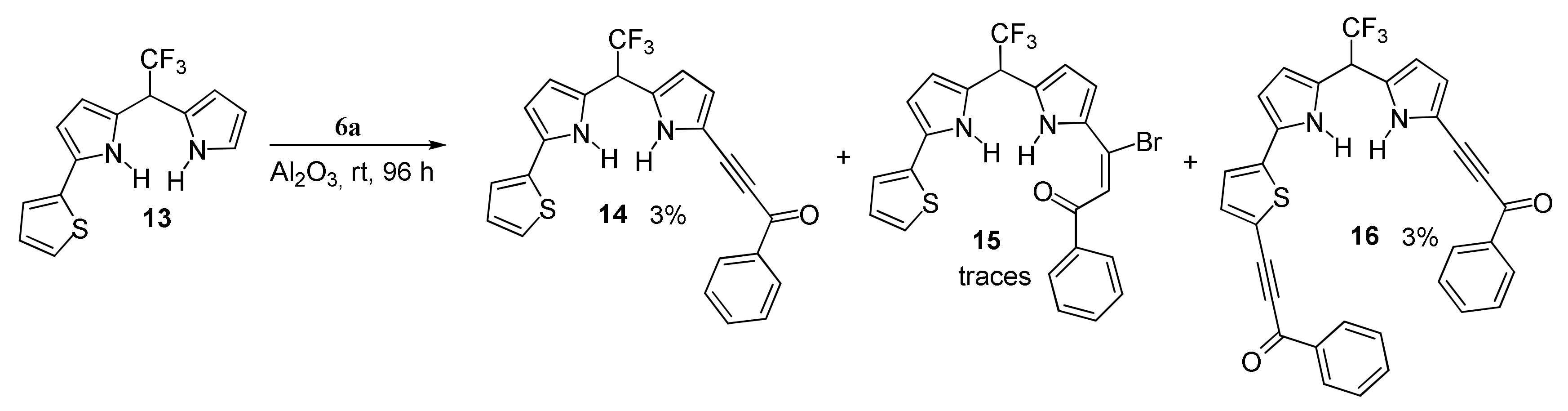
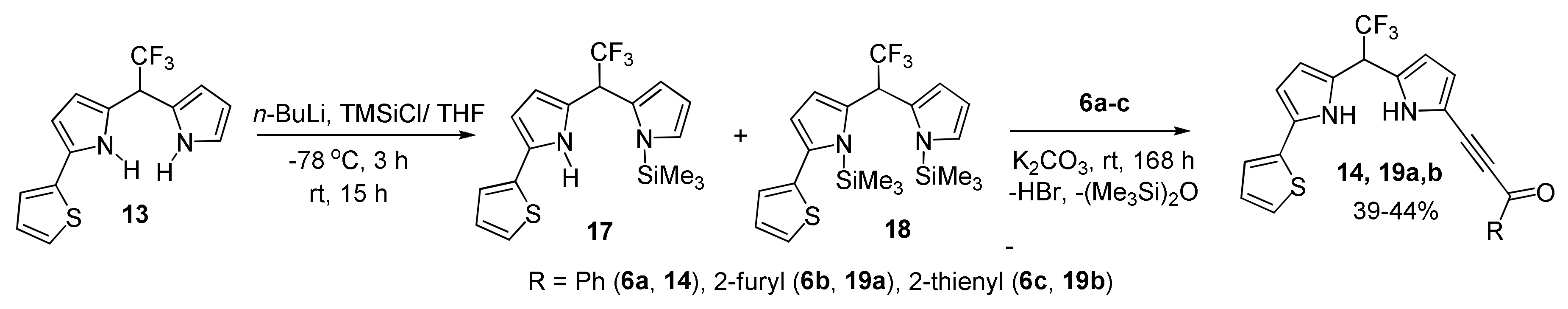
With Dipyrromethanes
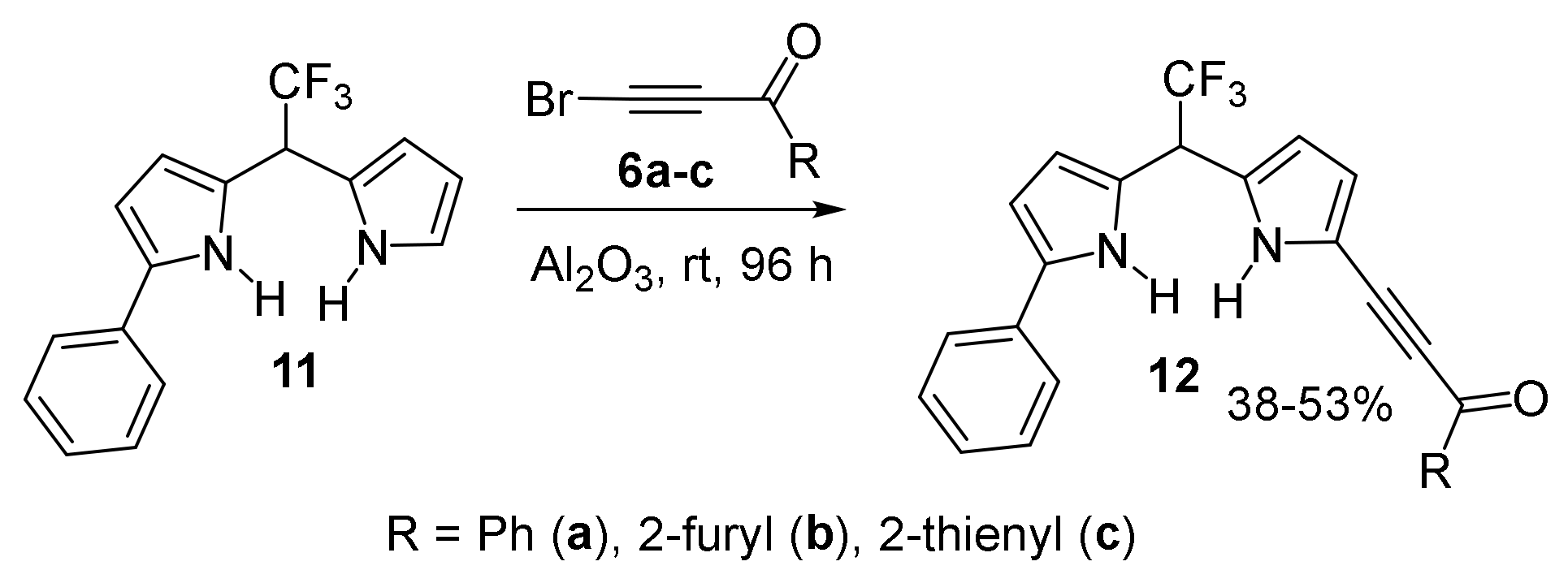
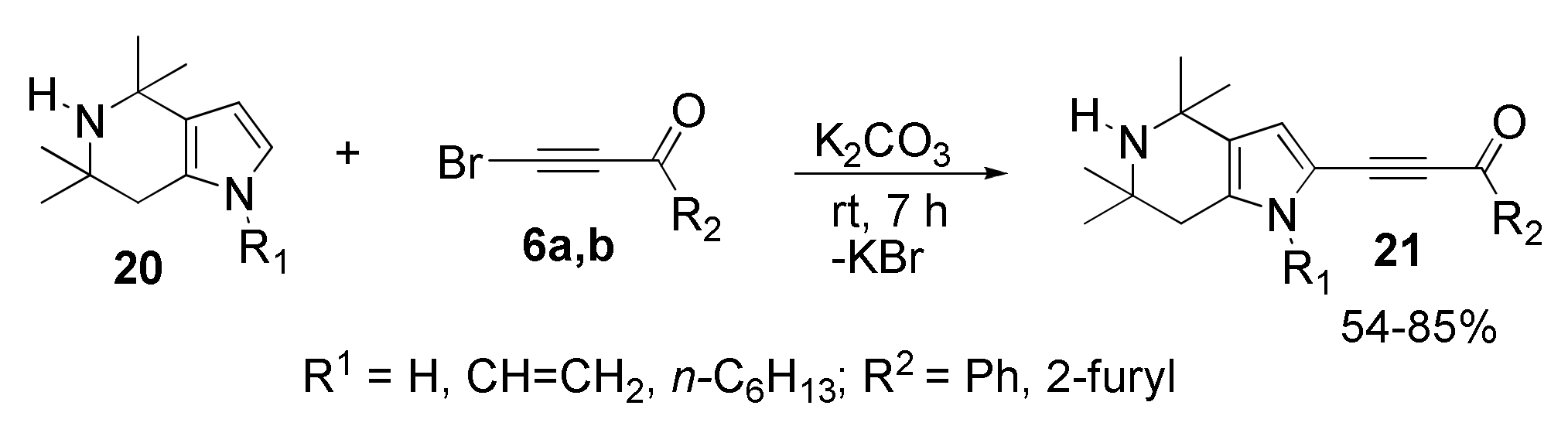
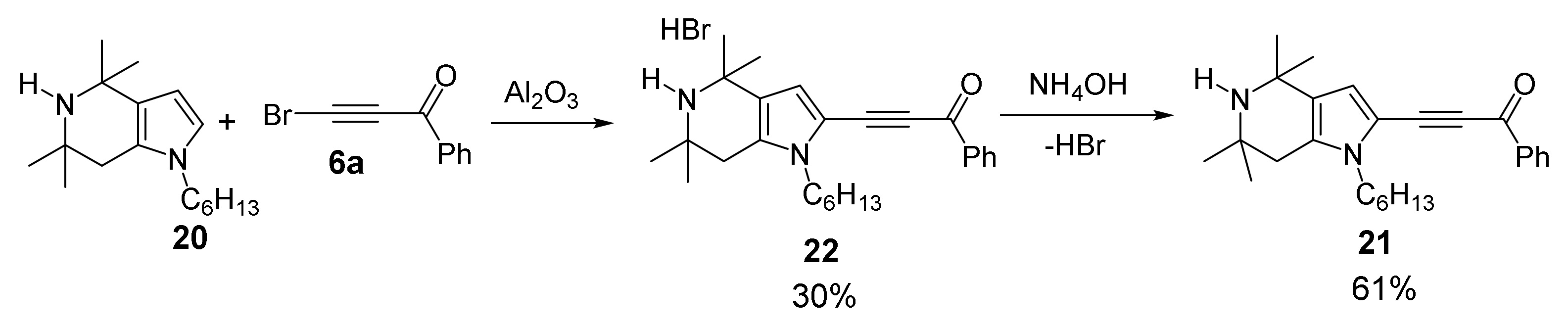
With Tetrahydropyrrolo [3,2-c]pyridines
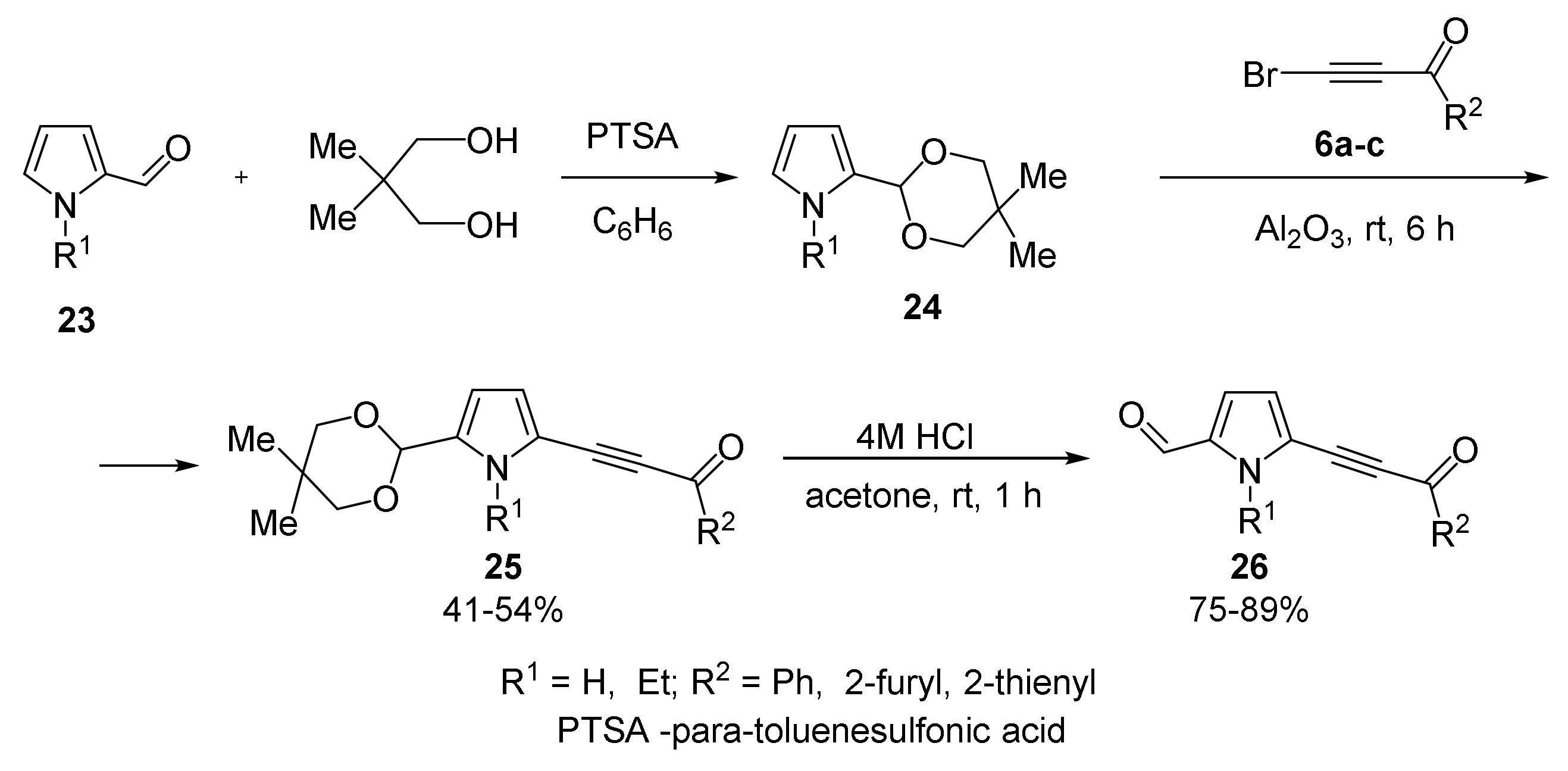
With Pyrrole-2-carbaldehydes
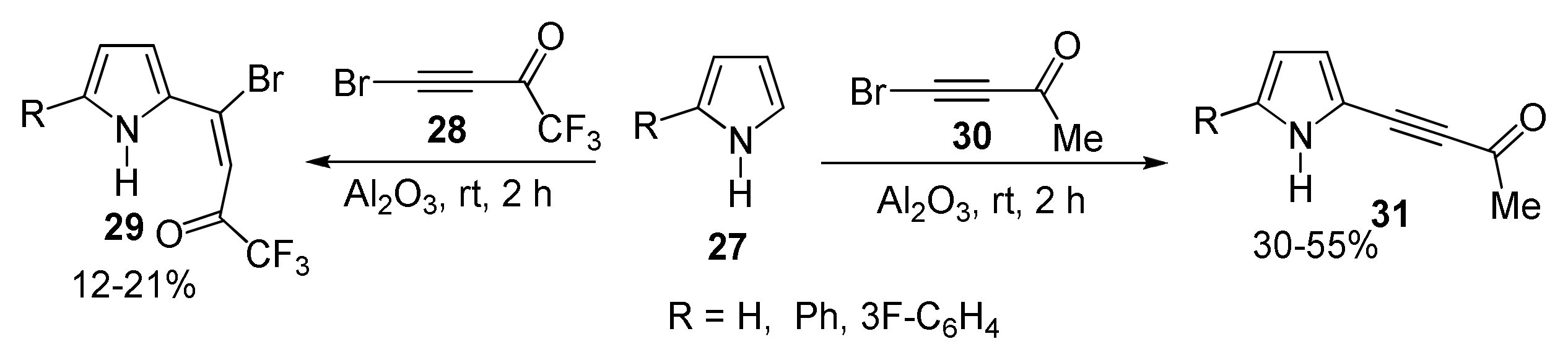
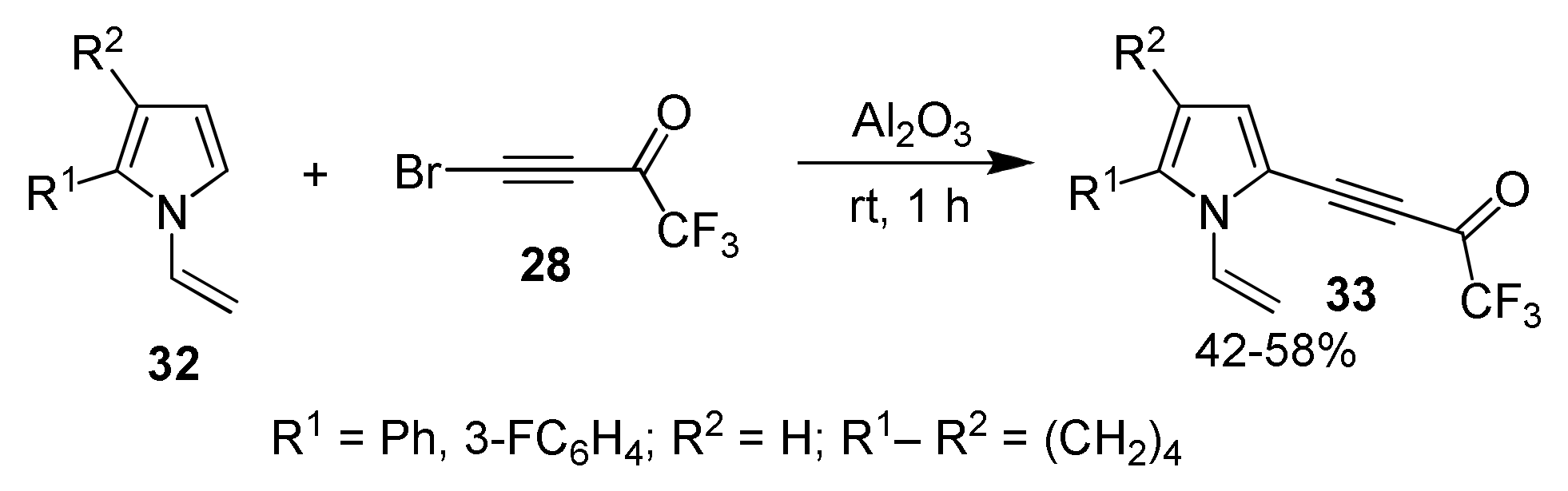
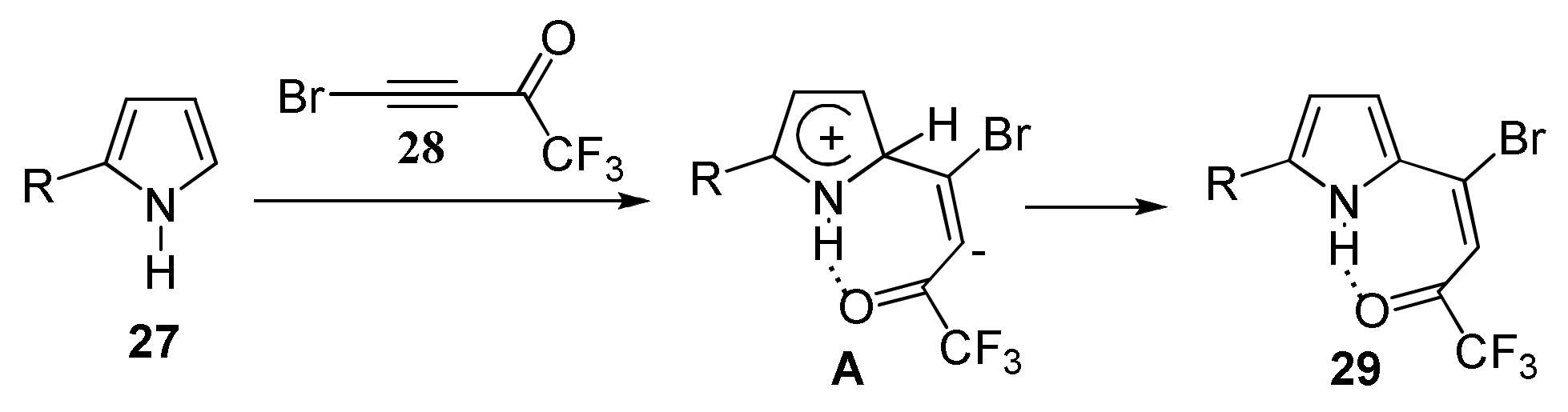
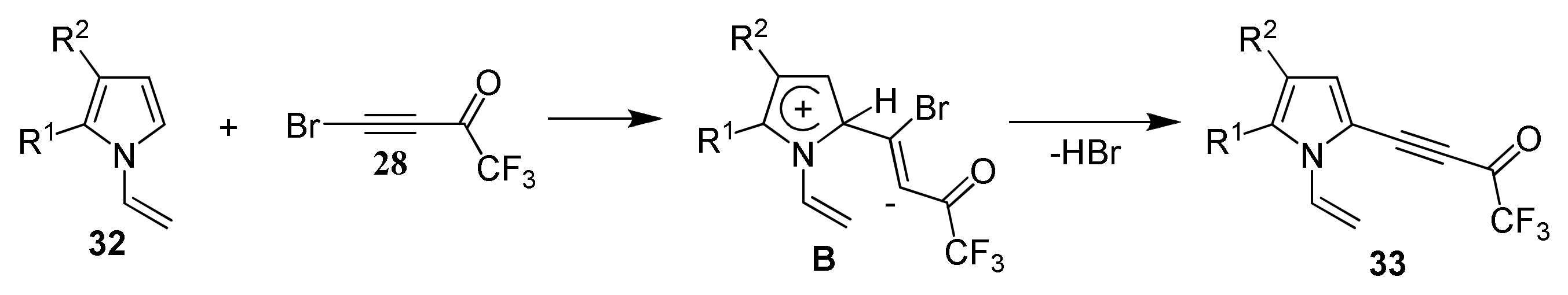
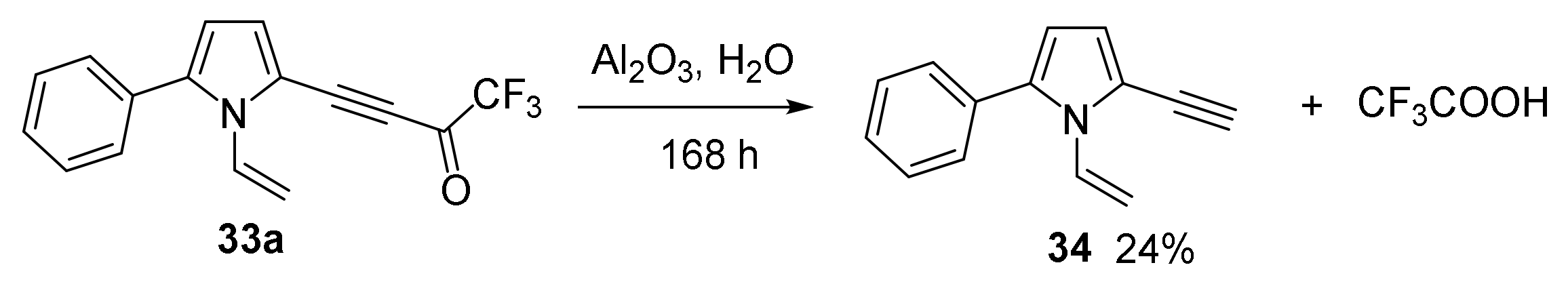
2.1.3. Cross-Coupling of Bromotrifluoroacetylacetylene with Pyrroles
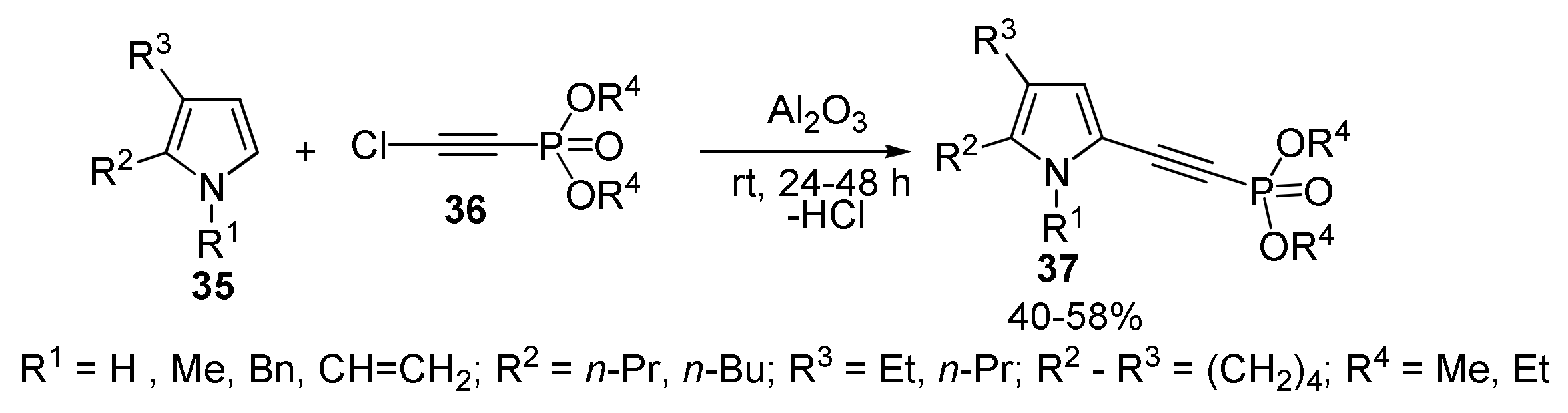
2.1.4. Cross-Coupling of Chloroethynylphosponates with Pyrroles
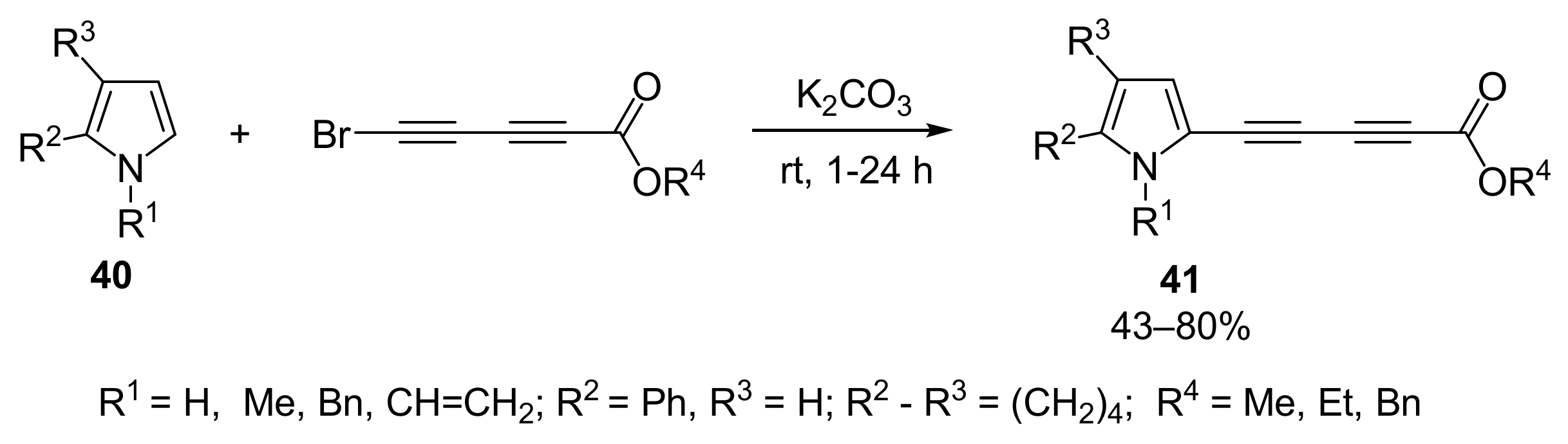
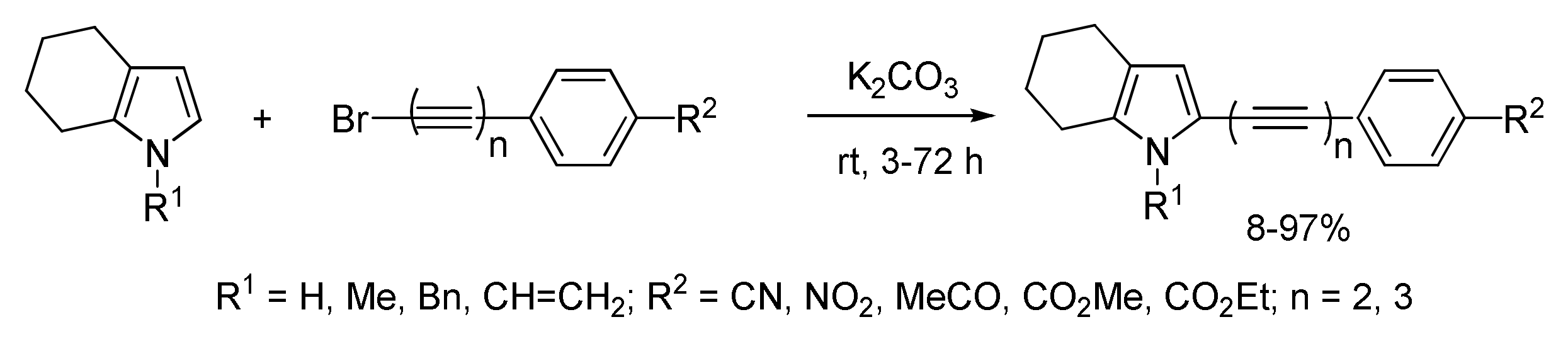
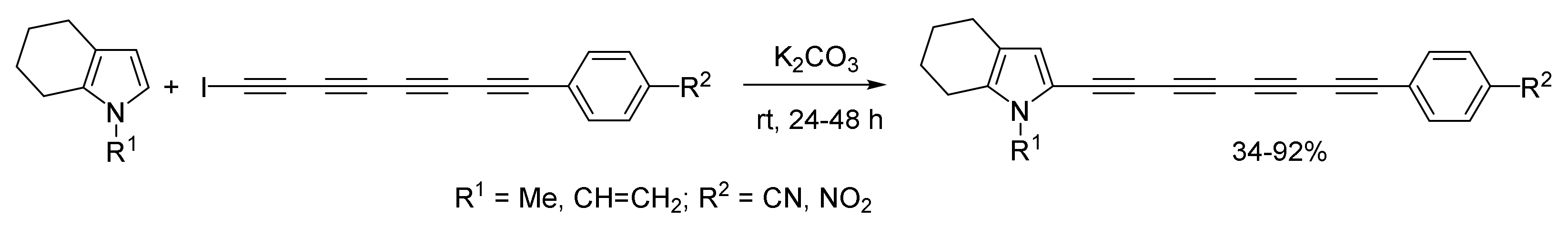
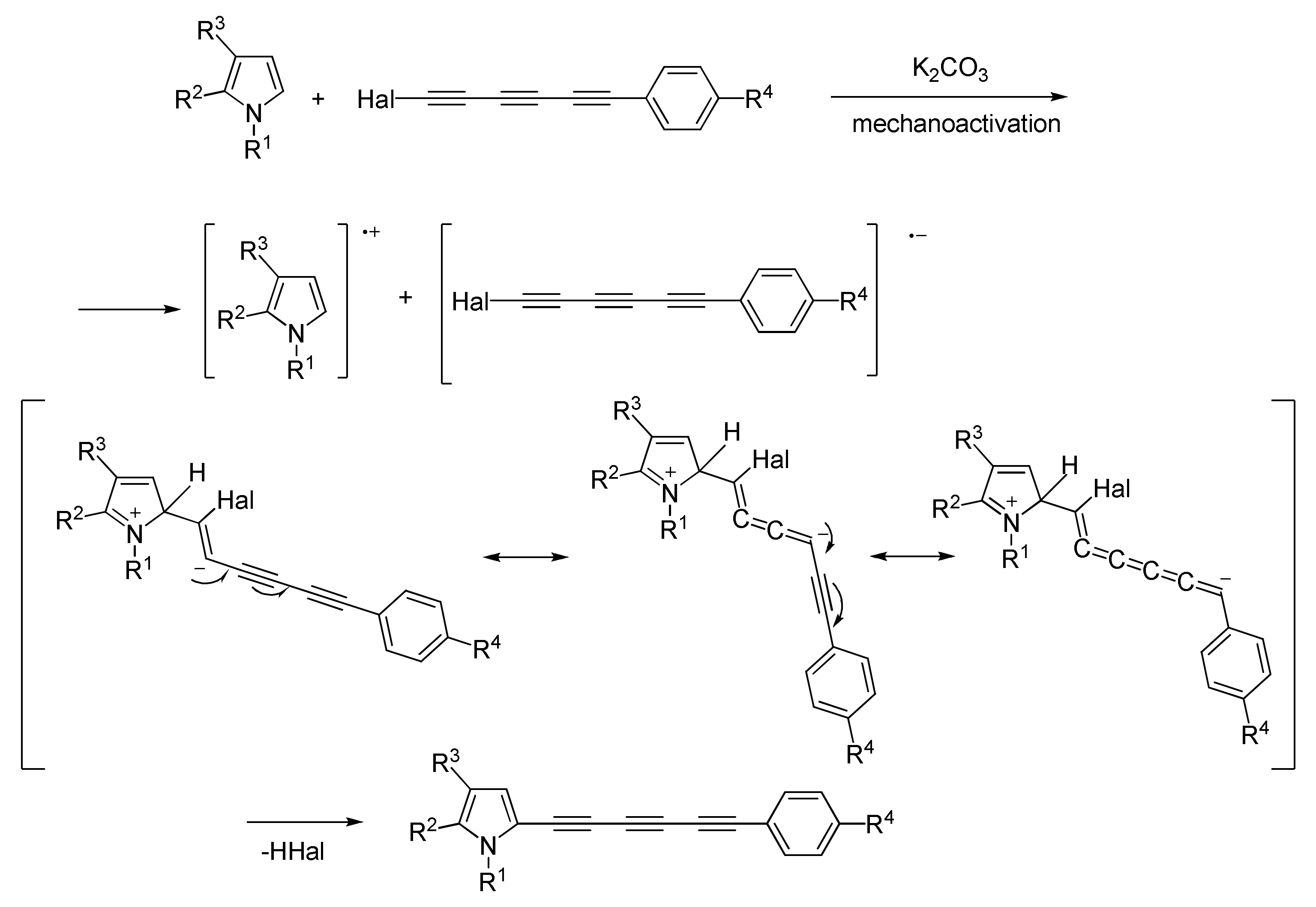
2.1.5. Cross-Coupling of Halopolyynes with Pyrroles
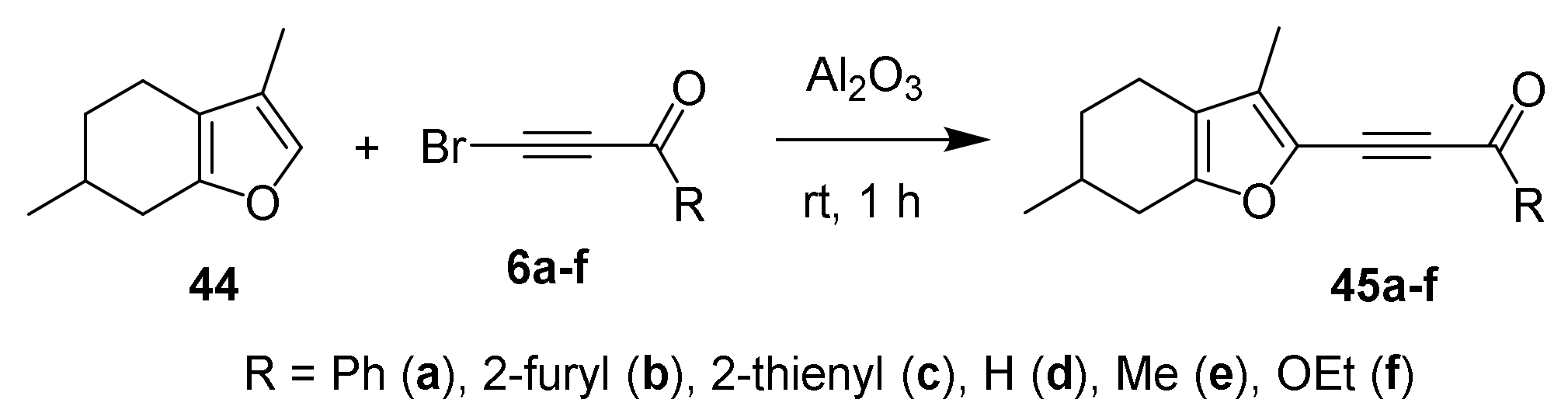
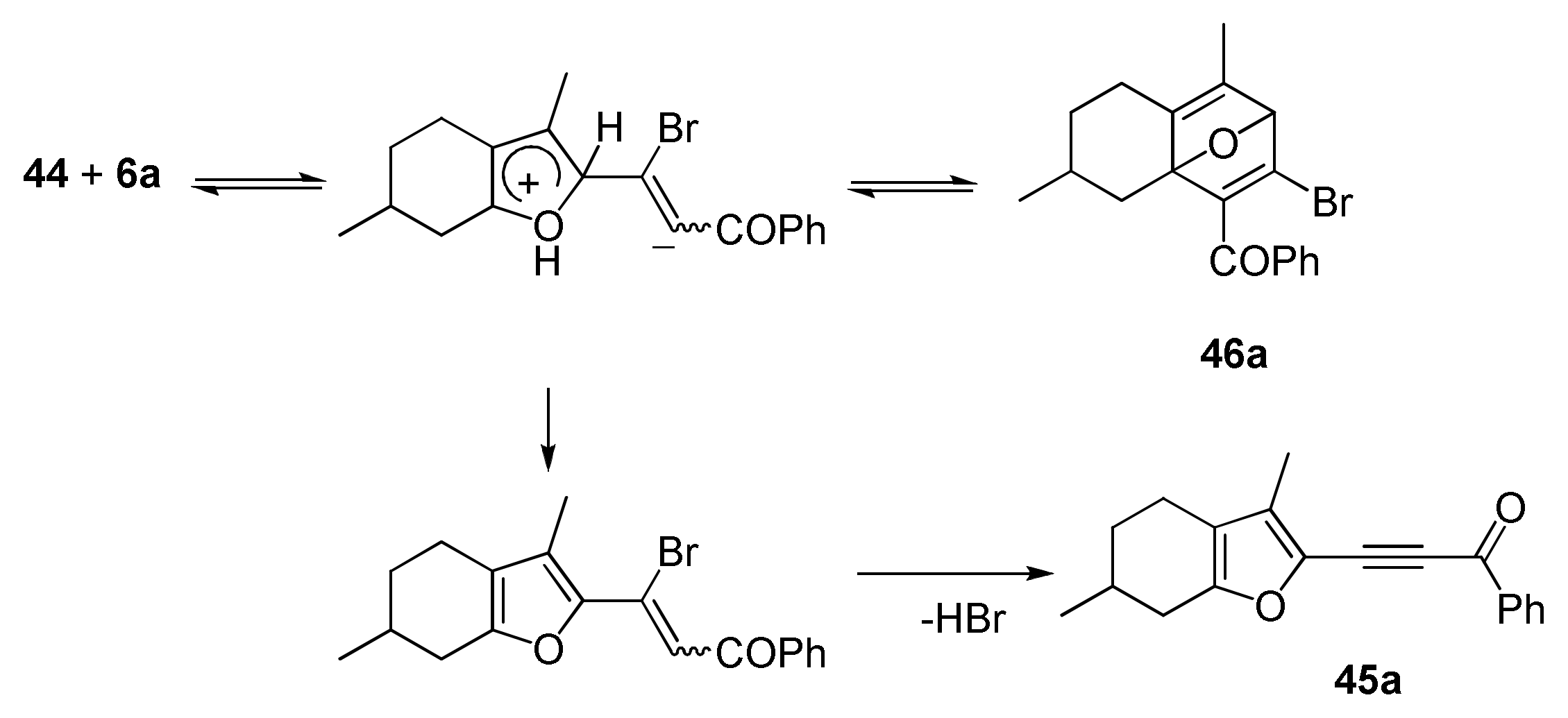
3. Reaction of Acylhaloacetylenes with Furans
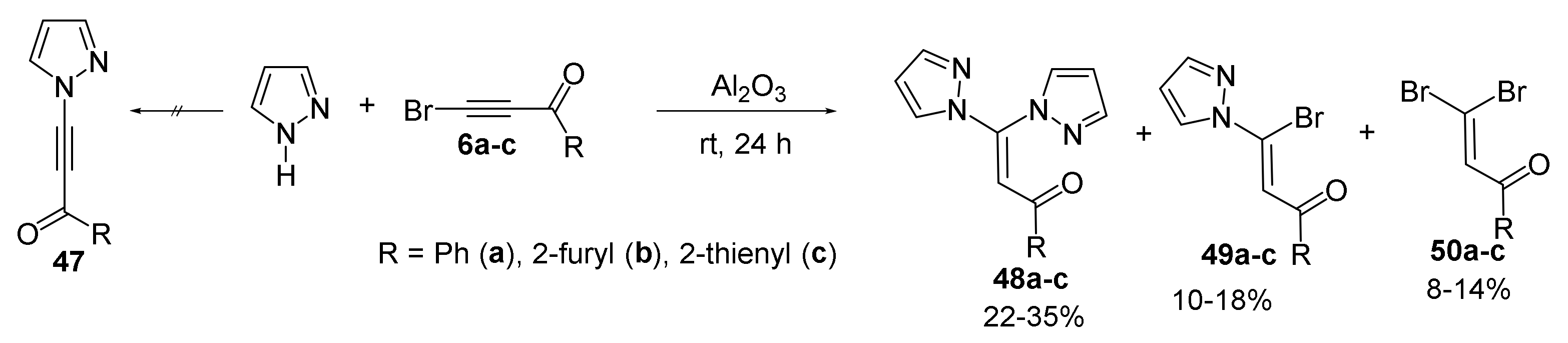
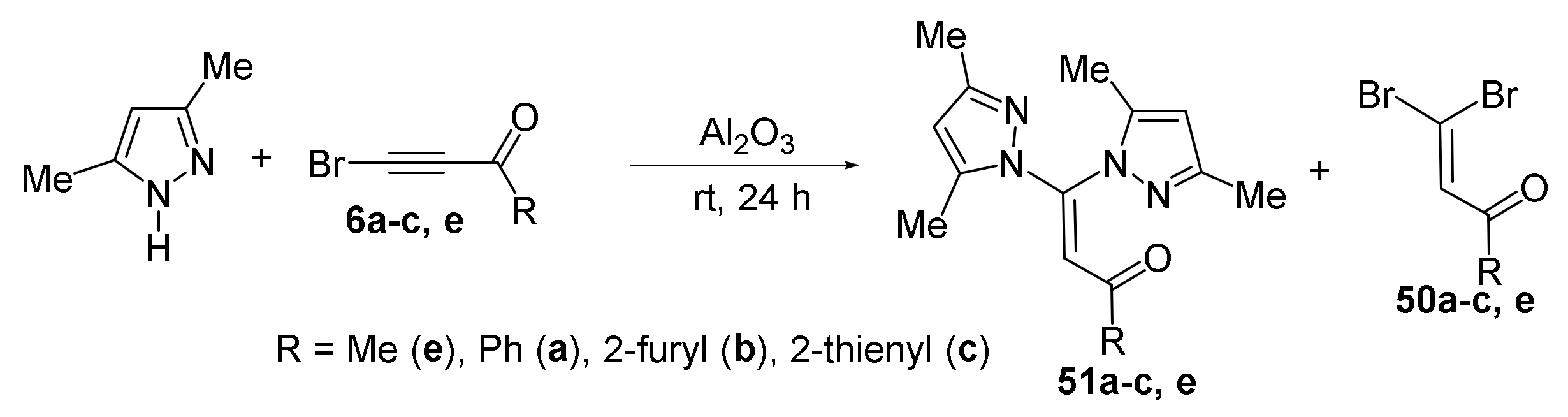
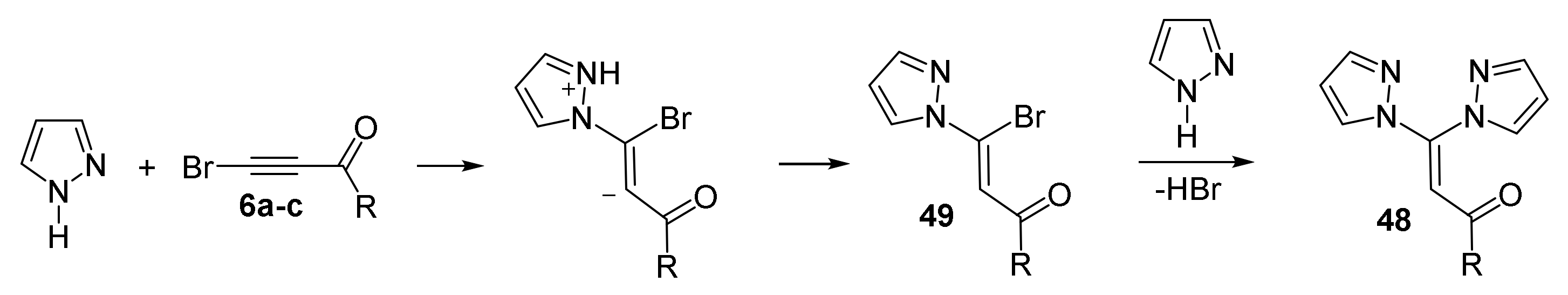
4. Reaction of Acylhaloacetylenes with Pyrazoles
5. Selected Reactions of Acylethynylpyrroles and Their Analogs
5.1. Cyclizations with Propargylamine
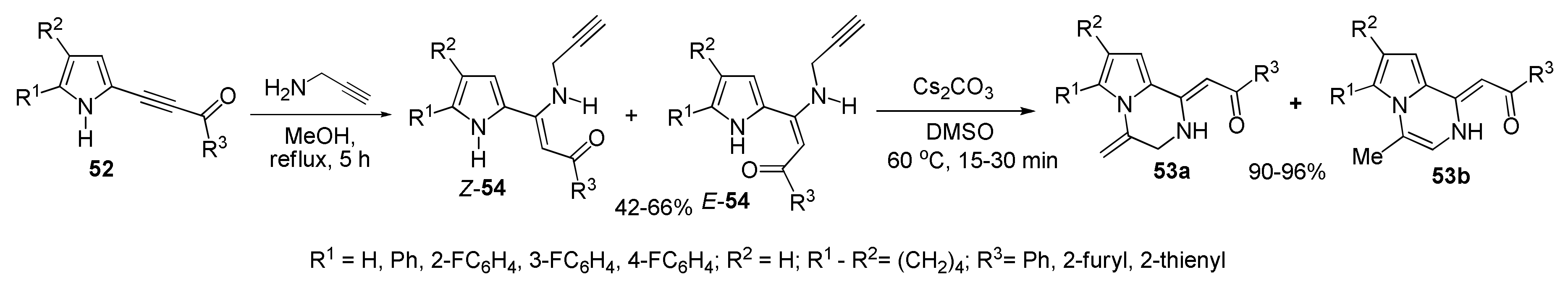
5.1.1. Synthesis of Pyrrolo[1,2-a]pyrazines
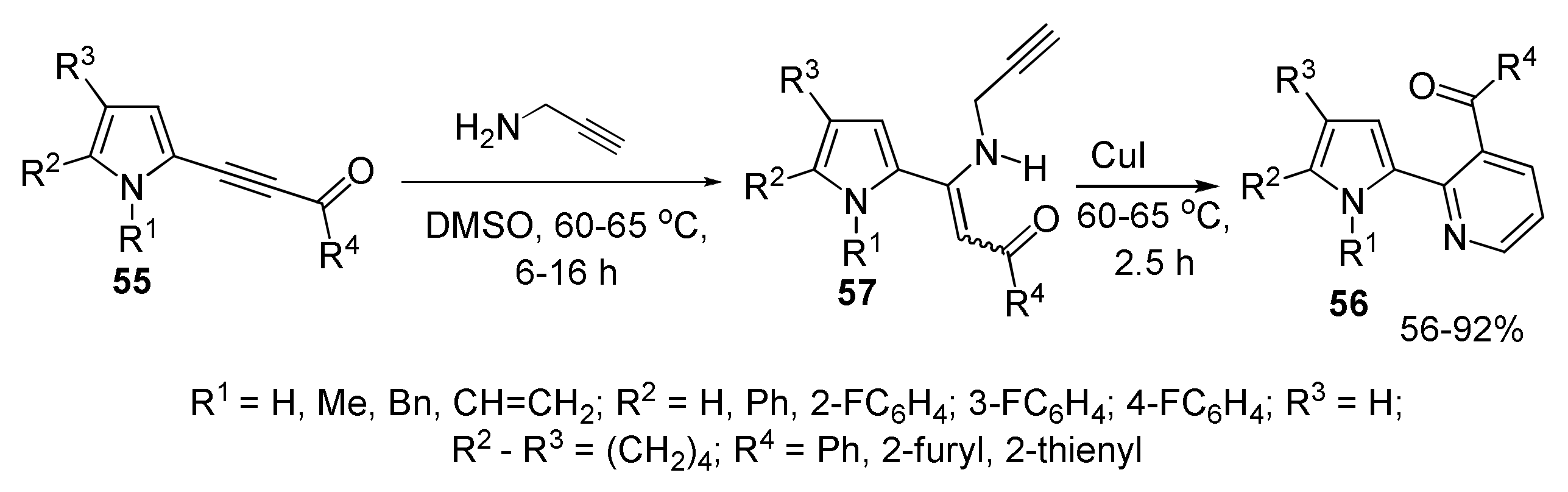
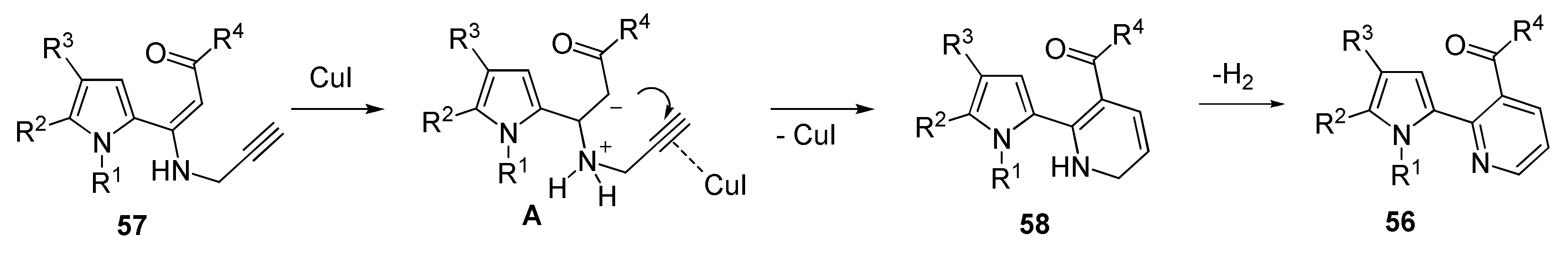
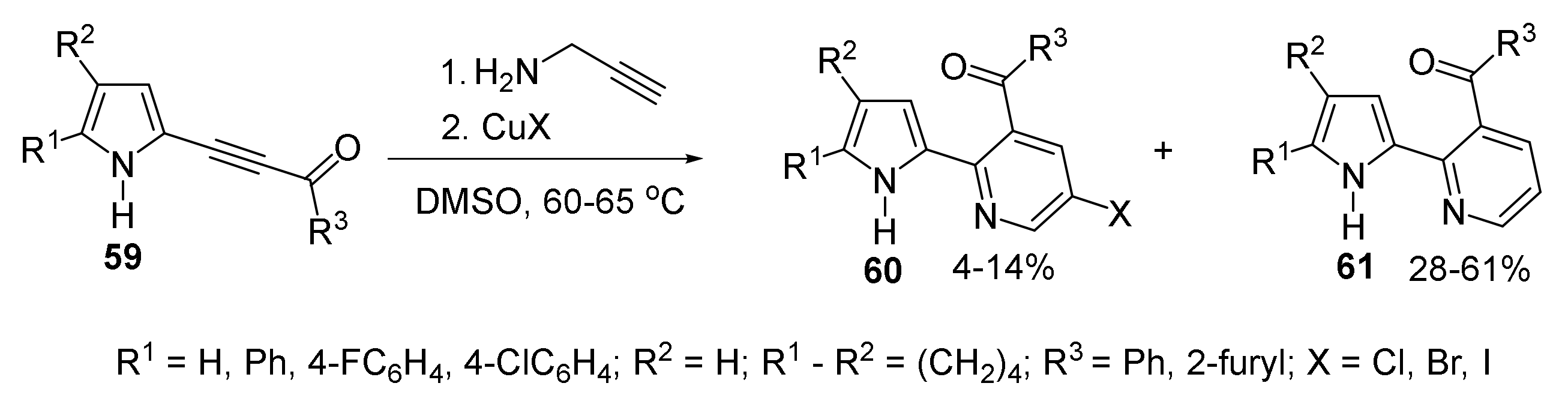
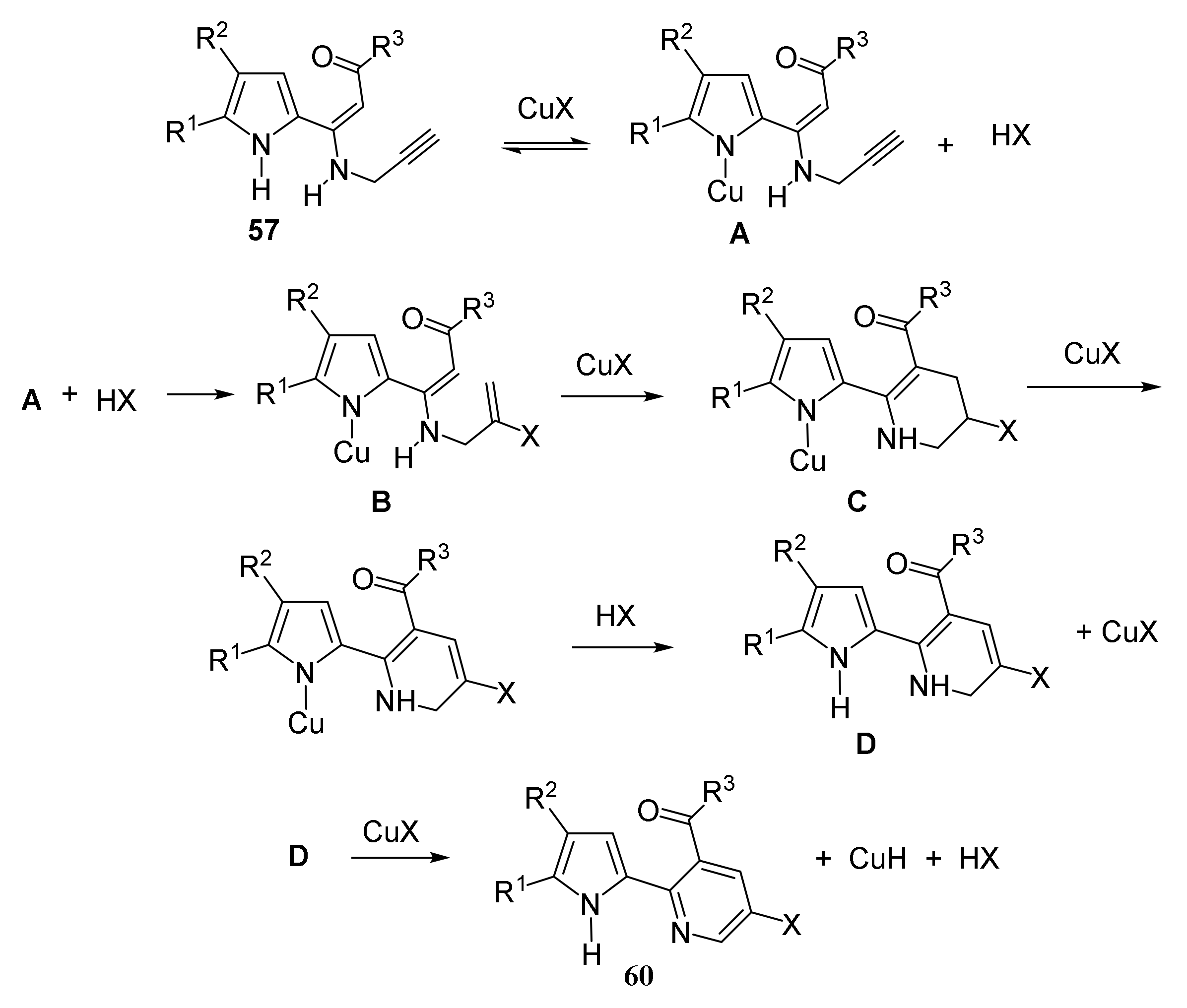
5.1.2. Synthesis of Pyrrolyl Pyridines
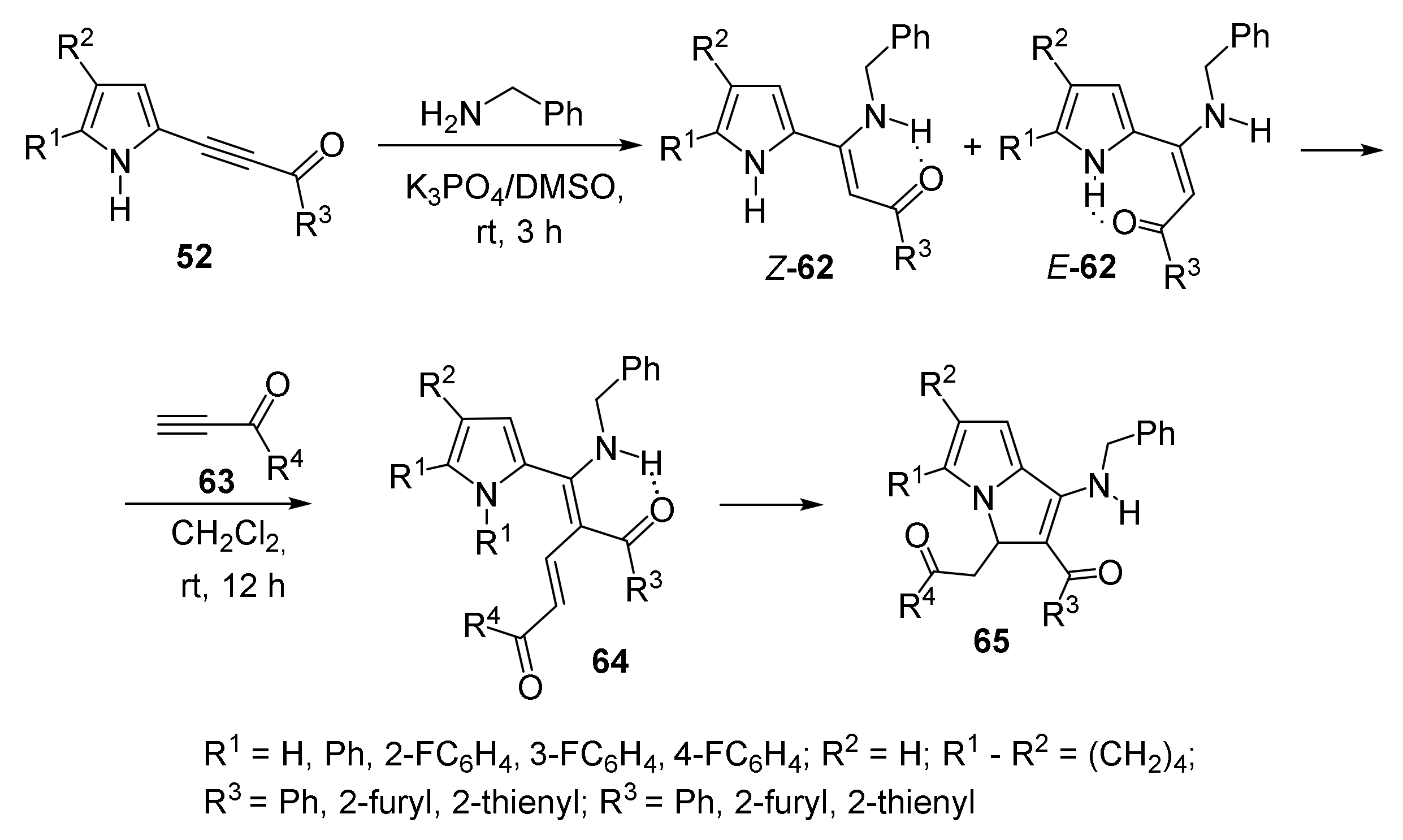
5.2. Synthesis of Pyrrolizines via Three-Component Cyclization with Benzylamine and Acylacetylenes
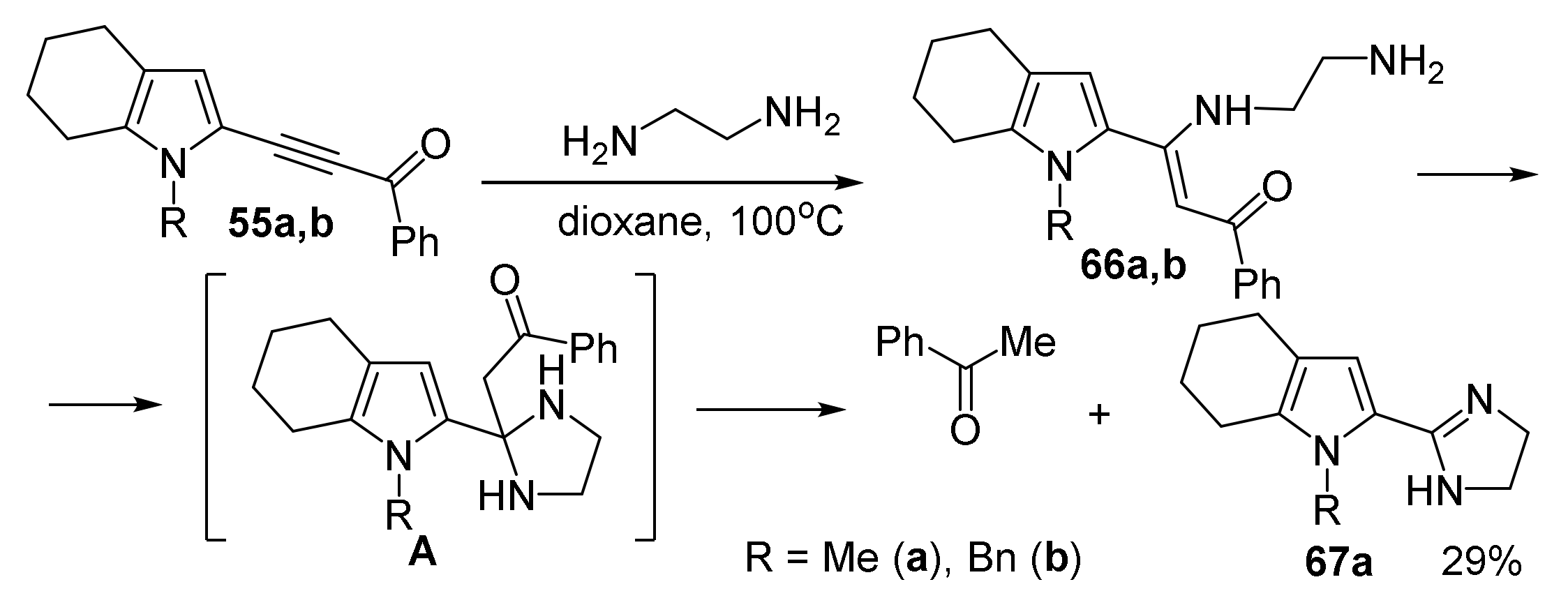
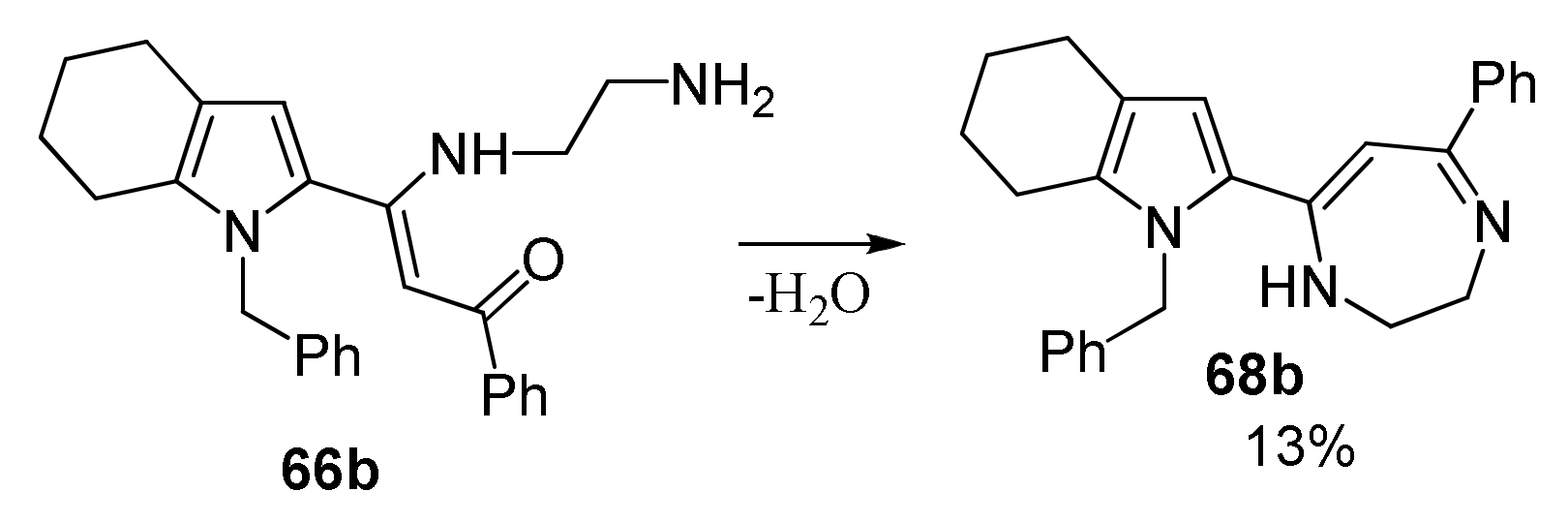
5.3. Reactions with Ethylenediamine
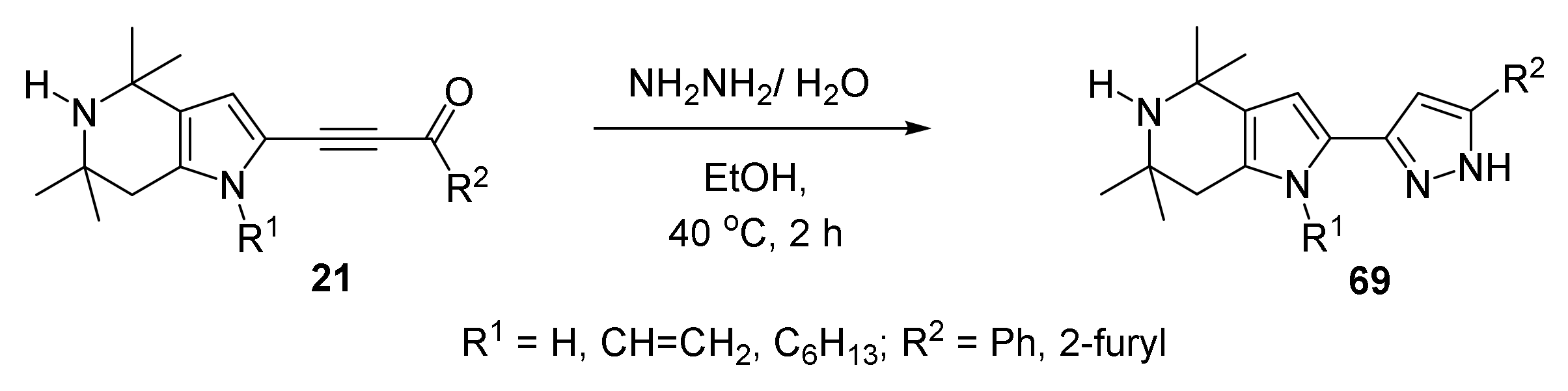
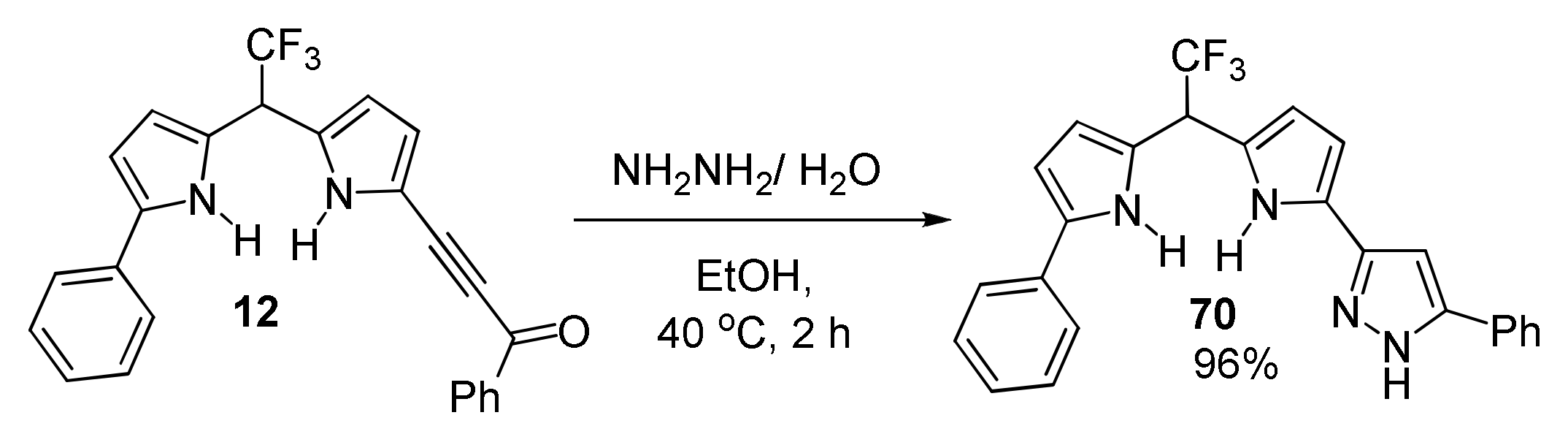
5.4. Cyclization with Hydrazine: Synthesis of Pyrrolyl Pyrazoles
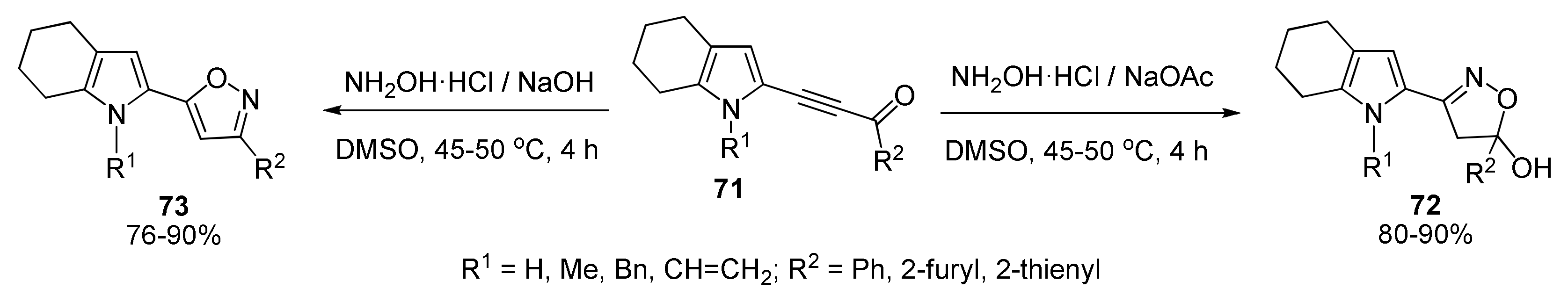
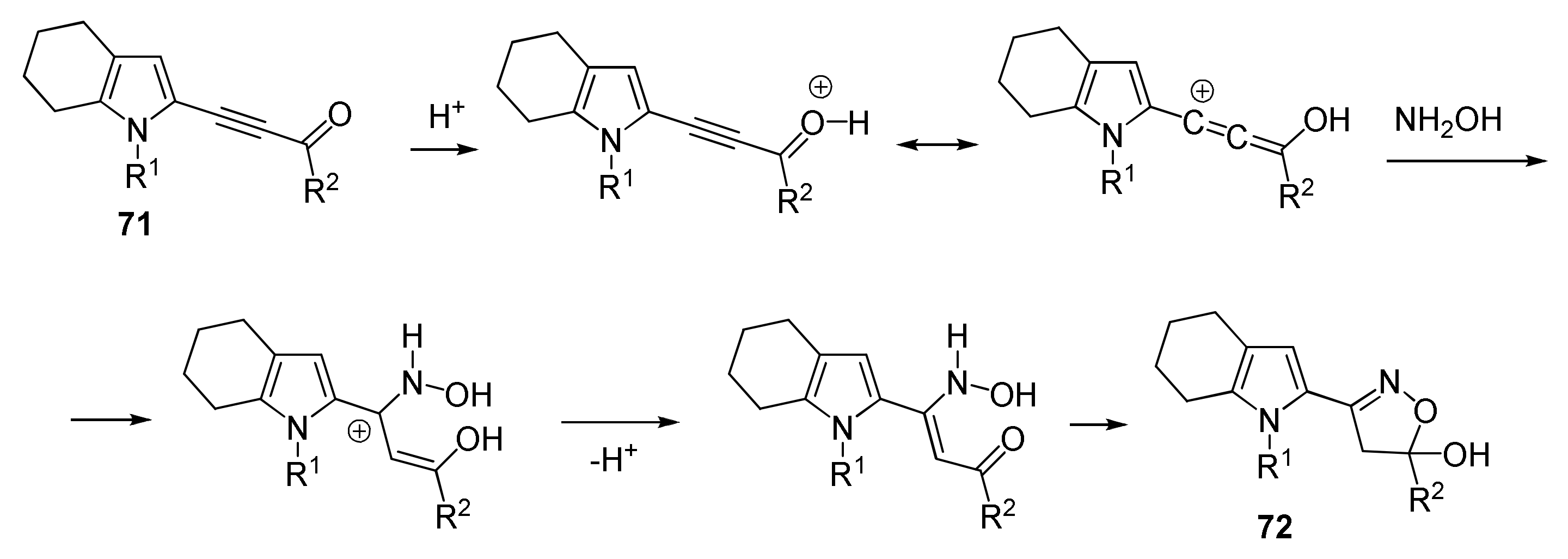
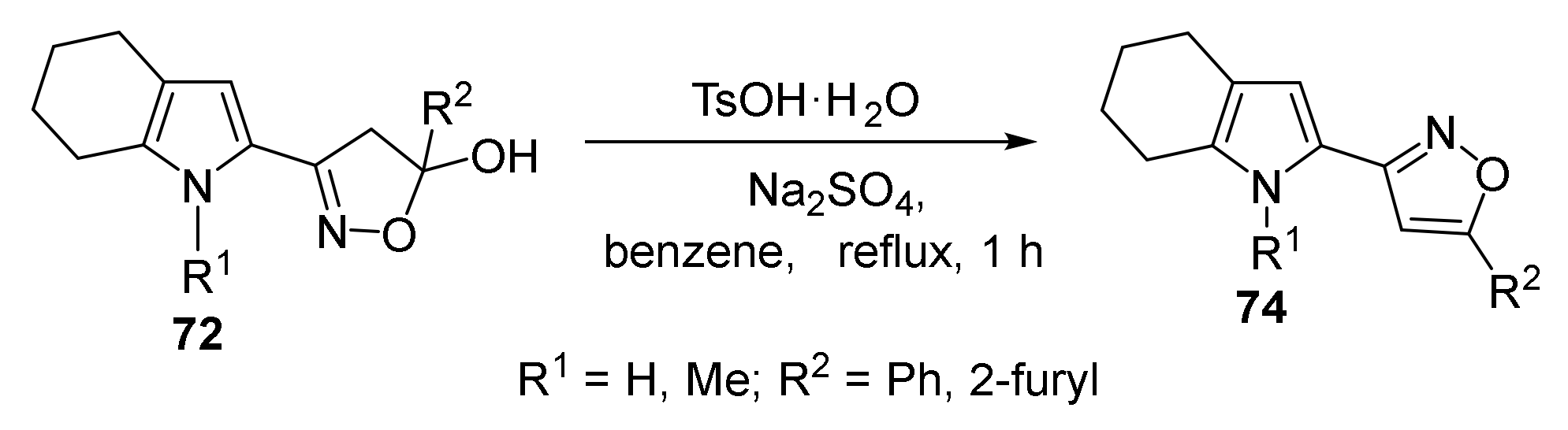
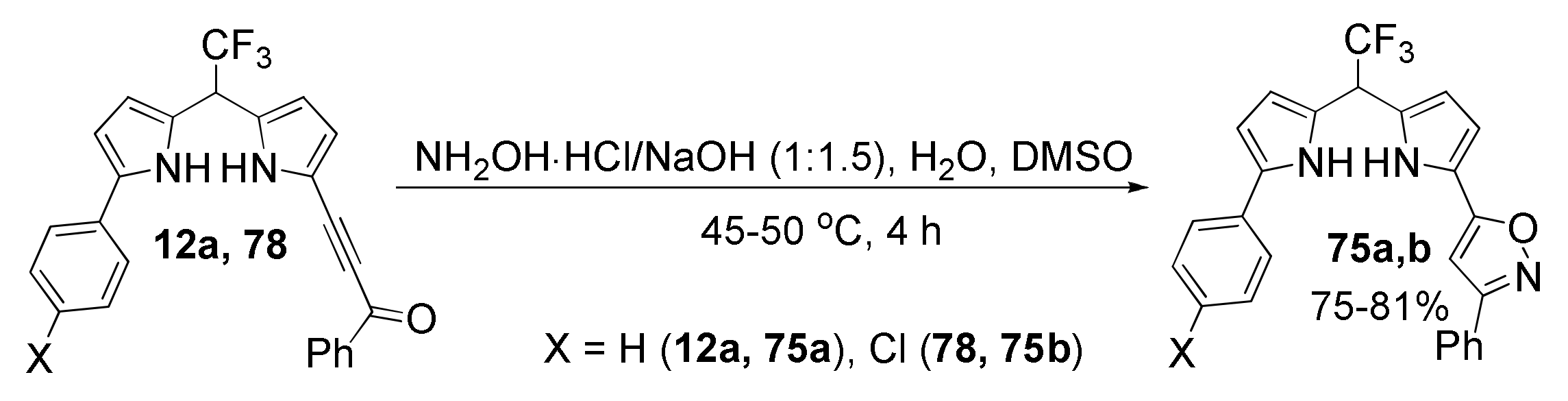
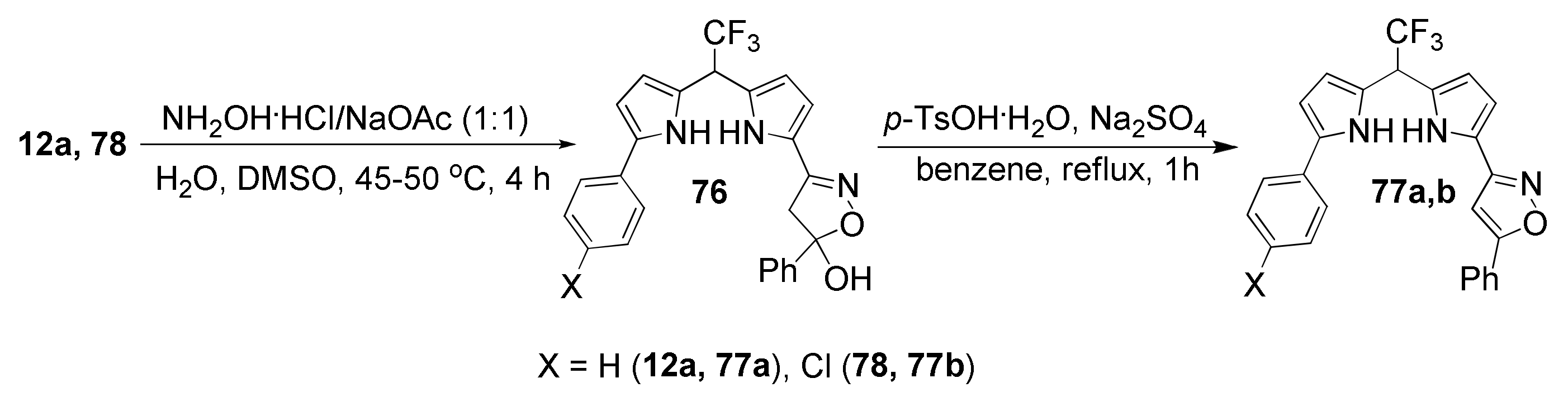
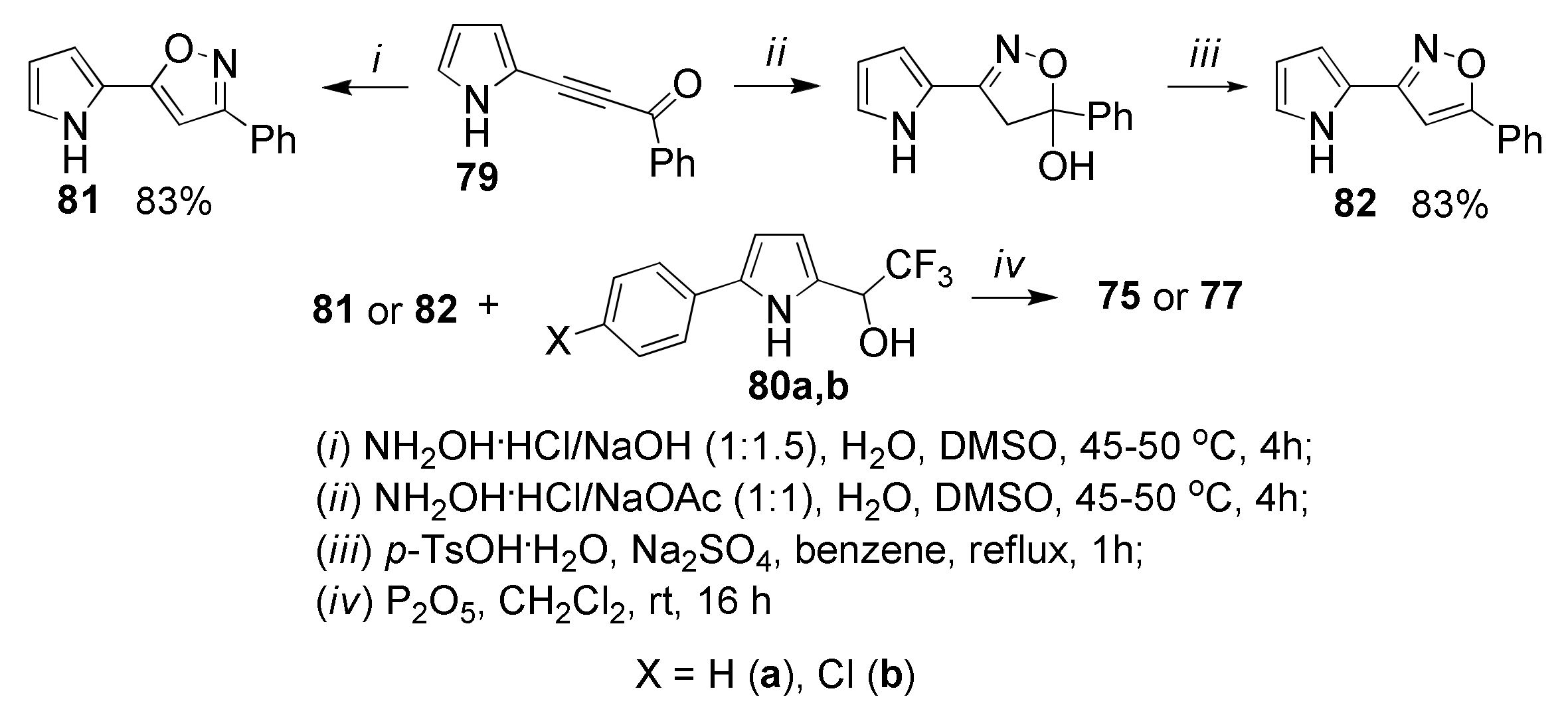
5.5. Cyclization with Hydroxylamine: Synthesis of Pyrrolyl Isoxazoles
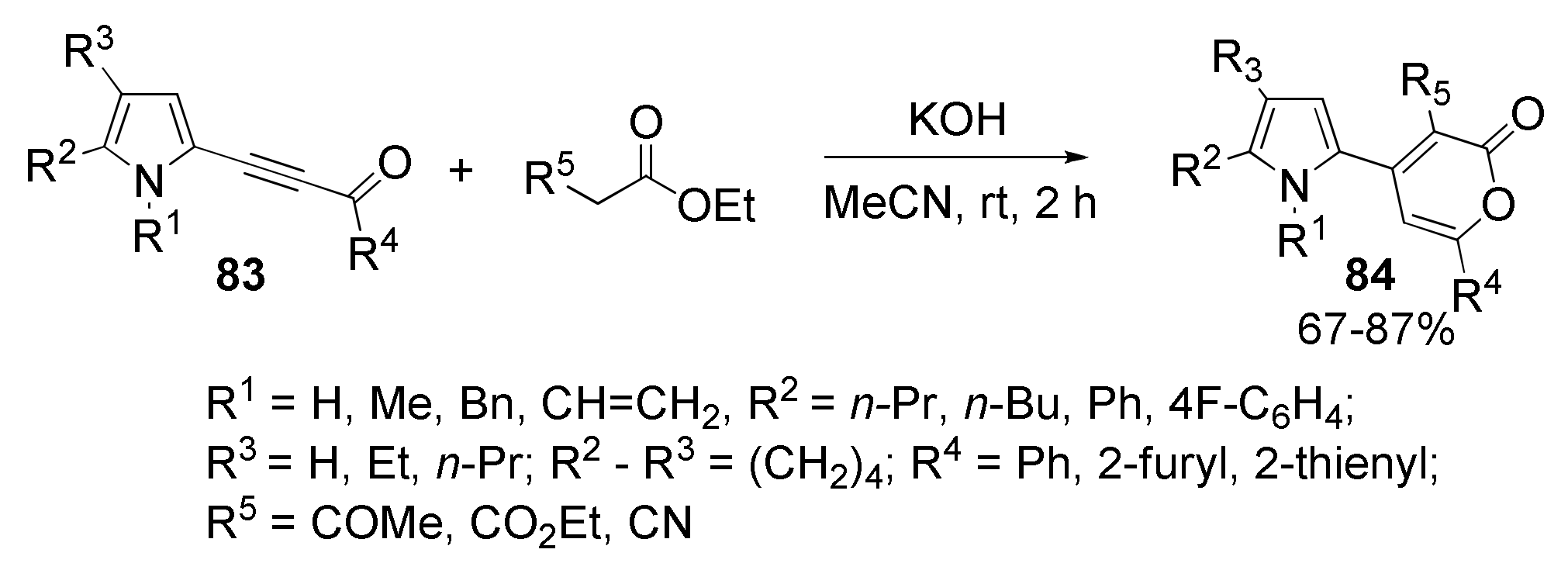
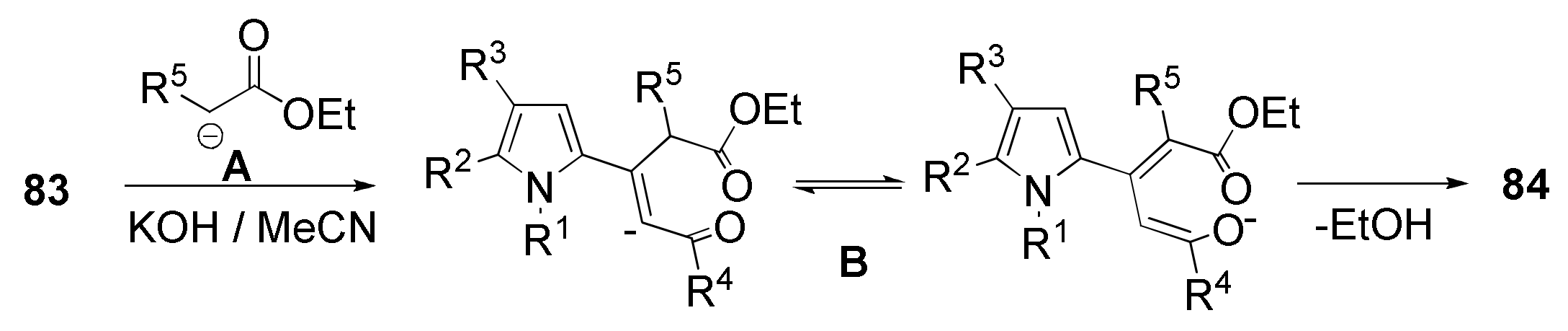
5.6. Cyclization with Methylene Active Esters: Synthesis of Pyrrolyl Pyrones
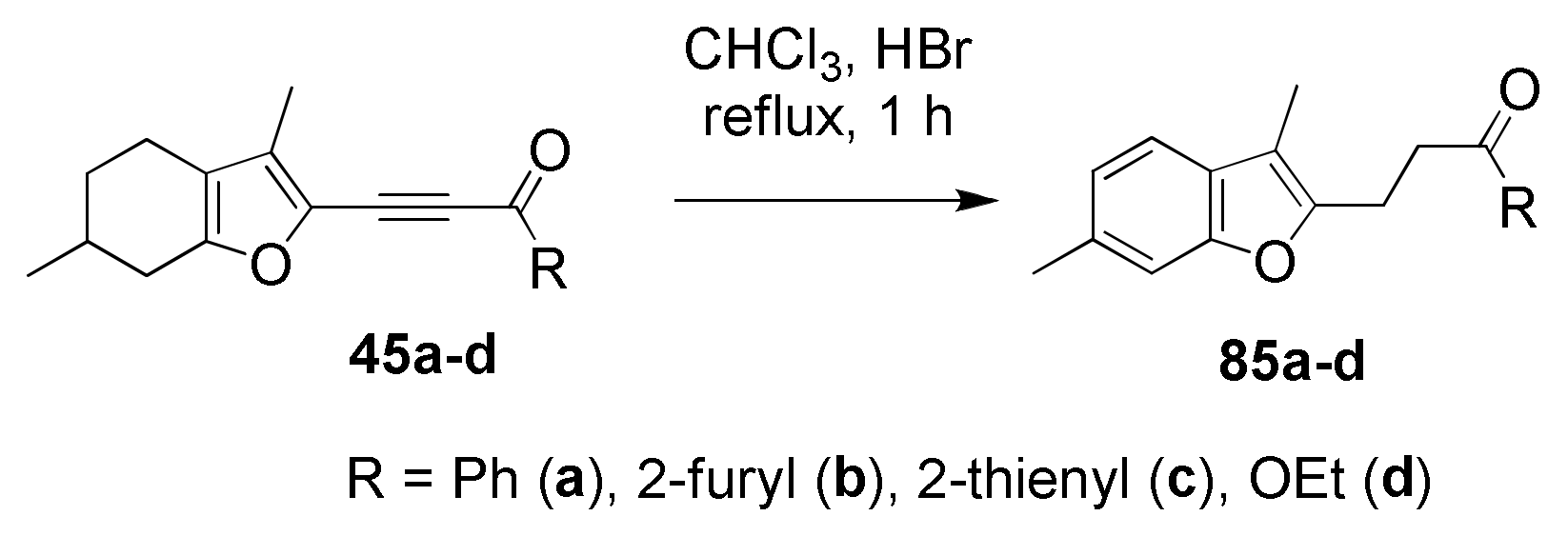
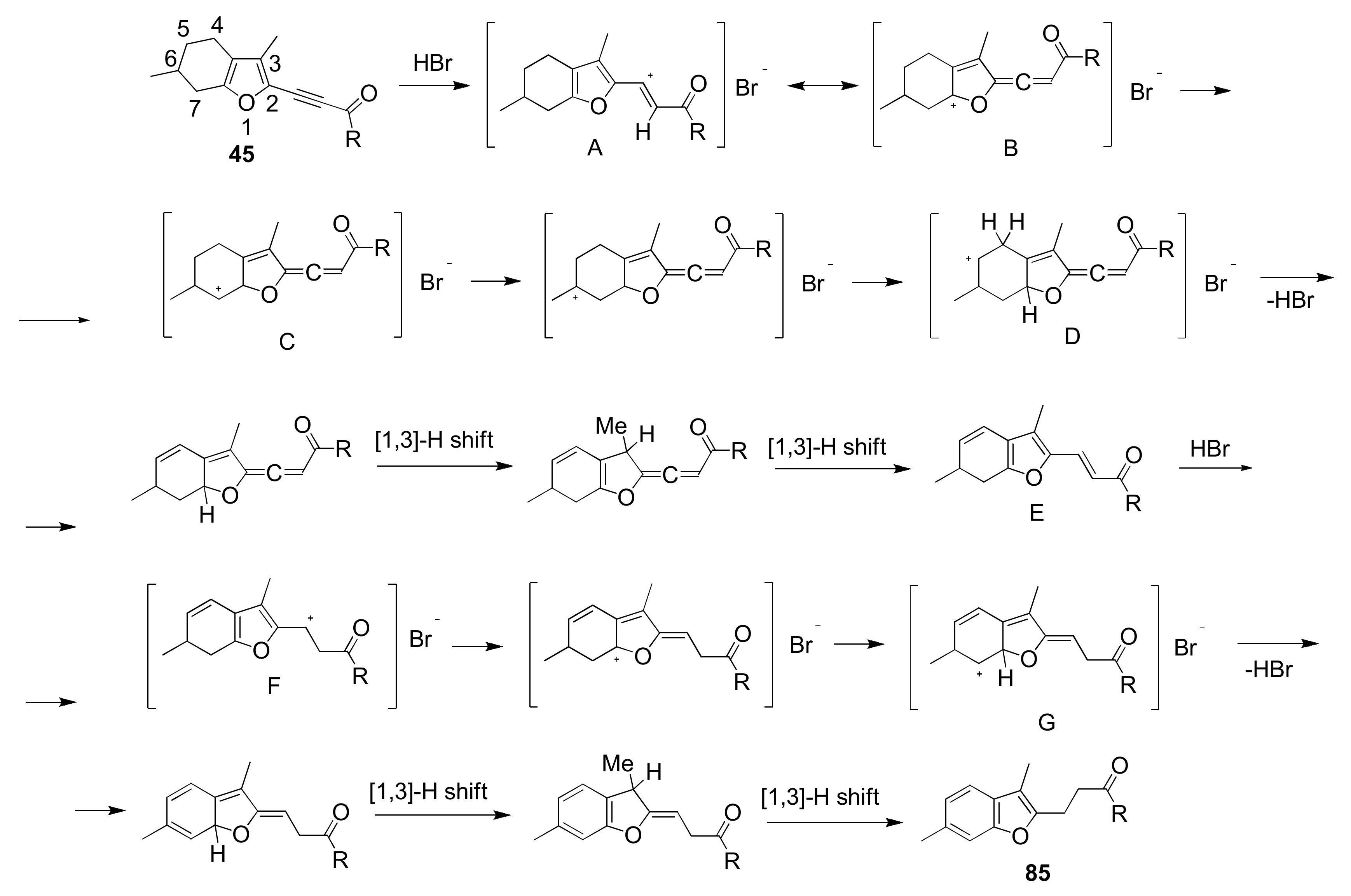
5.7. Unprecedented Four-Proton Migration in Acylethynylmenthofurans: “A Proton Pump”
6. Concluding Remarks and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Mishra, R.; Jha, K.K.; Kumar, S.; Tomer, I. Synthesis, properties and biological activity of thiophene: A review. Der. Pharm. Chem. 2011, 3, 38–54. [Google Scholar]
