
Development, Optimization, and Comparison of Different Sample Pre-Treatments for Simultaneous Determination of Vitamin E and Vitamin K in Vegetables

 , ,
, ,  ,
,  and
and 
Abstract
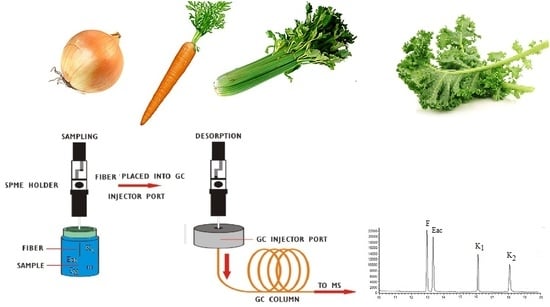
:
1. Introduction
2. Results
2.1. Vegetable Samples
2.2. Programmed Temperature Vaporization (PTV) and GC Parameter Optimization Using Direct Injection
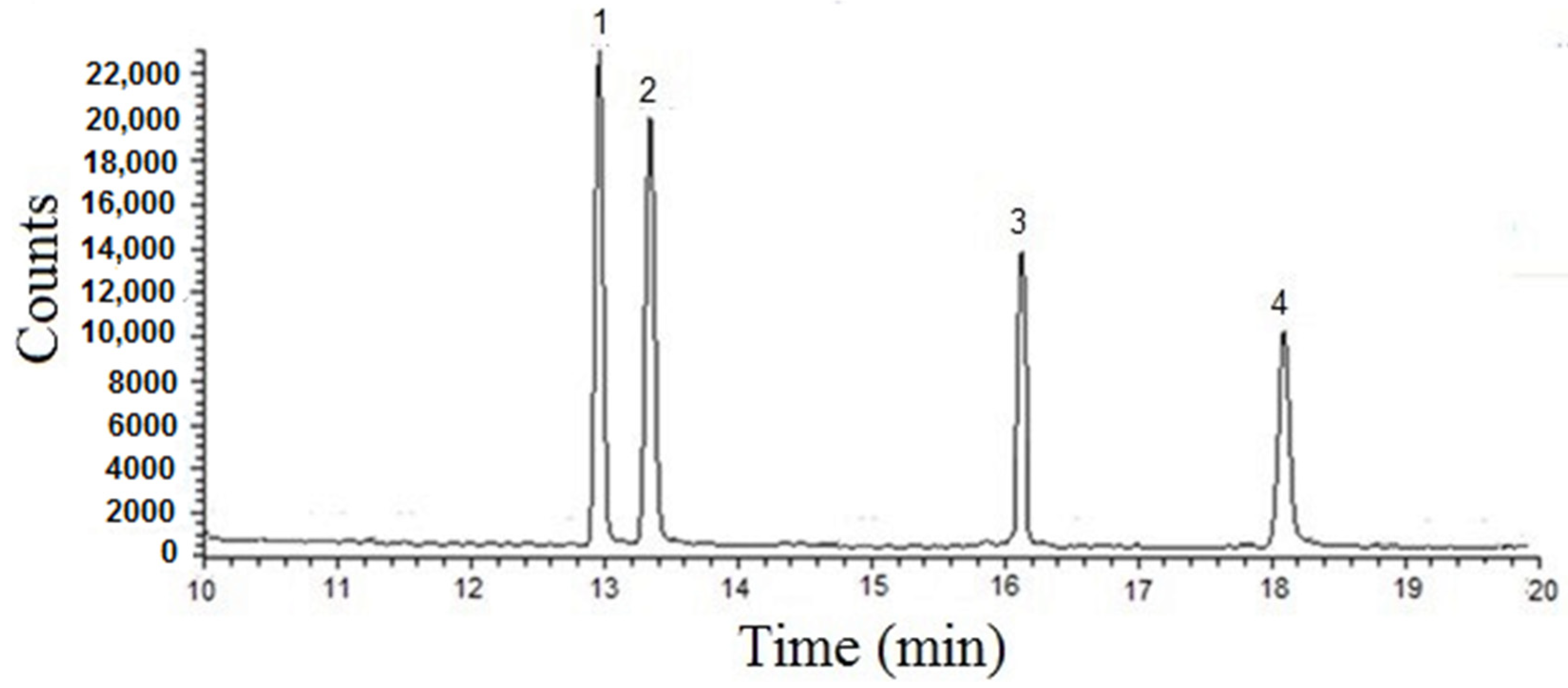
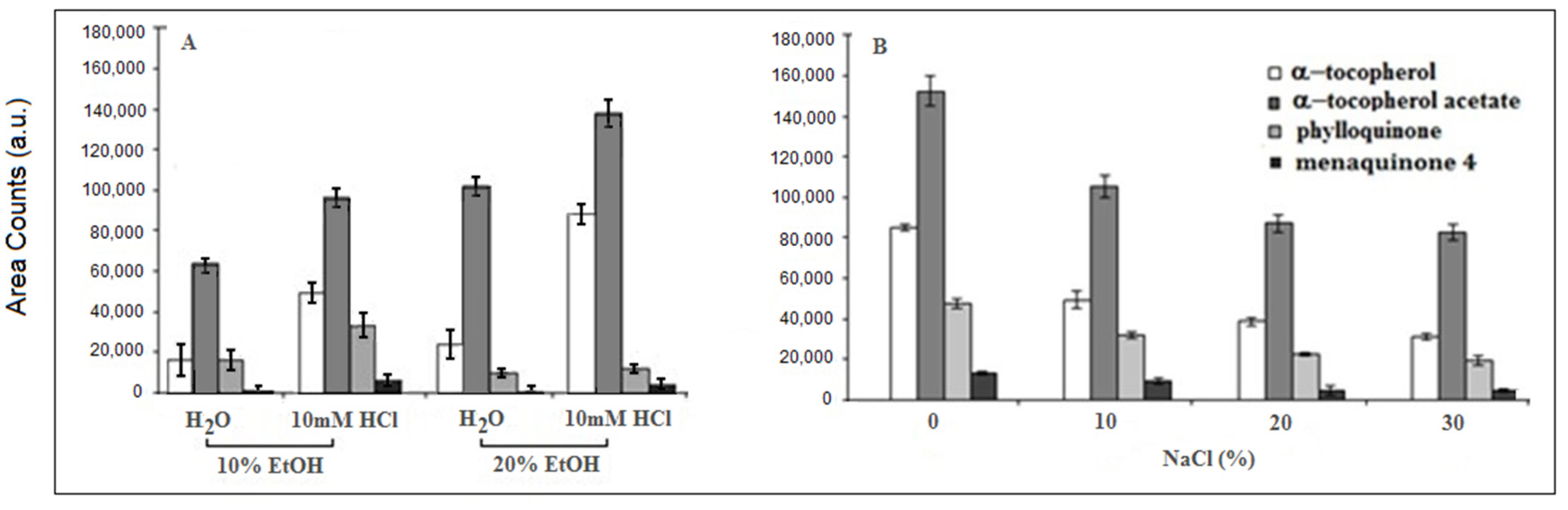
2.3. SPME Procedure Optimization
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Sample Pre-Treatment
4.3. Apparatus
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Krutz, L.J.; Senseman, S.A.; Sciumbato, A. Solid-phase microextraction for herbicide determination in environmental samples. J. Chromatogr. A 2003, 999, 103–121. [Google Scholar] [CrossRef]
- Godage, N.H.; Gionfriddo, E. A critical outlook on recent developments and applications of matrix compatible coatings for solid phase microextraction. TrAC Trends Anal. Chem. 2019, 111, 220–228. [Google Scholar] [CrossRef]
- Zingg, J.-M. Vitamin E: An overview of major research directions. Mol. Asp. Med. 2007, 28, 400–422. [Google Scholar] [CrossRef]
- Zingg, J.-M.; Meydani, M.; Azzi, A. α-Tocopheryl phosphate-An activated form of vitamin E important for angiogenesis and vasculogenesis? BioFactors 2012, 38, 24–33. [Google Scholar] [CrossRef]
- Ferland, G. Vitamin K and the Nervous System: An Overview of its Actions. Adv. Nutr. 2012, 3, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Commission. Regulation EC No. 1924/2006 on Nutrition and Health Claims Made on Foods. Off. J. Eur. Union 2012, L310. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02006R1924-20121129&from=EN (accessed on 4 April 2019).
