2.2. Energy Profiles
Using the equilibrium geometry as reference, we changed, in regular steps and up to a distance of 4 Å, the intermolecular distance in each complex between the two atoms involved in the interaction (and connected to the intermolecular BCP). In addition, distances shorter than the equilibrium were considered too. The properties of the intermolecular BCP along the dissociation profile were analysed independently for the two N···F contacts and the five N···Cl contacts. In both sets, ρ
BCP and ∇
2ρ
BCP increase as the intermolecular distance decreases and follow an exponential relationship. The Laplacian values, obtained for N···Cl contacts close to 2.1 Å, show the location of the maximum of this property, and confirms previous reports [
50]. The total energy density, H
BCP, steadily increases as the N···F distance decreases, while in the N···Cl contacts, H
BCP increases up to around 2.7 Å but for shorter distances tends to become smaller and even negative for values smaller than 2.4 Å (see
Figure S2).
Level 1: Total Atomic Energies
Table S2 shows the complete REG analysis of the energy profile at the first level considered (i.e., total atomic energies) for all seven complexes.
Table 3 lists the most positive and most negative REG value in each complex for both segments. In principle, a REG analysis of only its positively-valued energy contributions may suffice to explain what drives the formation of a complex. However, in order to expand the REG analysis, a complementary explanation can be drawn from the most
negative REG values. Such an alternative explanation can sometimes be more intuitive. A second and very important criterion for deriving a good explanation from a REG analysis is the uniformity across all complexes. Indeed, the most powerful explanation is one that encompasses all complexes. Finally, we point out that an explanation is always linked to the direction of analysis: from left to right, or
vice versa. However, we note that the relationship between the profile of E
i and that of E
tot is actually intrinsic, that is, independent of the direction of the explanation. Sometimes it is more natural to explain a segment from right to left, sometimes from left to right. Throughout this work we will always analyse a segment from right to left. For example, the segment between the equilibrium distance and infinity (SEG2) is best thought of as the formation of a complex (rather than its destruction), because one usually wonders which type of force pulls the monomers together. Similarly, it also makes sense to see SEG1 as a compression energy profile, rather than one relaxing from a very short-range complex to the equilibrium one.
We first look for overall patterns emerging from