- Chaudhary, A.; Jha, K.K.; Kumar, S. Biological diversity of thiophene: A review. J. Adv. Sci. Res. 2012, 3, 3–10. [Google Scholar]
- Kharb, R.; Birla, S.; Sharma, A.K. Recent updates on antimicrobial potential of novel furan derivatives. Int. J. Pharm. Phytopharmacol. Res. 2014, 3, 451–459. [Google Scholar]
- Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 2015, 5, 15233–15266. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Gkermani, A.; Hadjipavlou-Litina, D. Synthesis and pharmacochemistry of new pleiotropic pyrrolyl derivatives. Molecules 2015, 20, 16354–16374. [Google Scholar] [CrossRef]
- Mishra, R.; Sharma, P.K. A review on synthesis and medicinal importance of thiophene. Int. J. Eng. Al. Sci. 2015, 1, 46–59. [Google Scholar]
- Heravi, M.M.; Zadsirjan, V. Recent advances in the synthesis of biologically active compounds containing benzo[b]furans as a framework. Curr. Org. Syn. 2016, 13, 780–833. [Google Scholar] [CrossRef]
- Gholap, S.S. Pyrrole: An emerging scaffold for construction of valuable therapeutic agents. Eur. J. Med. Chem. 2016, 110, 13–31. [Google Scholar] [CrossRef]
- Yet, L. Privileged Structures in Drug Discovery: Medicinal Chemistry and Synthesis, 1st ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2018. [Google Scholar]
- Shah, R.; Verma, P.K. Therapeutic importance of synthetic thiophene. Chem. Centr. J. 2018, 12, 137–158. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Alam, O.; Naim, M.J.; Shaquiquzzaman, M.; Alam, M.M.; Iqbal, M. Pyrrole: An insight into recent pharmacological advances with structure activity relationship. Eur. J. Med. Chem. 2018, 157, 527–561. [Google Scholar] [CrossRef]
- Chiurchiù, E.; Gabrielli, S.; Ballini, R.; Palmieri, A. A new valuable synthesis of polyfunctionalized furans starting from β-nitroenones and active methylene compounds. Molecules 2019, 24, 4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeni, G.; Lüdtke, D.S.; Nogueira, C.W.; Panatieri, R.B.; Braga, A.L.; Silveira, C.C.; Stefani, H.A. New acetylenic furan derivatives: Synthesis and anti-inflammatory activity. Tetrahedron Lett. 2001, 42, 8927–8930. [Google Scholar] [CrossRef]
- Acetylene Chemistry: Chemistry, Biology and Material Science; Diederich, F.; Stang, P.J.; Tykwinski, R.R. (Eds.) Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Li, Y.; Waser, J. Platinum-catalyzed domino reaction with benziodoxole reagents for accessing benzene-alkynylated indoles. Angew. Chem. Int. Ed. 2015, 54, 5438–5442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonogashira, K. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Pattenden, G., Eds.; Pergamon Press: Oxford, UK, 1991; pp. 521–561. [Google Scholar]
- Chinchilla, R.; Nájera, C. The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef]
- Chinchilla, R.; Njera, C. Recent advances in Sonogashira reactions. Chem. Soc. Rev. 2011, 40, 5084–5121. [Google Scholar] [CrossRef]
