
Hydrogen Bond-Mediated Conjugates Involving Lanthanide Diphthalocyanines and Trifluoroacetic Acid (Lnpc2@TFA): Structure, Photoactivity, and Stability


 and
and 
Abstract
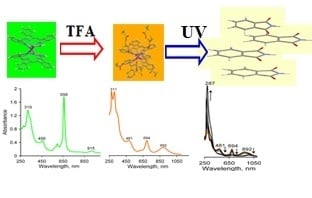
:
1. Introduction
2. Results and Discussion
2.1. Formation of LnPc2@TFA Conjugates
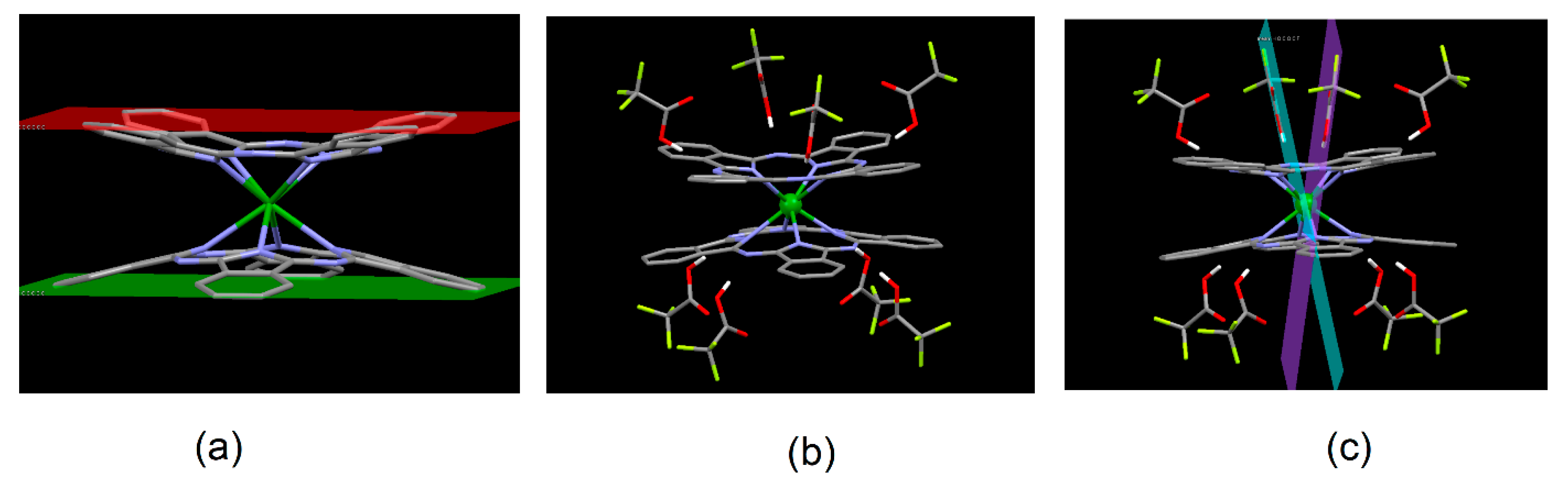
2.2. Molecular Structure of the LnPc2@TFA Conjugates (DFT Approach)
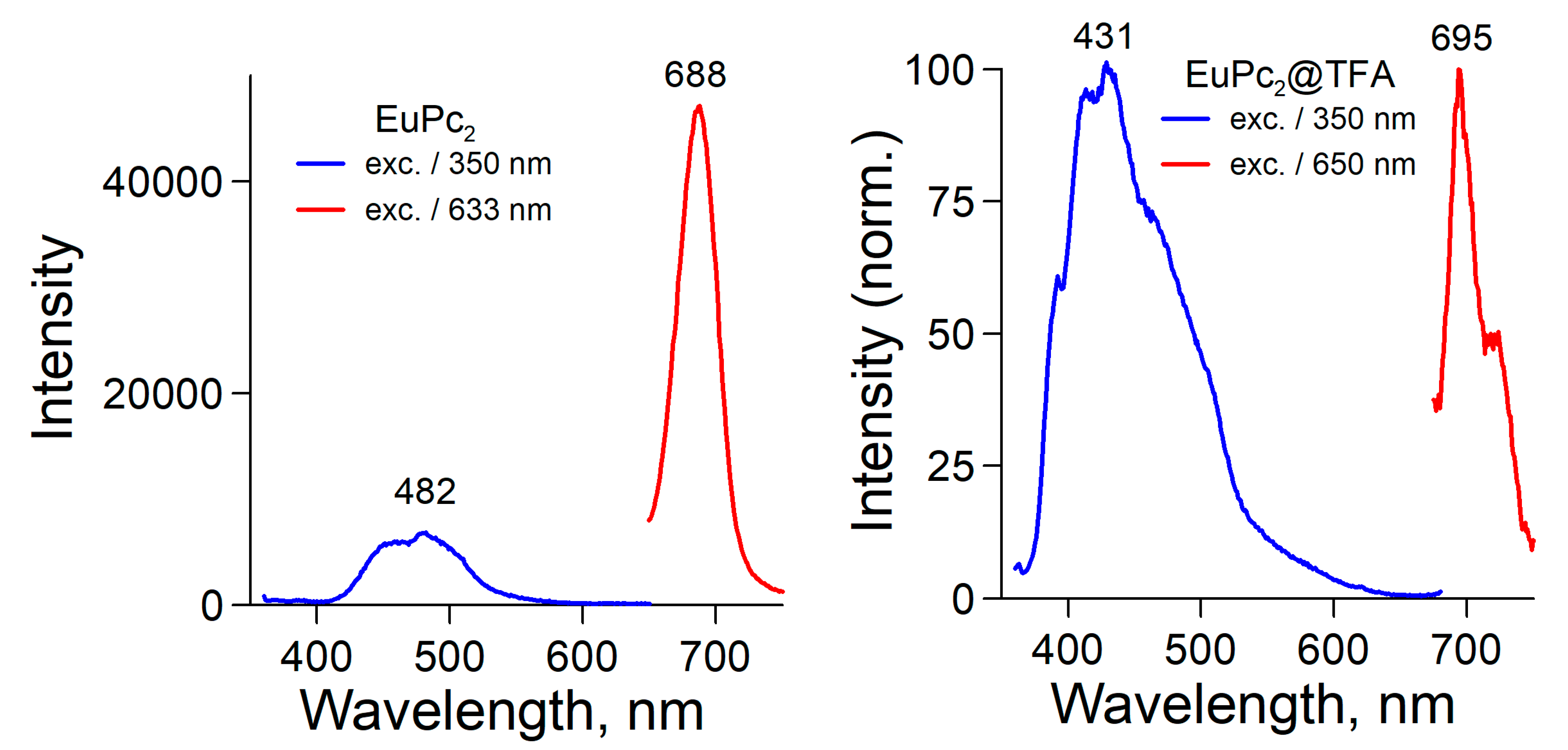
2.3. Fluorescence Spectra and Singlet Oxygen Generation
2.4. Photostability
3. Materials and Methods
3.1. Materials
3.2. Spectrophotometric Measurements
3.3. Phthalocyanine Protonation by TFA
3.4. Photostability Studies
3.5. Theoretical Structural Studies
4. Summary
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Castaneda, F.; Plichon, V.; Clarisse, C.; Riou, M.T. Electrochemistry of a lutetium diphthalocyanine film in contact with an acidic aqueous medium. J. Electroanal. Chem. 1987, 233, 77–85. [Google Scholar] [CrossRef]
- Daniels, R.B.; Peterson, J.; Porter, W.C.; Wilson, Q.D. The electrochromic behaviour of lanthanide bisphthalocyanines: The anomalous nature of the green lutetium species. J. Coord. Chem. 1993, 30, 357–366. [Google Scholar] [CrossRef]
- Simon, J.; Andre, J.J. Molecular Semiconductors: Photoelectrical Properties and Solar Cells; Springer Science & Business Media: Berlin, Germany, 2012; ISBN 978-3-642-70012-5. [Google Scholar]
- Kratochvílová, I.; Šebera, J.; Paruzel, B.; Pfleger, J.; Toman, P.; Marešová, E.; Havlová, Š.; Hubík, P.; Buryi, M.; Vrňata, M.; et al. Electronic functionality of Gd-bisphthalocyanine: Charge carrier concentration, charge mobility, and influence of local magnetic field. Synth. Met. 2018, 236, 68–78. [Google Scholar] [CrossRef]
- Schnurpfeil, G.; Sobbi, A.; Spiller, W.; Kliesch, H.; Wöhrle, D. Photo-oxidative stability and its correlation with eemi-empirical MO calculations of various Tetraazaporphyrin derivatives in solution. J. Porphyr. Phthalocyanines 1997, 1, 159–167. [Google Scholar] [CrossRef]
- Sobbi, A.; Wöhrle, D.; Schlettwein, D. Photochemical stability of various Porphyrins in solution and as thin film electrodes. J. Chem. Soc. Perkin Trans. 1993, 2, 481–488. [Google Scholar] [CrossRef]