Table 3. One such pattern is that the atoms, one for each complex, associated with the most positive REG values of SEG1, are the same as those associated with the most negative REG values of SEG2. This finding means that the two segments mirror each other in the following sense. For each complex, the atom that most works
against the total energy profile (negative REG) in one segment, also works most
with the total energy (positive REG) in the other segment.
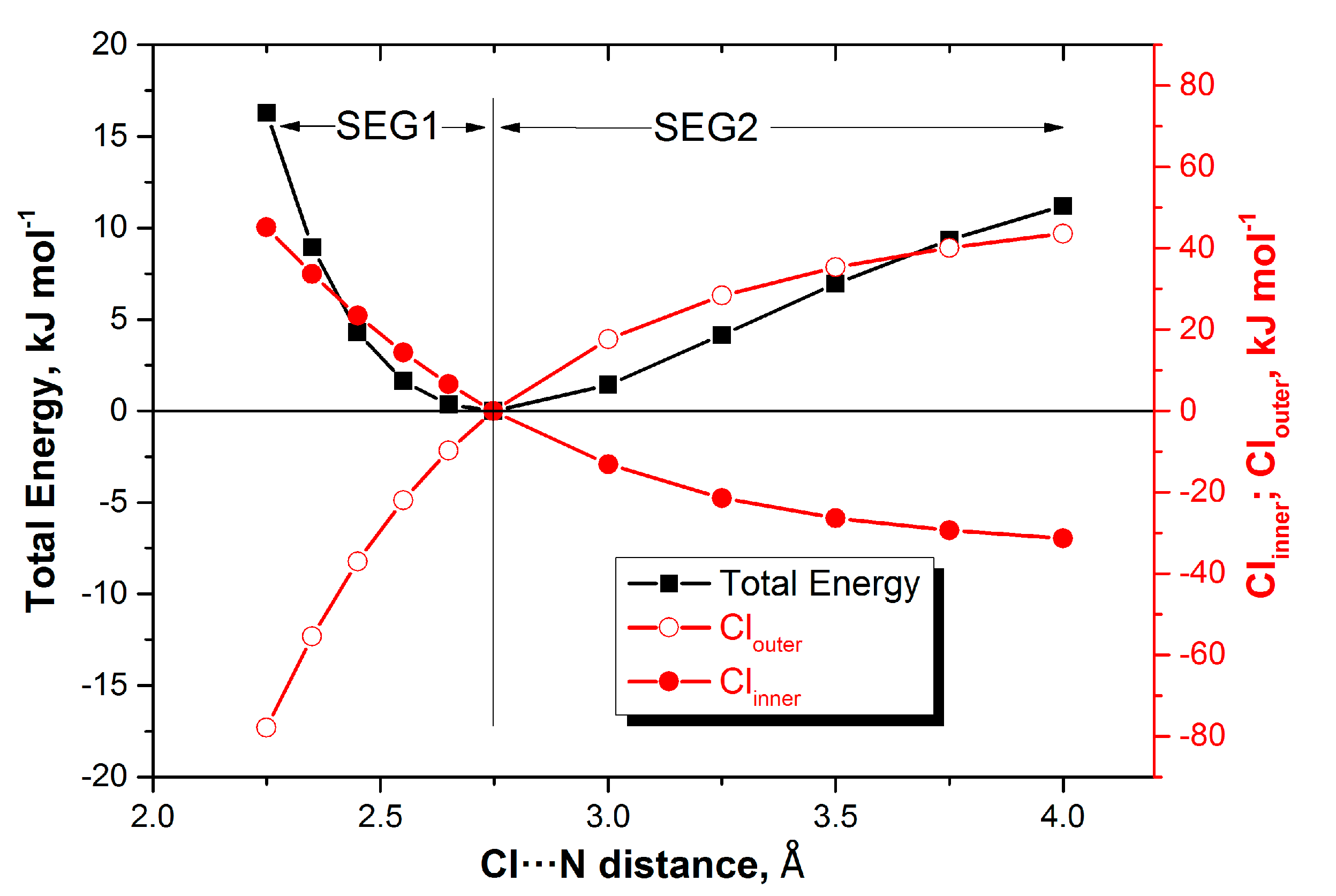
Figure 2 makes this behaviour clear for one representative complex (H
3N:Cl
2). Here, atom Cl
inner works most
with the total energy in SEG1, and very much
against the total energy in SEG2, so in each complex, there is a single atom that fulfills such an overall role over the whole energy profile. The same is true for Cl
outer but then vice versa.
Still, one could question the chemical and practical significance of finding such a single atom explaining the
whole energy profile, without segmentation. Indeed, there is another, more fruitful way of looking at
Figure 2, but then focusing only on the positive REG values.
Figure 2 shows that the coming together of H
3N and Cl
2, from infinity to equilibrium (SEG2), is governed by the total atomic stabilisation of the outer Cl. Indeed, this atom has the most positive REG values in SEG2 (see
Table 3). On the other hand, in SEG1, H
3N and Cl
2 are brought together further and compressed beyond the equilibrium geometry. This time, the total energy profile is ruled by the total atomic destabilisation of the other Cl, i.e., the inner Cl, directly involved in the halogen bond. So, which energy contribution, either in SEG1 or SEG2, that best explains the total energy profile is learnt from the most positive REG value, respectively.
We look again at
Table 3 and focus on the positive REG values only. When the X
2 complexes are formed (SEG2) then X
outer dominates. On the other hand, when complexes with F
2 and Cl
2 are compressed (SEG1) then the total atomic energy of X
inner dominates. These conclusions can also be drawn
Figure 2, which shows a X
2 complex. However, the three complexes with ClF reveal a more complex picture than for the four X
2 complexes. It is difficult to summarise the observations. However, the clearest common thread is the role of the “contact N” or inner nitrogen (i.e., N
3), whose stabilisation drives the formation of all three complexes (SEG2). The compression of the complexes (SEG1), up the repulsive barrier of the total energy profile, is predominately caused by non-contact atoms in the partner molecule of ClF. This finding can be conveniently depicted by the bold underlined atoms in
NN:ClF, H
CN:ClF and
H3N:ClF.