- Heravi, M.; Dehghani, M.; Zadsirjan, V.; Ghanbarian, M. Alkynes as privileged synthons in selected organic name reactions. Curr. Org. Syn. 2019, 16, 205–243. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Stepanova, Z.V.; Sobenina, L.N.; Mikhaleva, A.I.; Ushakov, I.A. Ethynylation of pyrroles with 1-acyl-2-bromoacetylenes on alumina: A formal-inverse Sonogashira coupling. Tetrahedron Lett. 2004, 34, 6513–6516. [Google Scholar] [CrossRef]
- Seregin, I.V.; Ryabova, V.; Gevorgyan, V. Direct palladium-catalyzed alkynylation of N-fused heterocycles. J. Am. Chem. Soc. 2007, 129, 7742–7743. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Wang, X. Direct palladium-catalyzed C-3 alkynylation of indoles. Tetrahedron Lett. 2009, 50, 763–766. [Google Scholar] [CrossRef]
- Matsuyama, N.; Hirano, K.; Satoh, T.; Miura, M. Nickel-catalyzed direct arylation of azoles with aryl bromides. Org. Lett. 2009, 11, 4156–4159. [Google Scholar] [CrossRef]
- Matsuyama, N.; Kitahara, M.; Hirano, K.; Satoh, T.; Miura, M. Nickel- and copper- direct alkynylation of azoles and polyfluoroarenes with terminal alkynes under O2 or atmospheric conditions. Org. Lett. 2010, 12, 2358–2361. [Google Scholar] [CrossRef] [PubMed]
- Besselievre, F.; Piguel, S. Copper as a powerful catalyst in the direct alkynylation of azoles. Angew. Chem. Int. Ed. 2009, 48, 9553–9556. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.P.; Charpentier, J.; Waser, J. Direct alkynylation of indole and pyrrole heterocycles. Angew. Chem. Int. Ed. 2009, 48, 9346–9349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, Z.V.; Sobenina, L.N.; Mikhaleva, A.I.; Ushakov, I.A.; Chipanina, N.N.; Elokhina, V.N.; Voronov, V.K.; Trofimov, B.A. Silica-assisted reactions of pyrroles with 1-acyl-2-bromoacetylenes. Synthesis 2004, 2736–2742. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Stepanova, Z.V.; Demenev, A.P.; Mikhaleva, A.I.; Ushakov, I.A.; Vakul’skaya, T.I.; Petrova, O.V. Synthesis of 2-benzoylethynylpyrroles by cross-coupling of 2-arylpyrroles with 1-benzoyl-2-bromoacetylene over aluminium oxide. Russ. J. Org. Chem. 2006, 42, 1348–1355. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Stepanova, Z.V.; Sobenina, L.N.; Mikhaleva, A.I.; Sinegovskaya, L.M.; Potekhin, K.A.; Fedyanin, I.V. 2-(2-Benzoylethynyl)-5-phenylpyrrole: Fixation of cis- and trans-rotamers in a crystal state. Mendeleev Commun. 2005, 15, 229–232. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Demenev, A.P.; Mikhaleva, A.I.; Ushakov, I.A.; Vasil’tsov, A.M.; Ivanov, A.V.; Trofimov, B.A. Ethynylation of indoles with 1-benzoyl-2-bromoacetylene on Al2O3. Tetrahedron Lett. 2006, 47, 7139–7141. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Stepanova, Z.V.; Ushakov, I.A.; Petrova, O.V.; Tarasova, O.A.; Volkova, K.A.; Mikhaleva, A.I. Regioselective cross-coupling of 1-vinylpyrroles with acylbromoacetylenes on Al2O3: A synthesis of 1-vinyl-2-(2-acylethynyl)pyrroles. Synthesis 2007, 39, 447–451. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Demenev, A.P.; Stepanova, Z.V.; Petrova, O.V.; Ushakov, I.A.; Mikhaleva, A.I. A palladium- and copper-free cross-coupling of ethyl 3-halo-2-propynoates with 4,5,6,7-tetrahydroindoles on alumina. Tetrahedron Lett. 2007, 48, 4661–4664. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Stepanova, Z.V.; Vakul’skaya, T.I.; Kazheva, O.N.; Aleksandrov, G.G.; Dyachenko, O.A.; Mikhaleva, A.I. Reactions of 2-phenylpyrrole with bromobenzoylacetylene on metal oxides active surfaces. Tetrahedron 2008, 64, 5541–5544. [Google Scholar] [CrossRef]
- Petrova, O.V.; Sobenina, L.N.; Ushakov, I.A.; Mikhaleva, A.I. Reaction of indoles with ethyl bromopropynoate over Al2O3 surface. Russ. J. Org. Chem. 2008, 44, 1512–1516. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Stepanova, Z.V.; Petrova, O.V. Chemo- and regioselective ethynylation of 4,5,6,7-tetrahydroindoles with ethyl 3-halo-2-propynoates. Tetrahedron Lett. 2008, 49, 3946–3949. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Petrova, O.V.; Gulia, N.; Osowska, K.; Szafert, S.; Mikhaleva, A.I.; Trofimov, B.A. Cross-coupling of 4,5,6,7-tetrahydroindole with functionalized haloacetylenes on active surfaces of metal oxides and salts. Russ. J. Org. Chem. 2010, 46, 1373–1377. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Petrova, O.V.; Ushakov, I.A.; Mikhaleva, A.I.; Trofimov, B.A. Hydroamination of 2-ethynyl-4,5,6,7-tetrahydroindoles: Towards 2-substituted aminoderivatives of indole. Synthesis 2010, 42, 2468–2474. [Google Scholar]
- Trofimov, B.A.; Sobenina, L.N.; Stepanova, Z.V.; Ushakov, I.A.; Mikhaleva, A.I.; Tomilin, D.N.; Kazheva, O.N.; Alexandrov, G.G.; Chekhlov, A.N.; Dyachenko, O.A. Peculiar rearrangement of the [2+2]-cycloadducts of DDQ and 2-ethynylpyrroles. Tetrahedron Lett. 2010, 51, 5028–5031. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Stepanova, Z.V.; Ushakov, I.A.; Sinegovskaya, L.M.; Vakul’skaya, T.I.; Mikhaleva, A.I. Facile [2+2]-cycloaddition of DDQ to acetylenic moiety: Synthesis of pyrrole-(indole)bicyclo[4.2.0]octadiene ensembles from C-ethynylpyrroles or –indoles. Synthesis 2010, 42, 470–477. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Stepanova, Z.V.; Ushakov, I.A.; Mikhaleva, A.I.; Tomilin, D.N.; Kazheva, O.N.; Alexandrov, G.G.; Dyachenko, O.A.; Trofimov, B.A. From 4,5,6,7-tetrahydroindole to functionalized furan-2-one-4,5,6,7-tetrahydroindole-cyclobutene sequence in two steps. Tetrahedron 2011, 67, 4832–4837. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Ushakov, I.A.; Mikhaleva, A.I.; Trofimov, B.A. Transition-metal-free stereoselective and regioselective hydroamination of 2-benzoylethynyl-4,5,6,7-tetrahydroindoles with amino acids. Synthesis 2012, 44, 2084–2090. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Ushakov, I.A.; Mikhaleva, A.I.; Ma, J.S.; Yang, G.; Trofimov, B.A. The “Click-Chemistry” with 2-ethynyl-4,5,6,7-tetrahydroindoles: Towards a functionalized tetrahydroindole-triazole ensembles. Synthesis 2013, 45, 678–682. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Stepanova, Z.V.; Petrova, O.V.; Ma, J.S.; Yang, G.; Tatarinova, A.A.; Mikhaleva, A.I.; Trofimov, B.A. Synthesis of 3-[5-(biphenyl-4-yl)pyrrol-2-yl]-1-phenylprop-2-yn-1-ones by palladium-free cross-coupling between pyrroles and haloalkynes on aluminum oxide. Russ. Chem. Bull. Int. Ed. 2013, 62, 88–92, [Izv. Akad. Nauk Ser. Khim. 2013, 88–92]. [Google Scholar] [CrossRef]