- European Commission. Regulation EC No. 432/2012 Establishing a List of Permitted Health Claims Made on Foods, Other than those Referring to the Reduction of Disease Risk and to Children’s Development and Health. Off. J. Eur. Union 2012, L136. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32012R0432&from=EN (accessed on 11 February 2019).
- Roselli, L.; Clodoveo, M.L.; Corbo, F.; De Gennaro, B. Are health claims a useful tool to segment the category of extra-virgin olive oil? Threats and opportunities for the Italian olive oil supply chain. Trends Food Sci. Technol. 2017, 68, 176–181. [Google Scholar] [CrossRef]
- Harshman, L.C.; Drake, C.G.; Wargo, J.A.; Seliger, B.; Bhardwaj, N. Cancer Immunotherapy Highlights from the 2014 ASCO Meeting. Cancer Immunol. Res. 2014, 2, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramps, T.; Probst, J. Messenger RNA-based vaccines: Progress, challenges, applications. Wiley Interdiscip. Rev. RNA 2013, 4, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.W.; Traber, M.G. Methods for assessment of Vitamin E. In Laboratory Assessment of Vitamin Status; Elsevier BV: Oxford, UK, 2019; pp. 79–105. [Google Scholar]
- Beldean-Galea, M.S.; Horga, C.; Coman, M.V. Separation and determination of tocopherols in vegetable oils by solid phase extraction on porous polymers SPE cartridges and capillary gas chromatography analysis. Open Chem. 2010, 8, 1110–1116. [Google Scholar] [CrossRef]
- Grigoriadou, D.; Androulaki, A.; Psomiadou, E.; Tsimidou, M.Z. Solid phase extraction in the analysis of squalene and tocopherols in olive oil. Food Chem. 2007, 105, 675–680. [Google Scholar] [CrossRef]
- Sunarić, S.; Lalić, J.; Spasić, A. Simultaneous Determination of Alpha-Tocopherol and Alpha-Tocopheryl Acetate in Dairy Products, Plant Milks and Health Supplements by Using SPE and HPLC Method. Food Anal. Methods 2017, 10, 3886–3901. [Google Scholar] [CrossRef]