In summary, we have learnt that: (i) the formation of X2 complexes is driven by the stabilisation of the outer X, but (ii) their compression by the inner X, while (iii) the formation of ClF complexes is driven by the stabilisation of the contact N, and (iv) their compression by non-contact atoms in the partner molecule of ClF.
Level 2: Total Interatomic and Intra-Atomic Energies
The next levels of REG analysis operate on more resolved energy data and may help to shed light on the nature of the (de)stablisation of the total atomic energies.
Table S2 shows the REG analysis at Level 2 (total interatomic and intra-atomic energies). From
Table S2,
Table 4 now extracts the
two most important terms instead of only one, as in
Table 3. This relaxation serves the purpose of ideally recovering a single explanation across all complexes. In other words, allowing a second close competitor to take the role of the most important contribution increases the chance of generating a uniform interpretation across complexes. Since there are now many more energy contributions to consider than at Level 1, REG values should be forced to be higher than a certain threshold, which was set to 2.0. Few complexes did not meet this threshold causing the absence of a second entry in
Table 4.
We analyse the whole energy profile from right to left, starting with SEG2. The Xinner–Ninner interaction is the major driving force (i.e., positive REG) to complex formation. In other words, the two contact atoms at the heart of each complex exert the “clearest pull”. This observation is true for all seven complexes, so there is no need to distinguish the X2 complexes from the ClF complexes, as had to be done at Level 1. Moving further to the left we then study the compression segment (SEG1). This time the intra-atomic energy of the inner nitrogen (Ninner or N3) governs the total energy barrier. Chemically, this finding means that it is the steric “discomfort” of the contact N that explains the compression energy barrier. Note that in order to reach this conclusion for all seven complexes we had to invoke the second most important term for one complex.
Interpreting the positive REG values suffices to understand the chemical behaviour of the complexes but for completeness one may inspect the most negative REG values as well. The good news is again that one single type of energy contribution is in charge for all complexes without exception: the interatomic X
inner–N
inner energy. We have to wait for the more resolved analysis of Level 3 to know which type of energy is behind X