- Banwell, M.G.; Goodwin, T.E.; Ng, S.; Smith, J.A.; Wong, D.J. Palladium-catalysed cross-coupling and reactions involving pyrroles. Eur. J. Org. Chem. 2006, 3043–3060. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Nedolya, N.A. Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 3, p. 45. [Google Scholar]
- Trofimov, B.A.; Sobenina, L.N. Targets in Heterocyclic Chemistry; Attanasi, O.A., Spinelli, D., Eds.; Societa Chimica Italiana: Rome, Italy, 2009; pp. 92–119. [Google Scholar]
- Tanaka, K.; Kaupp, G. Solvent-Free Organic Synthesis; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; 468p. [Google Scholar]
- Trofimov, B.A.; Mikhaleva, A.I.; Schmidt, E.Y.; Sobenina, L.N. Pyrroles and N-vinylpyrroles from ketones and acetylenes: Recent strides. Adv. Heterocycl. Chem. 2010, 99, 209–254. [Google Scholar]
- Dudnik, A.S.; Gevorgyan, V. Formal inverse Sonogashira reaction: Direct alkynylation of arenes and heterocycles with alkynyl halides. Angew. Chem. Int. Ed. 2010, 49, 2096–2098. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Brion, J.-D.; Alami, M. Transition-metal-catalyzed direct C–H alkenylation, alkynylation, benzylation, and alkylation of (hetero)arenes. Eur. J. Org. Chem. 2010, 6495–6516. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Mikhaleva, A.I.; Schmidt, E.Y.; Sobenina, L.N. Chemistry of Pyrroles; CRC Press Inc: Boca-Raton, LA, USA, 2014. [Google Scholar]
- Sobenina, L.N.; Tomilin, D.N.; Trofimov, B.A. C-Ethynylpyrroles: Synthesis and reactivity. Russ. Chem. Rev. 2014, 83, 475–501. [Google Scholar] [CrossRef]
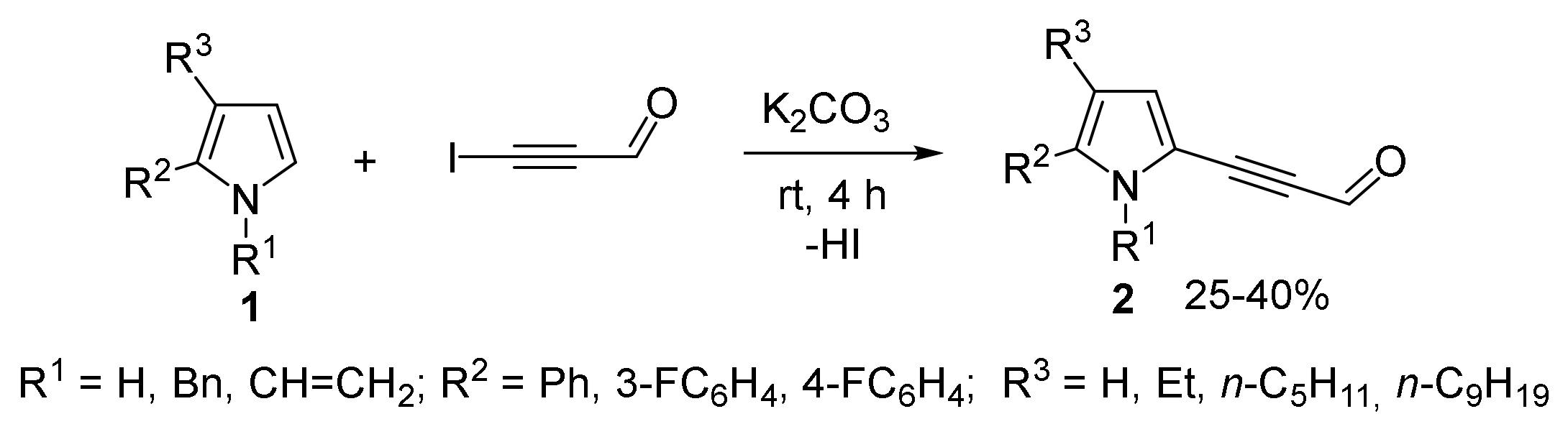
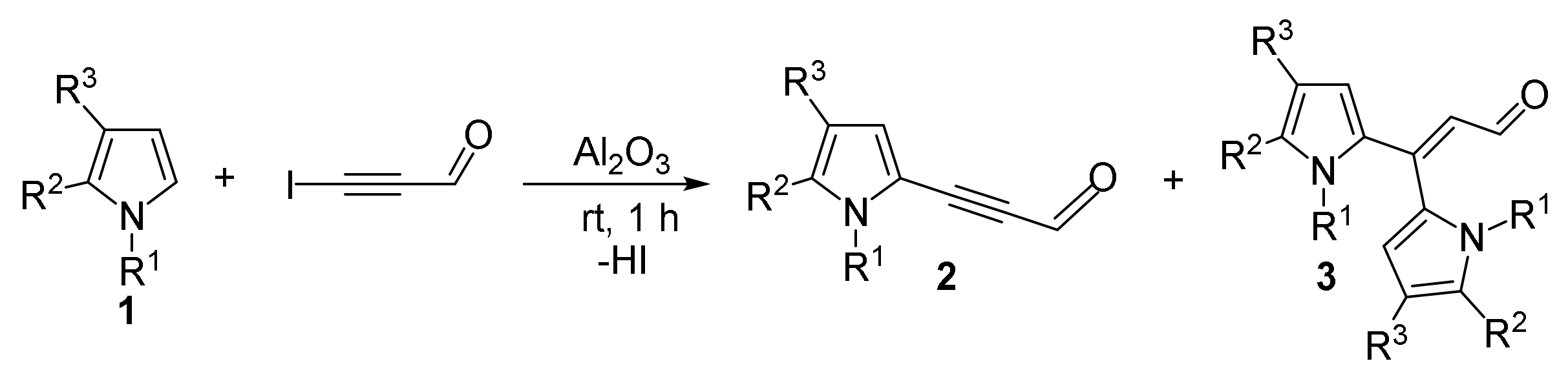
- Gotsko, M.D.; Sobenina, L.N.; Tomilin, D.N.; Markova, M.V.; Ushakov, I.A.; Vakul’skaya, T.I.; Khutsishvili, S.S.; Trofimov, B.A. Pyrrole acetylenecarbaldehydes: An entry to a novel class of functionalized pyrroles. Tetrahedron Lett. 2016, 57, 4961–4964. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Petrova, O.V.; Tomilin, D.N.; Gotsko, M.D.; Ushakov, I.A.; Klyba, L.V.; Mikhaleva, A.I.; Trofimov, B.A. Ethynylation of 2-(furan-2-yl)- and 2-(thiophen-2-yl)pyrroles with acylbromoacetylenes in the Al2O3 medium: Relative reactivity of heterocycles. Tetrahedron 2014, 70, 9506–9511. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Sobenina, L.N.; Mikhaleva, A.I.; Ushakov, I.A.; Vakul’skaya, T.I.; Stepanova, Z.V.; Toryashinova, D.-S.D.; Mal’kina, A.G.; Elokhina, V.N. N- and C-Vinylation of pyrroles with disubstituted activated acetylenes. Synthesis 2003, 35, 1272–1279. [Google Scholar] [CrossRef]