- Viñas, P.; Bravo-Bravo, M.; López-García, I.; Pastor-Belda, M.; Hernández-Córdoba, M. Pressurized liquid extraction and dispersive liquid–liquid microextraction for determination of tocopherols and tocotrienols in plant foods by liquid chromatography with fluorescence and atmospheric pressure chemical ionization-mass spectrometry detection. Talanta 2014, 119, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Aresta, A.; Di Grumo, F.; Zambonin, C. Determination of Major Isoflavones in Soy Drinks by Solid-Phase Micro Extraction Coupled to Liquid Chromatography. Food Anal. Methods 2015, 9, 925–933. [Google Scholar] [CrossRef]
- Aresta, A.; Zambonin, C. Determination of α-Tocopherol in Olive Oil by Solid-Phase Microextraction and Gas Chromatography—Mass Spectrometry. Anal. Lett. 2017, 50, 1580–1592. [Google Scholar] [CrossRef]
- Calvello, R.; Aresta, A.; Trapani, A.; Zambonin, C.; Cianciulli, A.; Salvatore, R.; Clodoveo, M.L.; Corbo, F.; Franchini, C.; Panaro, M.A. Bovine and soybean milk bioactive compounds: Effects on inflammatory response of human intestinal Caco-2 cells. Food Chem. 2016, 210, 276–285. [Google Scholar] [CrossRef]
- Salvo, A.; La Torre, G.L.; Rotondo, A.; Mangano, V.; Casale, K.E.; Pellizzeri, V.; Clodoveo, M.L.; Corbo, F.; Cicero, N.; Dugo, G. Determination of Squalene in Organic Extra Virgin Olive Oils (EVOOs) by UPLC/PDA Using a Single-Step SPE Sample Preparation. Food Anal. Methods 2016, 10, 1377–1385. [Google Scholar] [CrossRef]
- Sadrykia, F.; Shayanfar, A.; Valizadeh, H.; Nemati, M. A Fast and Simple Method for Determination of Vitamin E in Infant Formula by Dispersive Liquid-Liquid Microextraction Combined with HPLC-UV. Food Anal. Methods 2018, 12, 23–31. [Google Scholar] [CrossRef]
- Górska, R.M. Methods for assessment of Vitamin K. In Laboratory Assessment of Vitamin Status; Elsevier BV: Oxford, UK, 2019; pp. 107–147. [Google Scholar] [CrossRef]