- Słota, R.; Dyrda, G. UV Photostability of Metal Phthalocyanines in Organic Solvents. Inorg. Chem. 2003, 42, 5743–5750. [Google Scholar] [CrossRef]
- Słota, R.; Dyrda, G.; Hofer, M.; Mele, G.; Bloise, E.; Sole, R. Novel Lipophilic Lanthanide Bis-Phthalocyanines Functionalized by Pentadecylphenoxy Groups: Synthesis, Characterization and UV-Photostability. Molecules 2012, 17, 10738–10753. [Google Scholar] [CrossRef]
- Stuzin, P.A.; Khelevina, O.G.; Berezin, B.D. Phthalocyanines: Properties and Applications; Leznoff, C.C., Lever, A.B.P., Eds.; VCH: New York, NY, USA, 1996; Volume 4, pp. 19–78. [Google Scholar]
- Liu, J.; Zhao, Y.; Zhang, F.; Zhao, F.; Tang, Y.; Song, X.; Chau, F.T. Effects of protonation and deprotonatiion on Phthalocyanine’s spectra. Acta Phys. Chim. Sin. 1996, 12, 202–207. [Google Scholar]
- Stuzhin, P.A. Azaporphyrins and Phthalocyanines as Multicentren Conjugated Ampholites. J. Porphyr. Phthalocyanines 1999, 3, 500–513. [Google Scholar] [CrossRef]
- Ledson, D.L.; Twigg, M.V. Acid-base behaviour of Phthalocyanine. Inorg. Chim. Acta 1975, 13, 43–46. [Google Scholar] [CrossRef]
- Bernstein, P.A.; Lever, A.B.P. Protonation of cobalt tetraneopentoxyphthalocyanine as a function of oxidation state. Inorg. Chim. Acta 1992, 198, 543–555. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Honda, T.; Kojimad, T. Structures and photoinduced electron transfer of protonated complexes of porphyrins and metallophthalocyanines. Coord. Chem. Rev. 2012, 256, 2488–2502. [Google Scholar] [CrossRef]
- Kociscakova, L.; Senipek, M.I.; Zimcik, P.; Npvakova, V. Non-peripherally alkylamino-substituted phthalocyanines: Synthesis, spectral, photophysical and acid-base properties. J. Porphyr. Phthalocyanines 2019, 23, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Beeby, A.; FitzGerald, S.; Stanley, C.F. Protonation of Tetrasulfonated Zinc Phthalocyanine in Aqueous Acetonitrile Solution. Photochem. Photobiol. 2001, 74, 566–569. [Google Scholar] [CrossRef]