- Trofimov, A.B.; Zaitseva, I.L.; Moskovskaya, T.E.; Vitkovskaya, N.M. Theoretical investigation of photoelectron spectra of furan, pyrrole, thiophene, and selenole. Chem. Heterocycl. Compd. 2008, 44, 1101–1112. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Petrushenko, K.B.; Sobenina, L.N.; Gotsko, M.D.; Ushakov, I.A.; Skitnevskaya, A.D.; Trofimov, A.B. Synthesis and optical properties of meso-CF3-BODIPY with acylethynyl substituents in the position 3 of the indacene core. Asian J. Org. Chem. 2016, 5, 1288–1294. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Vasil’tsov, A.M.; Petrova, O.V.; Petrushenko, K.B.; Ushakov, I.A.; Clavier, G.; Meallet-Renault, R.; Mikhaleva, A.I.; Trofimov, B.A. A general route to symmetric and asymmetric meso-CF3-3(5)-aryl(hetaryl)- and 3,5-diaryl(dihetaryl)-BODIPY dyes. Org. Lett. 2011, 13, 2524–2527. [Google Scholar] [CrossRef] [PubMed]
- Sagitova, E.F.; Tomilin, D.N.; Petrova, O.V.; Budaev, A.B.; Sobenina, L.N.; Trofimov, B.A.; Yang, G.Q.; Hu, R. Acetylene-based short-cut from oxime of 2,2,6,6-tetramethylpiperidine-4-one to 4,4,6,6-tetramethyl-4,5,6,7-tetrahydropyrrolo[3,2-c]pyridine-pyrazole ensembles. Mendeleev Commun. 2019, 29, 658–660. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Markova, M.V.; Tomilin, D.N.; Tret’yakov, E.V.; Ovcharenko, V.I.; Mikhaleva, A.I.; Trofimov, B.A. First example of the synthesis of pyrrolecarbaldehyde with electron-deficient acetylene substituents. Russ. J. Org. Chem. 2013, 49, 1241–1243. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Sobenina, L.N.; Markova, M.V.; Gotsko, M.D.; Ushakov, I.A.; Smirnov, V.I.; Vaschenko, A.V.; Mikhaleva, A.I.; Trofimov, B.A. Expediant strategy for the synthesis of 5-acylethynylpyrrole-2-carbaldehydes. Synth. Commun. 2015, 45, 1652–1661. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Soshnikov, D.Y.; Trofimov, A.B.; Gotsko, M.D.; Sobenina, L.N.; Ushakov, I.A.; Afonin, A.V.; Koldobsky, A.B.; Vitkovskaya, N.M.; Trofimov, B.A. A peculiarity in the Al2O3-mediated cross-coupling of pyrroles with bromotrifluoroacetylacetylene: A quantum-chemical insight. Mendeleev Commun. 2016, 26, 480–482. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Gotsko, M.D.; Sobenina, L.N.; Ushakov, I.A.; Afonin, A.V.; Soshnikov, D.Y.; Trofimov, A.B.; Koldobsky, A.B.; Trofimov, B.A. N-Vinyl-2-(trifluoroacetylethynyl)pyrroles and E-2-(1-bromo-2-trifluoroacetylethenyl)pyrroles: Cross-coupling vs. addition during C-H-functionalization of pyrroles with bromotrifluoroacetylacetylene in solid Al2O3 medium. H-bonding control. J. Fluor. Chem. 2016, 186, 1–6. [Google Scholar] [CrossRef]
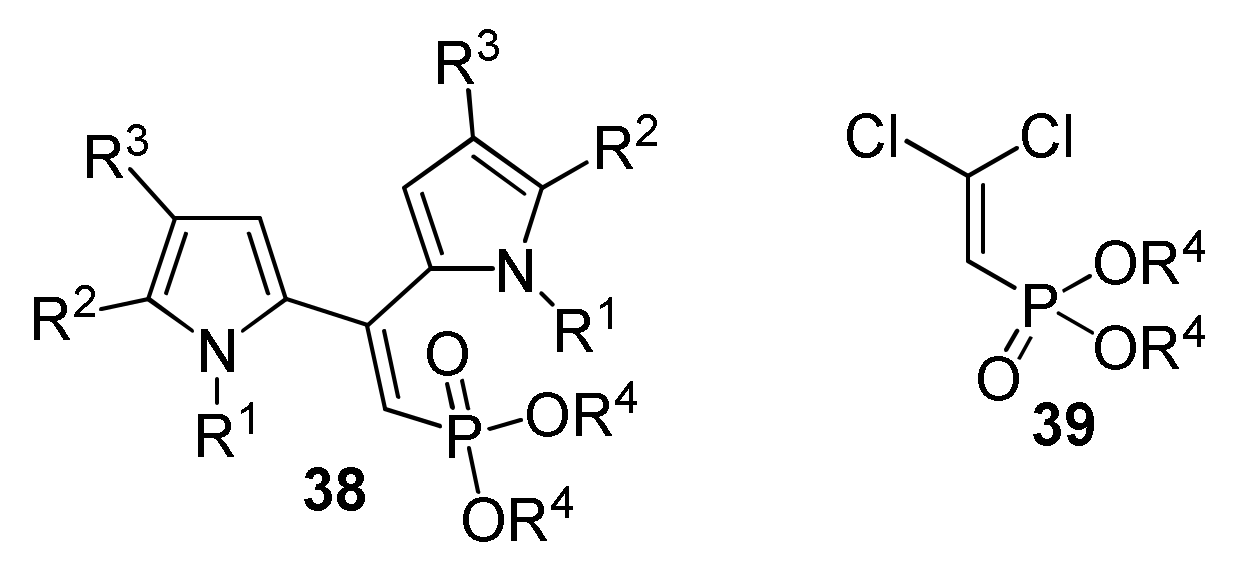
- Gotsko, M.D.; Sobenina, L.N.; Tomilin, D.N.; Ushakov, I.A.; Dogadina, A.V.; Trofimov, B.A. Topochemical mechanoactivated phosphonylethynylation of pyrroles with chloroethynylphosphonates in solid Al2O3 or K2CO3 media. Tetrahedron Lett. 2015, 57, 4961–4964. [Google Scholar] [CrossRef]