- Huo, J.Z.; Nelis, H.J.; Lavens, P.; Sorgeloos, P.; De Leenheer, A.P. Simultaneous determination of α-tocopheryl acetate and tocopherols in aquatic organisms and fish feed. J. Chromatogr. B: Biomed. Sci. Appl. 1999, 724, 249–255. [Google Scholar] [CrossRef]
- Xiao, Z.; Lester, G.E.; Luo, Y.; Wang, Q. Assessment of Vitamin and Carotenoid Concentrations of Emerging Food Products: Edible Microgreens. J. Agric. Food Chem. 2012, 60, 7644–7651. [Google Scholar] [CrossRef] [PubMed]
- Viñas, P.; Bravo-Bravo, M.; López-García, I.; Hernández-Córdoba, M. Dispersive liquid–liquid microextraction for the determination of vitamins D and K in foods by liquid chromatography with diode-array and atmospheric pressure chemical ionization-mass spectrometry detection. Talanta 2013, 115, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Tarvainen, M.; Fabritius, M.; Yang, B. Determination of vitamin K composition of fermented food. Food Chem. 2019, 275, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Pokkanta, P.; Sookwong, P.; Tanang, M.; Setchaiyan, S.; Boontakham, P.; Mahatheeranont, S. Simultaneous determination of tocols, γ-oryzanols, phytosterols, squalene, cholecalciferol and phylloquinone in rice bran and vegetable oil samples. Food Chem. 2019, 271, 630–638. [Google Scholar] [CrossRef]
- Reto, M.; Figueira, M.E.; Mota-Filipe, H.; Almeida, C.M.M. Analysis of vitamin K in green tea leafs and infusions by SPME–GC-FID. Food Chem. 2007, 100, 405–411. [Google Scholar] [CrossRef]
- Dubbs, M.D.; Gupta, R.B. Solubility of Vitamin E (α-Tocopherol) and Vitamin K3(Menadione) in Ethanol−Water Mixture. J. Chem. Eng. Data 1998, 43, 590–591. [Google Scholar] [CrossRef]
- Aresta, A.; Calvano, C.D.; Trapani, A.; Zambonin, C.; De Giglio, E. α-Tocopherol/chitosan-based nanoparticles: Characterization and preliminary investigations for emulsion systems application. J. Nanoparticle Res. 2014, 16, 2230. [Google Scholar] [CrossRef]