inner–N
inner but, whichever its nature, it vehemently works against the compression energy barrier.
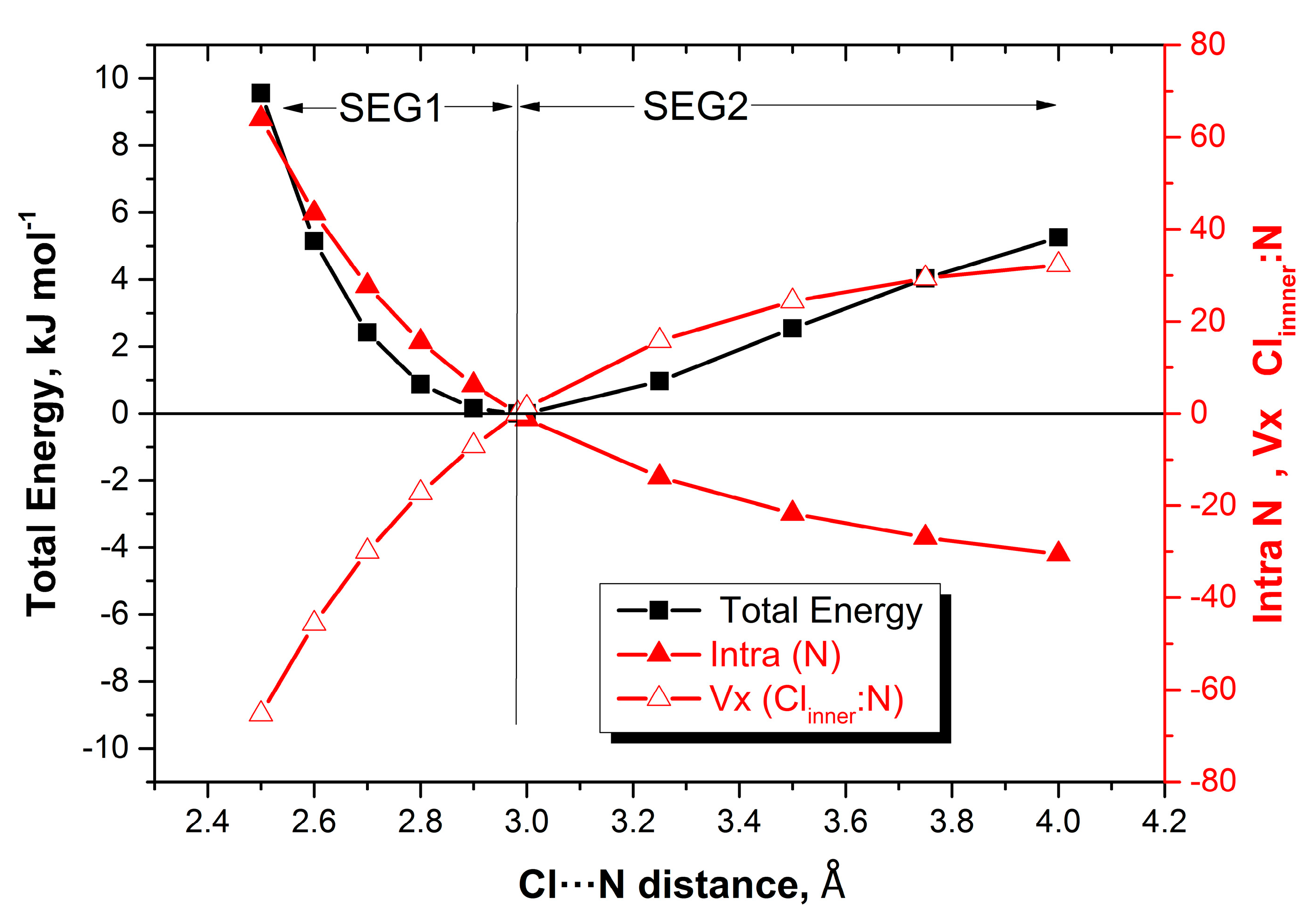
Figure 3 shows the evolution of the intra-atomic energy of N
inner and that of the X
inner–N
inner energy alongside the total energy profile of NH
3:ClF.
In summary, at Level 2 we have learnt that, for all seven complexes, (i) the interatomic Xinner–Ninner energy drives their formation (i.e., energy of contact atom…contact atom), and that (ii) their compression is dominated by the destabilisation of Ninner (i.e., contact N).
Before we move on the next level of REG analysis, Level 3, we briefly contrast Level 2 and Level 1. Level 2 firmly associates the driving forces for both formation and compression with the inner (contact) atoms, and does so for all seven complexes. In contrast, the total atomic energy analysis of Level 1, brings in non-contact atoms as drivers and differentiates two different types of complex (X2 and ClF). So, the explanation at Level 2 is more minimal than that at Level 1. In short, Level 2 is thus more powerful, and therefore preferred. We deduce that a too coarse REG analysis (Level 1) spoils the uniformity of interpretation and thus its power.
Level 3: Total Intra-Atomic (Eintra), Electrostatic (Vcl), Exchange (Vx) and Correlation (Vc) Energies.
It remains to be seen if the even more resolved Level 3 analysis can repeat the uniformity of Level 2 but now also reveal the type of interaction energy.
Table S4 gathers all data in connection with Level 3, which is the most resolved (i.e., the least coarse) REG analysis that we apply here. Level 3 takes into account four possible types of energy:
, the classical electrostatic energy
, the exchange energy
, and the correlation energy
. Similarly to Level 2,
Table 5 skims from
Table S4 the
two most important energy terms, by absolute REG value.
Again we analyse the whole energy profile from right to left, starting with SEG2, and focus on the most positive values. We find that the covalent energy contribution V
x (X
inner–N
inner) is dominant. This is true for all seven complexes although for HCN:ClF the third most positive REG value had to be invoked, as the two energy terms with higher REG values were electrostatic in nature. Note that there is a sudden drop (
Table S4) in REG value after the third term, which we allowed to slip in order to make the interpretation uniform throughout all complexes. How can we contextualise the conclusion that the predominant interaction forming a halogen-bonded complex is covalent in nature?