- Gaspard, S.; Verdaquer, M.; Viovy, R. Etude des phtalocyanines en solution dans l’acide sulfurique. J. Chim. Phys. 1972, 69, 1740–1747. [Google Scholar] [CrossRef]
- Iodko, S.S.; Kaliya, O.L.; Lebedev, O.L.; Luk’yanets, E.A. Absorption spectra of complexes of aluminum bromide with phthalocyanines. Koord. Khim. 1979, 5, 611–617. [Google Scholar] [CrossRef]
- Ahrens, U.; Kuhn, H. Lichtabsorption und Assoziations- und Protonierungsgleichgewichte von Lösungen eines Cu-Phthalocyaninsulfonats. Z. Phys. Chem. 1963, 37, 1–32. [Google Scholar] [CrossRef]
- Petrik, P.; Zimcik, P.; Kopecky, K.; Musil, Z.; Miletin, M.; Loukotova, V. Protonation and deprotonation of nitrogens in tetrapyrazinoporphyrazine macrocycles. J. Porphyr. Phthalocyanines 2007, 11, 487–495. [Google Scholar] [CrossRef]
- Ogunsipe, A.; Nyokong, T. Effects of substituents and solvents on the photochemical properties of zinc phthalocyanine complexes and their protonated derivatives. J. Mol. Struct. 2004, 689, 89–97. [Google Scholar] [CrossRef]
- Ghani, F.; Kristen, J.; Riegler, H. Solubility Properties of Unsubstituted Metal Phthalocyanines in Different Types of Solvents. J. Chem. Eng. Data 2012, 57, 439–449. [Google Scholar] [CrossRef]
- Lin, M.J.; Fang, X.; Xu, M.B.; Wang, J.D. The effect of protonation on the spectra and stabilities of alkoxyl substituted phthalocyaninatometals. Spectrochim. Acta A 2008, 71, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, T.N.; Lomova, T.N.; Zaitseva, S.V.; Zdanovich, S.A.; Suslova, E.E.; Maizlish, V.E. Acid-base properties and stability of (hydroxo) tetra (carboxy) phthalocyaninatoaluminum (III). Russ. J. Gen. Chem. 2005, 50, 476–482. [Google Scholar]
- Strelkova, T.I.; Gurinovich, G.P.; Sinyakov, G.N. Investigation of the ionization of phthalocyanines by luminescence spectra. Zh. Prik. Spektr. 1966, 4, 429–433. [Google Scholar] [CrossRef]
- Makarov, D.A.; Derkacheva, V.M.; Kuznetsova, N.A.; Kaliya, O.L.; Lukyanets, E.A. Octa-3,6-hexoxyphthalocyanines: Effect of Protonation on Spectral and Photochemical Properties. Macroheterocycles 2013, 6, 371–378. [Google Scholar] [CrossRef]