- Zambonin, C.; Quinto, M.; De Vietro, N.; Palmisano, F. Solid-phase microextraction—Gas chromatography mass spectrometry: A fast and simple screening method for the assessment of organophosphorus pesticides residues in wine and fruit juices. Food Chem. 2004, 86, 269–274. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
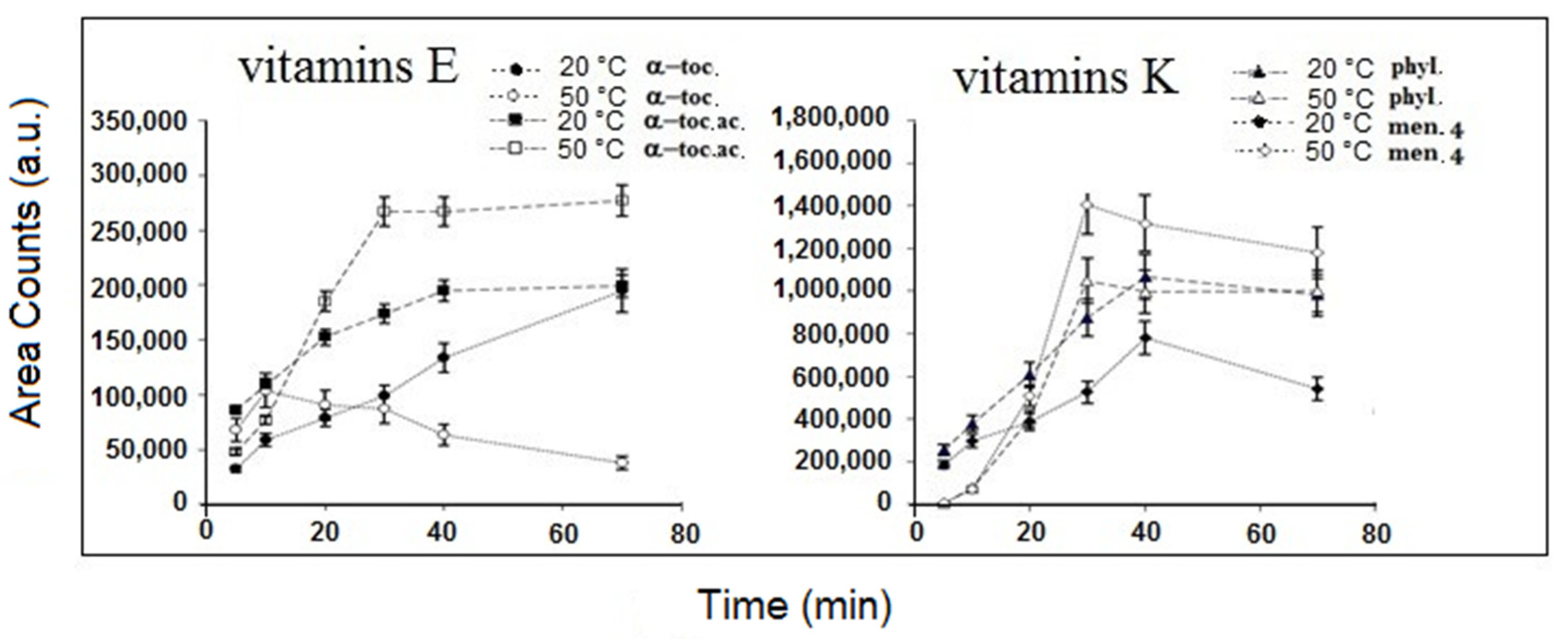{kind=link}
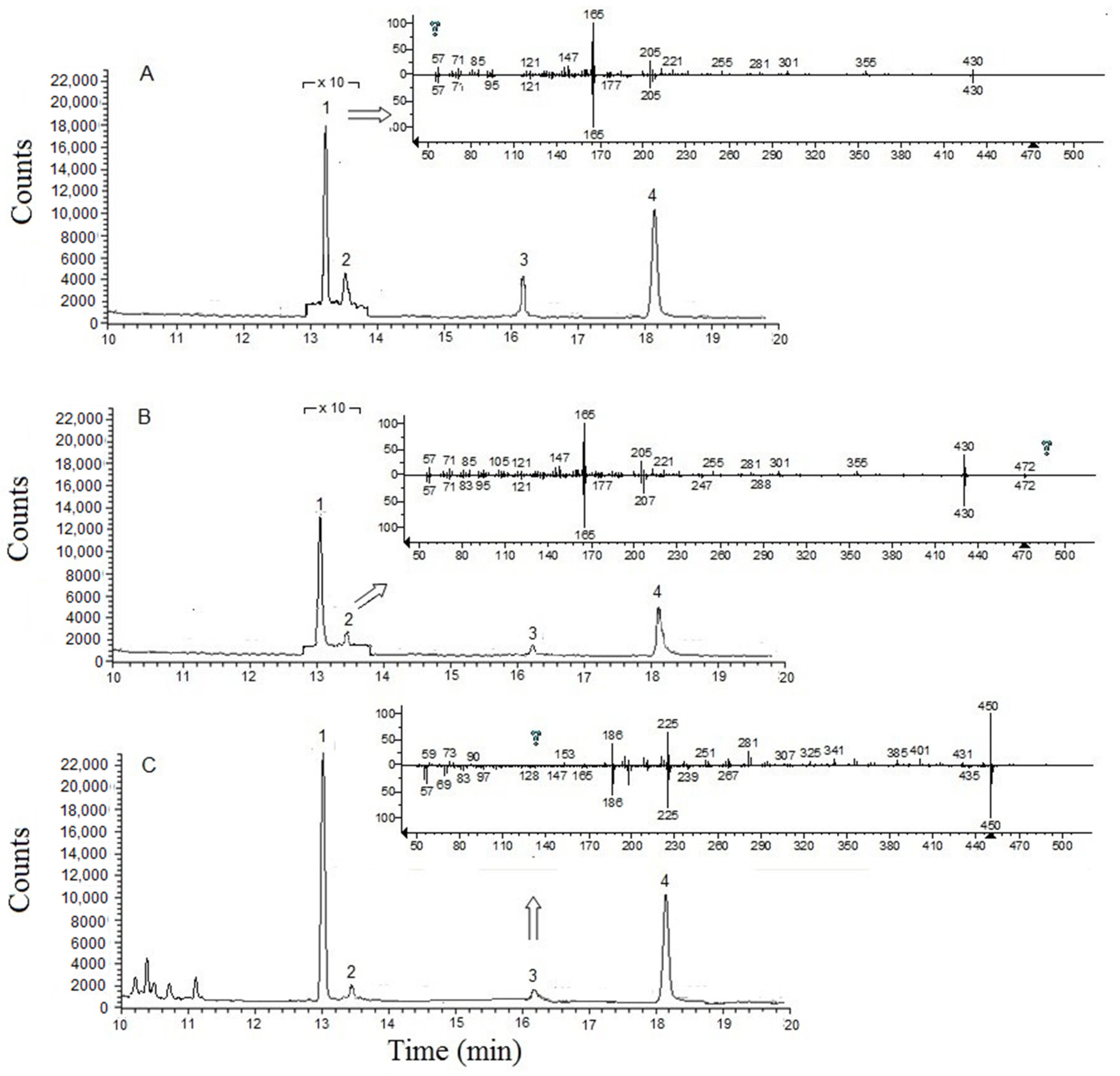{kind=link}
| Analyte | Linearity Range (µg mL−1) | R2 | LOD (µg mL−1) | LOQ (µg mL−1) | Within-Day (RSD%, n = 3) | Between-Days (RSD%, n = 15) |
|---|---|---|---|---|---|---|
| α-tocopherol | 0.4–100 | 0.999 | 0.1 | 0.4 | 2.8 | 3.4 |
| α-tocopherol acetate | 0.4–100 | 0.999 | 0.1 | 0.4 | 2.9 | 3.6 |
| Phylloquinone | 0.7–100 | 0.999 | 0.2 | 0.7 | 3.9 | 4.4 |
| Menaquinone | 1.9–100 | 0.999 | 0.6 | 1.9 | 4.0 | 5.9 |
| Analyte | Linearity Range (µg mL−1) | R2 | LOD (µg mL−1) | LOQ (µg mL−1) | Within-Day (RSD%, n = 3) | Between-Days (RSD%, n = 15) |
|---|---|---|---|---|---|---|
| α-tocopherol | 0.004–1.0 | 0.9999 | 0.001 | 0.004 | 4.3 | 8.8 |
| α-tocopheryl acetate | 0.006–1.0 | 0.9999 | 0.002 | 0.006 | 4.8 | 7.5 |
| phylloquinone | 0.036–5.0 | 0.9999 | 0.011 | 0.036 | 4.7 | 8.0 |
| menaquinone | 0.038–5.0 | 0.9990 | 0.011 | 0.038 | 4.6 | 6.9 |
| Method | A | B | ||||
|---|---|---|---|---|---|---|
| [Vitamin] | [5] (µg mL−1) | [50] (µg mL−1) | [100] (µg mL−1) | [5] (µg mL−1) | [50] (µg mL−1) | [100] (µg mL−1) |
| α-tocopherol | 59 ± 16% | 61 ± 18% | 63 ± 7% | 84 ± 15% | 93 ± 11% | 85 ± 6% |
| α-tocopheryl acetate | 55 ± 7% | 58 ± 10% | 60 ± 6% | 83 ± 18% | 77 ± 6% | 80 ± 7% |
| phylloquinone | 64 ± 15% | 66 ± 6% | 60 ± 12% | 86 ± 12% | 85 ± 13% | 81 ± 9% |
| menaquinone | 65 ± 24% | 66 ± 10% | 66 ± 23% | 93 ± 21% | 88 ± 18% | 95 ± 10% |
| Vegetable | Analyte Concentration (µg g−1) | ||||||
|---|---|---|---|---|---|---|---|
| [α-tocopherol] | [α-tocopheryl acetate] | [phylloquinone] | |||||
| carrot | fresh | dry | fresh | dry | Fresh | dry | |