It is instructive to briefly look at hydrogen bonding first. Our first ever REG study illustrated this newly proposed method
37 on the water dimer, in which two monomers form the global energy minimum. In this classic hydrogen-bonded system, REG shows that the formation of the dimer is dominated by the electrostatic energy between the hydrogen-bond hydrogen atom and the acceptor oxygen. All subsequent terms are also electrostatic in nature, thereby confirming the well-known Buckingham-Fowler model [
51]. However, the sixth term in that sequence is of the type exchange-correlation (as the study was carried out using DFT). The exchange energy typically dominates the correlation energy in this combined energy term, which means that there is some covalent character in a hydrogen bond.
Now, Wolters and Bickelhaupt compared [
52] hydrogen-bonded systems with halogen-bonded ones from a molecular orbital perspective. From their extensive analysis on DX…A
− and on DH…A
− (D, X, A = F, Cl, Br, I) systems they concluded that halogen bonds and hydrogen bonds have a very similar bonding mechanism consisting of both electrostatic and covalent contributions. However, they also stated that the electrostatic attraction is less favourable in the halogen bonds, adding that halogen bonds can become stronger than hydrogen bonds because of a more stabilising covalent component in the former. In summary, their conclusions agree with our own. To strengthen this main point, we mention that Li et al. concluded [
53] from their combined QTAIM/NBO study on complexes with formaldehyde and hypohalous acids that the energy decomposition analyses indicate that the contribution from the electrostatic interaction energy is larger in the hydrogen-bonded complexes than that in the halogen-bonded complexes. Next, in the rather vague and future-work-directed conclusions of their review of computer modelling of halogen bonds, Kolář and Hobza unfortunately do not offer the insight that we need to support our own findings or not. Still, on the basis of S66 and X40 benchmark data sets, they mention a difference between hydrogen-bonded and halogen-bonded systems as decided by the SAPT scheme. Whereas the electrostatic energy is the most attractive contribution to hydrogen-bonded systems, they state, the situation is different for halogen-bonded systems. For the latter, they quote a medley of energy types being responsible, short of declaring exchange though. In short, the clarity that REG offers is not matched with any statements found in this review.
The next discussion point is how the dominance of covalent energy is compatible with the σ-hole model, which is of course electrostatic in nature. Let us take the example of H
3N:ClF to understand how REG operates.
Figure S3 directly contrasts V
x (X
inner–N
inner) and V
cl (X
inner–N
inner), first for the full complex formation energy (SEG2, eight distances) and then for the long-range part of that segment (four longest distances). For the full segment it is clear that the electrostatic linear fit is better (R
2 = 0.99) than that of the exchange fit (R
2 = 0.88). However, the REG value (i.e., slope) of the exchange interaction is higher (5.33) than that of the electrostatic interaction (4.47). So, in spite of the worse fit, the exchange is singled out as the most pronounced interaction aiding the total energy profile. However, the situation flips for the long-range where the REG value (4.44) of the electrostatic interaction now dominates that of the exchange (2.08), with both fits being excellent (R
2 ~ 0.99). This REG-based conclusion is compatible with the σ-hole interpretation of electrostatics being in charge at long-range.
However, it is clear that a complex closer to its equilibrium geometry is not governed by electrostatics. In fact, a fit of the 4 shortest distances returns a REG value of 9.41 (R2 = 0.91) for exchange and 5.34 (R2 = 0.95) for electrostatics. Indeed, at short-range the Xinner–Ninner interaction becomes overwhelmingly covalent in nature, as the exchange REG value is then almost twice that over the whole SEG2 interval, and almost five times that at long-range. Tognetti and Joubert mention2 the “dual character” of the halogen bond, classical electrostatics being comparable to covalency. For their Cl3COX…NH3 complexes (X = F, Cl, and Br) they used IQA (but not REG) and QTAIM to show that electrostatics generally predominates at long-range, and that exchange becomes an important contribution around the equilibrium position.