- Garibina, B.A.; Dogadina, A.V.; Zakharov, V.I.; Ionin, B.I.; Petrov, A.A. Phosphorus containing ynamines. Zh. Obshch. Khim. 1979, 71, 1964–1973. [Google Scholar]
- Petrov, A.A.; Dogadina, A.V.; Ionin, B.I.; Garibina, B.A.; Leonov, A.A. The Arbuzov rearrangement with participation of halogenoacetylenes as a method of synthesis of ethynylphosphonates and other organophosphorus compounds. Russ. Chem. Rev. 1983, 52, 1030–1035. [Google Scholar] [CrossRef]
- Dogadina, A.V.; Svintsitskaya, N.I. New reactions of chloroethynylphosphonates. Russ. J. Gen. Chem. 2015, 85, 351–358. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Pigulski, B.; Gulia, N.; Arendt, A.; Sobenina, L.N.; Mikhaleva, A.I.; Szafert, S.; Trofimov, B.A. Direct synthesis of butadiynyl-substituted pyrroles under solvent and transition metal-free conditions. RSC Adv. 2015, 5, 73241–73248. [Google Scholar] [CrossRef]
- Pigulski, B.; Arendt, A.; Tomilin, D.N.; Sobenina, L.N.; Trofimov, B.A.; Szafert, S. Transition-metal free mechanochemical approach to polyyne substituted pyrroles. J. Org. Chem. 2016, 81, 9188–9198. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Joon, H.L.; Kim, B. An improved synthesis of 1,4-bis(3,4-dimethyl-5-formyl-2-pyrryl)butadiyne and 1,2-bis(3,4-dimethyl-5-formyl-2-pyrryl)ethyne. J. Org. Chem. 1999, 64, 8048–8050. [Google Scholar] [CrossRef]
- McDonagh, A.F.; Lightner, D.A. Influence of conformation and intramolecular hydrogen bonding on the acyl glucuronidation and biliary excretion of acetylenic bis-dipyrrinones related to bilirubin. J. Med. Chem. 2007, 50, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Hua, R.; Jiang, J.; Zhang, L. A general approach to arylated furans, pyrroles, and thiophenes. Tetrahedron 2014, 70, 8252–8256. [Google Scholar] [CrossRef]
- Nizami, T.A.; Hua, R. Cycloaddition of 1,3-butadiynes: Efficient synthesis of carbo- and heterocycles. Molecules 2014, 19, 13788–13802. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Gotsko, M.D.; Ushakov, I.A.; Trofimov, B.A. Transition metal-free cross-coupling of furan ring with haloacetylenes. Tetrahedron 2018, 74, 1565–1570. [Google Scholar] [CrossRef]
- Gotsko, M.D.; Sobenina, L.N.; Vashchenko, A.V.; Trofimov, B.A. Synthesis of 2,2-(pyrazol-1-yl)enones via 2:1 coupling of pyrazoles and acylbromoacetylenes in solid alumina. Tetrahedron Lett. 2018, 59, 4231–4235. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Sagitova, E.F.; Ushakov, I.A.; Trofimov, B.A. Transition-metal-free synthesis of pyrrolo[1,2-a]pyrazines via intramolecular cyclization of N-propargyl(pyrrolyl)enaminones. Synthesis 2017, 49, 4065–4081. [Google Scholar] [CrossRef] [Green Version]
- Sagitova, E.F.; Sobenina, L.N.; Trofimov, B.A. From acylethynylpyrroles to pyrrolo[1,2-a]pyrazines in one step. Russ. J. Org. Chem. 2020, 56, 225–233. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Sagitova, E.F.; Markova, M.V.; Ushakov, I.A.; Ivanov, A.V.; Trofimov, B.A. Acylethynylpyrroles as a platform for the one-pot access to 2-(pyrrol-2-yl)-3-acylpyridines via the dihydrogenative annelation with propargylamine. Tetrahedron Lett. 2018, 59, 4047–4949. [Google Scholar] [CrossRef]