| Pre-treatment | A | 14.5 ± 1.4 | 32.0 ± 1.1 | 2.4 ± 0.4 | 6.0 ± 0.4 | 2.6 | 8.1 ± 0.1 |
| B | 15.0 ± 2.5 | 30.6 ± 9.1 | nd | 5.9 ± 0.6 | nd | 7.8 ± 0.1 | |
| C | 19.3 ± 1.1 | 47.9 ± 3.8 | nd | 6.8 ± 0.5 | nd | 8.0 ± 0.1 | |
| celery | fresh | dry | fresh | dry | Fresh | dry | |
| Pre-treatment | A | 18.9 ± 0.9 | 32.8 ± 9.4 | 2.8 ± 0.4 | 4.7 ± 0.2 | 3.2 ± 0.2 | 8.0 ± 0.6 |
| B | 22.5 ± 1.1 | 46.1 ± 8.2 | 2.0 ± 0.6 | 4.9 ± 0.3 | 3.4 ± 0.1 | 8.6 ± 0.5 | |
| C | 20.0 ± 0.9 | 50 ± 1.2 | 2.0 ± 0.1 | 4.0 ± 0.3 | nd | 8.8 ± 0.7 | |
| curly kale | fresh | dry | fresh | dry | fresh | dry | |
| Pre-treatment | A | 17.9 ± 1.0 | 39.2 ± 2.3 | 1.9 ± 0.6 | 3.3 ± 0.3 | 6.9 ± 0.6 | 13.9 ± 0.8 |
| B | 18.9 ± 1.5 | 43.6 ± 7.6 | 2.0 ± 0.5 | 3.8 ± 1.3 | 8.1 ± 0.5 | 13.5 ± 0.6 | |
| C | 20.6 ± 1.2 | 35.2 ± 2.2 | 2.0 ± 0.5 | 4.1 ± 0.8 | 9.3 ± 0.8 | 14.6 ± 1.1 | |
| onion | fresh | dry | fresh | dry | fresh | dry | |
| Pre-treatment | A | 4.9 ± 1.1 | 10.4 ± 2.1 | 2.2 ± 0.6 | 4.1 ± 0.9 | 3.0 ± 0.9 | 7.9 ± 0.8 |
| B | 4.1 ± 0.9 | 11.5 ± 2.0 | 2.0 ± 0.1 | 4.7 ± 0.5 | 3.4 ± 0.9 | 8.5 ± 0.9 | |
| C | 3.3 ± 0.4 | 10.9 ± 1.1 | 2.0 ± 0.9 | 4.3 ± 0.9 | nd | 9.0 ± 1.1 | |
| Number Samples | Vegetable | Concentration Range (µg g−1) | |||||
|---|---|---|---|---|---|---|---|
| [α-tocopherol] | [α-tocopheryl acetate] | [phylloquinone] | |||||
| 10 | carrot | fresh | dry | fresh | dry | fresh | dry |
| Pre-treatment | A | 6.1–44.3 | 13.5–98.0 | nd−20.2 | nd−50.5 | nd−2.6 | nd−8.1 |
| B | 6.3–47.5 | 13.0–97.5 | nd−19.8 | nd−49.6 | nd−3.6 | nd−9.0 | |
| C | 5.6–39.8 | 13.9–98.8 | nd−19.9 | nd−52.2 | nd−3.2 | nd−8.0 | |
| 4 | celery | fresh | dry | fresh | dry | fresh | dry |
| Pre-treatment | A | 18.0–20.9 | 32.0 ± 44.4 | nd−2.5 | nd−4.9 | nd−3.6 | nd−9.1 |
| B | 17.8–22.5 | 31.9–43.8 | nd−2.7 | nd−4.8 | nd−3.6 | nd−9.0 | |
| C | 18.1–21.8 | 32.1–45.0 | nd−2.5 | nd−5.0 | nd−3.9 | nd−9.8 | |
| 6 | curly kale | fresh | dry | fresh | dry | fresh | dry |