- Ogunsipe, A.O.; Idowu, M.A.; Ogunbayo, T.B.; Akinbulu, I.A. Protonation of some non-transition metal phthalocyanines—Spectral and photophysicochemical consequences. J. Porphyr. Phthalocyanines 2012, 16, 1–10. [Google Scholar] [CrossRef]
- Mohan Kumar, T.M.; Achar, B.N. UV–visible spectral study on the stability of lead phthalocyanine complexes. J. Phys. Chem. Solids 2006, 67, 2282–2288. [Google Scholar] [CrossRef]
- Iodko, S.S.; Kaliya, O.L.; Kondratenko, N.V.; Luk’yanets, E.A.; Popov, V.I.; Yaguopol’skij, L.M. Quantitative characteristics of the stagewise protonation of phthalocyanines. Zh. Obshch. Khim. 1983, 53, 901–903. [Google Scholar]
- Białkowska, E.; Graczyk, A. The interaction of metallophthalocyanines with electron- acceptor metal chlorides. Org. Magn. Reson. 1978, 11, 167–171. [Google Scholar] [CrossRef]
- Lipatova, M.; Yusova, A.A.; Makarova, L.I.; Petrova, M.V. Effect of hyaluronic acid on the state and photoactivity of Zn(II) phthalocyanine cationic derivative in mixed aqueous solutions. J. Photochem. Photobiol. A Chem. 2019, 382, 111927. [Google Scholar] [CrossRef]
- Ovchenkova, E.N.; Lomova, T.N. Protonation Equilibria of (octakis(3_Trifluoromethylphenyl),(3-Trifluoromethylphenoxy), and (3,5 Di_tert_butylphenoxy)phthalocyaninato) manganese(III) acetate. Russ. J. Phys. Chem. A 2015, 89, 190–195. [Google Scholar] [CrossRef]
- Graczyk, A.; Białkowska, E. Magnetic properties of oxidation products of phthalocyanine complexes with metal chlorides with electron acceptor properties. Terahedron 1978, 34, 3505–3509. [Google Scholar] [CrossRef]
- Kagaya, Y.; Isago, H. Rapid reactions of Phthalocyanines with Tellurium Tetrachloride in non-aqueous solutions. J. Porphyr. Phthalocyanines 1999, 3, 537–543. [Google Scholar] [CrossRef]
- Beeby, A.; FitzGerald, S.; Stanley, C.F. A photophysical study of protonated (tetra-tert-butylphthalocyaninato) zinc. J. Chem. Soc. Perkin Trans. 2001, 2, 1978–1982. [Google Scholar] [CrossRef]
- Berezin, B.D. On the question of protonation of tetrapyrrole ligands and their complexes. Zh. Obshch. Khim. 1973, 43, 2738–2743. [Google Scholar]
- Isago, H.; Fujita, H.; Hirota, M.; Sugimori, T.; Kagaya, Y. Synthesis, spectral and electrochemical properties of a novel phosphorous(V)-phthalocyanine. J. Porphyr. Phthalocyanines 2013, 17, 763–771. [Google Scholar] [CrossRef]