Finally, we contrast the REG findings, which are based on global properties (i.e., obtained by integration over atomic volumes) with local properties (i.e., evaluated at a point, e.g., ρ
BCP, ∇
2ρ
BCP and H
BCP listed in
Table 2). The local properties suggest that halogen bonds are non-covalent but the REG analysis suggests that their behaviour at short-range (stretching the complex beyond the equilibrium by a modest amount) is covalent. There is a case to make (also done by others) that global properties are more trustworthy than local ones because they carry more information (given the volume integration, which covers a multitude of points). Otherwise, one may question to what extent the BCP properties describe the same entity as the REG-IQA data. The former is a quick characterisation of the nature of a bond in the minimum energy geometry only. This is a static picture that contrasts with the dynamic nature of REG, on top of the global nature of IQA. The REG-IQA data may point at an energy term that poorly describes, drives or explains a whole energy segment of complex formation, such as the electrostatic energy. But then, this term can have a significant value at the equilibrium geometry, and be linked to the non-covalency (i.e., “closed-shell interaction”) indicted by the local properties.
After this lengthy discussion of the nature of the SEG2 energy profile, we now look at the compression regime (SEG1). We attempt to find an explanation, common to all complexes, typically starting with the positive REG values. If we allow flipping between the most and second most important term, then again “Intra-n3” or the total intra-atomic energy of Ninner is the dominant term. This finding is compatible with the conclusion of Level 2.
Equally good news emerges when looking at the most negative REG values. They show a clear pattern for all seven complexes and highlight V
x (X
inner–N
inner) as the dominant energy. Thus, the covalent energy between the halogen atom and the Lewis base contact atom most opposes the compression. Level 2 already identified this interaction but could not determine its type. However, now we can, at Level 3. Indeed,
Figure 4 shows how this interaction emphatically lowers its energy upon compression of HCN:Cl
2 and therefore strongly opposes the compression energy barrier.
In summary, at Level 3 and for all seven complexes, we find that: (i) the exchange energy between Xinner and Ninner drives their formation, and that (ii) their compression is best described by the destabilisation of Ninner (i.e., contact N).
Level 4: Correlation/Dispersion Energies Only
We now move on to the final level of analysis. None of the correlation terms (V
c) plays a significant role in explaining the total energy profile because of their small absolute REG values in
Table S4. Still, given the attention that electron correlation received in the literature we can confine a REG analysis to correlation terms only. This is done in the fourth and final level of REG analysis, Level 4, which focuses only on electron correlation and thus dispersion. It is true that the correlation energy contribution seems to be small or relatively meaningless, compared to the much larger REG values seen for V
cl or V
x. Still, one should remember that the binding energies of the halogen complexes studied here (and in general) are relatively small, ranging from 5.5 to 39.6 kJ mol
−1, compared to those of covalent bonds. This means that if one fails to take into account the electron correlation contribution, the halogen bond could potentially cease to exist altogether, especially for those compounds at the weaker end of the energy spectrum. We now turn our sights to the correlation/dispersion part of the halogen bond invoking a final REG analysis.
Table 6 gives the REG analysis of the correlation (V
c) terms only, both intra- and interatomic. As always, we discuss the complex formation regime (SEG2) first. The most important positive REG contribution is V
c (X
inner–N
inner), i.e., the interaction between the two (contact) atoms involved in the halogen bond. This result is “clean” in the sense that there was no need to involve the second most important REG contribution, and V
c (X
inner–N
inner) showed up for all seven complexes. So, the correlation contribution mirrors that of the exchange, V
x (X
inner–N
inner), as shown in Level 3. Of course the exchange energy overwhelms the correlation one but if the latter is excluded (as it is in
Table 6) then correlation would be responsible for forming the complex.
For sake of completeness
Table 6 also lists the most negative REG values where, curiously, V
c (X
outer–N
inner) now dominates for all complexes (with the exception of N
2:ClF where the second most important contribution had to be invoked). This is a “through space” effect that is hard to interpret but it returns based on the most positive REG values for the compression segment (SEG1). In other words, if we focus on electron correlation only (and thus omit the intra-atomic interaction of N
inner) then the destabilisation of the correlation energy between X
outer and N
inner best explains the repulsive compression energy barrier. For completeness we add that V
c (X
inner–N
inner) most opposes the compression (with the only exception of H
3N:ClF). In other words, this contribution keeps becoming more stable as the complex is compressed.