| Pre-treatment | A | 8.6–20.5 | 18.0–43.1 | nd−13.0 | nd−22.1 | nd−7.6 | nd−15.7 |
| B | 8.9–19.8 | 18.3–42.2 | 2.0 ± 0.5 | nd−21.9 | nd−7.5 | nd−14.6 | |
| C | 8.2–20.6 | 19.5–43.6 | 2.0 ± 0.5 | 4.1 ± 0.8 | nd−9.3 | nd−16.0 | |
| 14 | onion | fresh | dry | fresh | dry | fresh | dry |
| Pre-treatment | A | 2.3–6.0 | 5.1–14.6 | nd−8.1 | nd−19.9 | nd−3.5 | nd−8.7 |
| B | 2.0–5.6 | 5.0–14.1 | nd−8.0 | nd−20.3 | nd−3.6 | nd−9.0 | |
| C | 2.1–6.3 | 4.9–14.9 | nd−7.9 | nd−19.7 | nd−3.5 | nd−9.5 | |
| Injection Phase | Pressure (KPa) | Rate (°C/sec) | Temperature (°C) | Time (min) |
|---|---|---|---|---|
| injection | 83 | 70 | 0.15 | |
| evaporation | 83 | 6.7 | 100 | 0.3 |
| transfer | 210 | 14.5 | 270 | 1 |
| cleaning | 14.5 | 300 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aresta, A.; Milani, G.; Clodoveo, M.L.; Franchini, C.; Cotugno, P.; Radojcic Redovnikovic, I.; Quinto, M.; Corbo, F.; Zambonin, C. Development, Optimization, and Comparison of Different Sample Pre-Treatments for Simultaneous Determination of Vitamin E and Vitamin K in Vegetables. Molecules 2020, 25, 2509. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112509
Aresta A, Milani G, Clodoveo ML, Franchini C, Cotugno P, Radojcic Redovnikovic I, Quinto M, Corbo F, Zambonin C. Development, Optimization, and Comparison of Different Sample Pre-Treatments for Simultaneous Determination of Vitamin E and Vitamin K in Vegetables. Molecules. 2020; 25(11):2509. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112509
Chicago/Turabian StyleAresta, Antonella, Gualtiero Milani, Maria Lisa Clodoveo, Carlo Franchini, Pietro Cotugno, Ivana Radojcic Redovnikovic, Maurizio Quinto, Filomena Corbo, and Carlo Zambonin. 2020. "Development, Optimization, and Comparison of Different Sample Pre-Treatments for Simultaneous Determination of Vitamin E and Vitamin K in Vegetables" Molecules 25, no. 11: 2509. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112509