- Sagitova, E.F.; Sobenina, L.N.; Tomilin, D.N.; Markova, M.V.; Ushakov, I.A.; Trofimov, B.A. Formation of 2-(3-acyl-5-halopyridin-2-yl)pyrroles during annulation/aromatization of NH-2-acylethynylpyrroles with propargylamine in the presence of copper(I) halide. Mendeleev Commun. 2019, 29, 252–253. [Google Scholar] [CrossRef]
- Sobenina, L.N.; Tomilin, D.N.; Sagitova, E.F.; Ushakov, I.A.; Trofimov, B.A. Metal-free, atom- and step-economic synthesis of aminoketopyrrolizines from benzylamine, acylethynylpyrroles and acylacetylenes. Org. Lett. 2017, 19, 1586–1589. [Google Scholar] [CrossRef] [PubMed]
- Vasilevsky, S.F.; Davydova, M.P.; Tomilin, D.N.; Sobenina, L.N.; Mamatuyk, V.I.; Pleshkova, N.V. Peculiarities of the cascade cleavage of the polarized C-C-fragment in α-ketoacetylenes on reaction with ethylene diamine. ARKIVOC 2014, 5, 132–144. [Google Scholar]
- Sobenina, L.N.; Tomilin, D.N.; Gotsko, M.D.; Ushakov, I.A.; Mikhaleva, A.I.; Trofimov, B.A. From 4,5,6,7-tetrahydroindoles to 3- or 5-(4,5,6,7-tetrahydroindol-2-yl)isoxazoles in two steps: A regioselective switch between 3- and 5-isomers. Tetrahedron 2014, 70, 5168–5174. [Google Scholar] [CrossRef]
- Tomilin, D.N.; Sobenina, L.N.; Petrushenko, K.B.; Ushakov, I.A.; Trofimov, B.A. Design of novel meso-CF3-BODIPY dyes with isoxazole substituents. Dyes Pigments 2018, 152, 14–18. [Google Scholar] [CrossRef]
- Gotsko, M.D.; Saliy, I.V.; Sobenina, L.N.; Ushakov, I.A.; Trofimov, B.A. From acylethynylpyrroles to pyrrole-pyrone ensembles in a one step. Tetrahedron Lett. 2019, 60, 151126. [Google Scholar] [CrossRef]
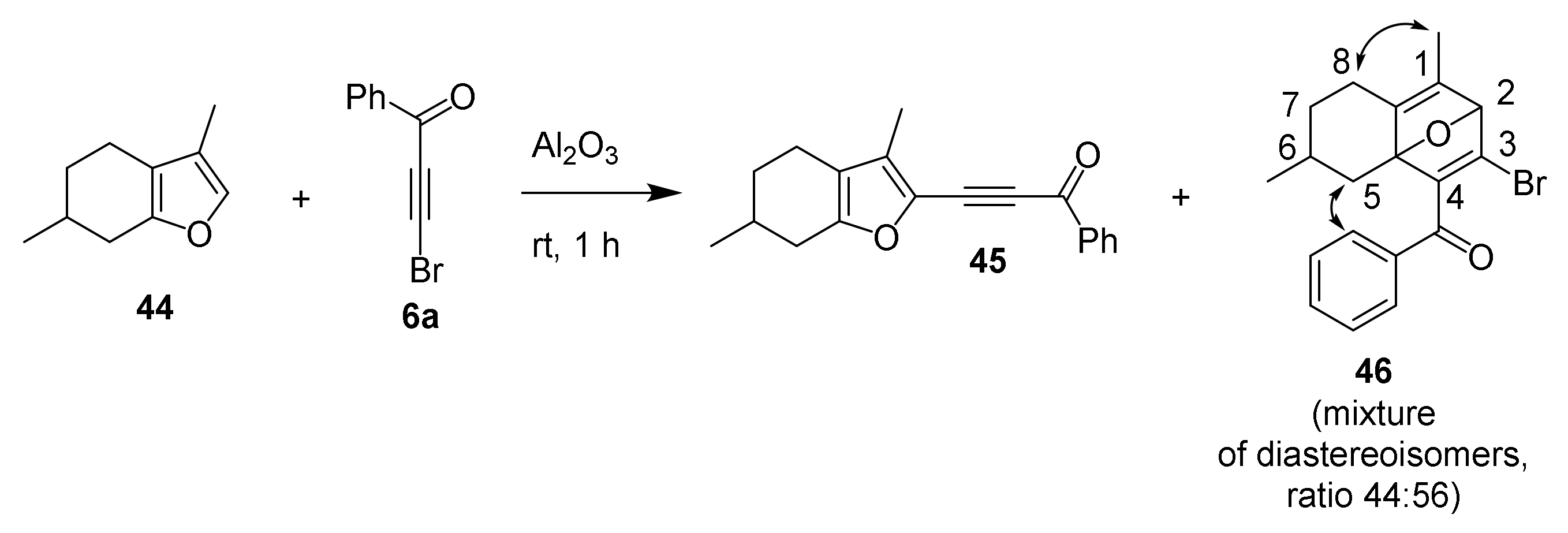
- Tomilin, D.N.; Gotsko, M.D.; Sobenina, L.N.; Ushakov, I.A.; Trofimov, B.A. A hydrogen pump: Transfer of four hydrogens from a cyclohexane ring to a triple bond during a menthofuran/bromoacetylene adduct rearrangement. Tetrahedron Lett. 2019, 60, 1864–1867. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |


















































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobenina, L.N.; Trofimov, B.A. Recent Strides in the Transition Metal-Free Cross-Coupling of Haloacetylenes with Electron-Rich Heterocycles in Solid Media. Molecules 2020, 25, 2490. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112490
Sobenina LN, Trofimov BA. Recent Strides in the Transition Metal-Free Cross-Coupling of Haloacetylenes with Electron-Rich Heterocycles in Solid Media. Molecules. 2020; 25(11):2490. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112490
Chicago/Turabian StyleSobenina, Lyubov’ N., and Boris A. Trofimov. 2020. "Recent Strides in the Transition Metal-Free Cross-Coupling of Haloacetylenes with Electron-Rich Heterocycles in Solid Media" Molecules 25, no. 11: 2490. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112490