A further remark concerns the importance of dispersion in halogen bonding. A study [
54] used DFT-SAPT on benzene…dihalogen and formaldehyde…compound halogen-bonded complexes, covering X…π and Cl…O contacts amongst others. It turned out that the dispersion energy is non-negligible and indeed of the same order of magnitude as the electrostatic energy. This statement should not be confused with our own findings and wrongly deemed to be contradictory. Earlier work
27 from our lab showed that the interatomic correlation energies of halogen bonds and hydrogen bonds are on a par, and of the order of 1 to 3 kJ mol
−1. This contribution is not negligible (see
Table 2) and similar to that found between the hydrogens in a single water molecule. Moreover, the intra-atomic correlation energy within a hydrogen in a single water is about 20 kJ mol
−1. The intra-atomic components are typically much larger than the interatomic ones, which is also true for the helium dimer. As a consequence, we showed
26 that the stability of this traditional van der Waals complex is best explained by (intra)atomic correlation energy lowering, rather than by a direct dispersion interaction between the helia. So, we are not claiming that the various electron correlation contributions in halogen-bonded systems are negligible. However, we do claim that the interatomic correlation energies cannot be used to explain what drives the formation or repulsive compression energy barrier of a complex, when these energies operate in the presence of much larger contributions such as exchange or steric energy (intra-atomic energy).
In summary, we learn from the exclusive electron correlation REG analysis at Level 4 that (i) the interatomic correlation energy between Xinner and Ninner drives the complex formation, and that (ii) complex compression is best described by the destabilisation of the through-space correlation energy between Xouter and Ninner.
Finally, we comment briefly on charge transfer, which is well-defined within the topological approach, which does not introduce any reference states and is parameter-free.
Figure S4 profiles for all seven complexes. The overall net charge of the halogen molecule is plotted versus the N…X distance, for the whole distance range (SEG1 and SEG2). It is convenient to express the charge transfer in milli-electron (m
e), a resolution well supported by the atomic integration error, which is typically two to three orders of magnitude smaller. It is clear that all halogen molecules (ClF, F
2, Cl
2) start off almost neutral (except in N
2:ClF where a very small positive charge built up, of less than 1 m
e). Subsequently, a negative net charge accumulates as each complex forms, increasing in magnitude to respectively −8, −22, −5, −9, −3, −18 and −11 m
e for HCN:F
2, H
3N:F
2, HCN:Cl
2, H
3N:Cl
2, N
2:ClF, HCN:ClF and H
3N:ClF. There is no correlation between the binding energy of the complex and its molecular charge transfer. Note that this charge transfer peaks (in absolute value) at the shortest complex distance, at values that can be about two to six times larger than at equilibrium.
The reason we mention charge transfer data here is because of the attention it received from researchers using non-topological energy decomposition analyses. For example, Thirman et al. argued [
11] that a picture of the halogen bond that excludes charge transfer cannot be complete. Based on their study on systems of the type CX
3I…Y
− (for X = F, Cl, Br, I and Y = F, Cl, Br) they went on to state that permanent and induced electrostatics do not always provide the dominant stabilising contributions to halogen bonds. Their work was triggered by an earlier study [
12] on these systems that showed that the trend in binding strength is exactly opposite to the trend determined by σ-hole size. From the point of view of the current study, charge transfer is connected to the electrostatic energy as the first term of its multipolar expansion. In that sense, charge transfer is only part of a more complete narrative based on the electrostatic energy itself, which already played a full role in our REG-IQA analysis. Indeed, we could link the excess charge on the halogen molecule (and the corresponding depletion on the other monomer in the complex) to a crude estimate of the electrostatic energy between the monomers. However, this severe approximation and the fact that there is no visible trend between the numerical values of charge transfer and the binding energy (see above), makes this an avenue not worth pursuing. Instead, we used the REG-IQA analysis itself to point out that the σ-hole model can be linked to the underlying atomic energy patterns only at long-range (N…Cl distances ≥3.25 Å in the H
3N:ClF complex). The importance of the covalent character of the halogen bond, that we discovered in the current study, automatically weakens the trustworthiness of the σ-hole when used at short-range.









{kind=link}
{kind=link}
{kind=link}
{kind=link}