- Fang, X.; Wang, J.D.; Lin, M.J. The chemical stabilities of phthalocyanine monomers vs. aggregations. J. Mol. Cat. A Chem. 2013, 372, 100–104. [Google Scholar] [CrossRef]
- Thamae, M.; Nyokong, T. Interaction of sulfur dioxide and cyanide with cobalt(II)tetrasulfophthalocyanine in aqueous media. Polyhedron 2002, 21, 133–140. [Google Scholar] [CrossRef]
- Strivastava, K.P.; Kumar, A. UV spectral studies in protonation of Cu-Phthalocyanine and Phthalocyanine in Sulfuric acid-solvent. Asian J. Chem. 2001, 13, 1539–1543. [Google Scholar]
- Stuzhin, P.A.; Ivanova, S.S.; Hamdoush, M.; Kirakosyan, G.A.; Kiselev, A.; Popov, A.; Sliznev, V.; Ercolani, C. Tetrakis(1,2,5-thiadiazolo)porphyrazines. 9. Synthesis and spectral and theoretical studies of the lithium(I) complex and its unusual behaviour in aprotic solvents in the presence of acids. Dalton Trans. 2019, 48, 14049–14061. [Google Scholar] [CrossRef]
- Claessens, C.; Hahn, U.; Torres, T. Phthalocyanines: From outstanding electronic properties to emerging applications. Chem. Rec. 2008, 8, 75–97. [Google Scholar] [CrossRef]
- M’sadak, M.; Roncali, J.; Garnier, F. Lanthanides—Phthalocyanines complexes: From a diphthalocyanine Pc2Ln to a super complex Pc3ln2. J. Chim. Phys. 1986, 83, 211–216. [Google Scholar] [CrossRef]
- Daniels, R.B.; Payne, G.L.; Peterson, J. The electrochromic behaviour of lanthanide bisphthalocyanines: The acid-base nature of the mechanism. J. Coord. Chem. 1993, 28, 23–31. [Google Scholar] [CrossRef]
- Słota, R.; Dyrda, G.; Szczegot, K. Sulfur dioxide oxidation catalyzed by photosensitized Ytterbium Diphthalocyanine. Catal. Lett. 2008, 127, 247–252. [Google Scholar] [CrossRef]
- Nensala, N.; Nyokong, T. Photosensization reactions of neodymium, dysprosium and ytterbium diphthalocyanines. Polyhedron 1997, 16, 2971–2976. [Google Scholar] [CrossRef] [Green Version]
- Nensala, N.; Nzimande, A.; Nyokong, T. Photochemically induced electron transfer between sulfur dioxide and tin(IV) mono- and di-phthalocyanines. J. Photochem. Photobiol. A Chem. 1996, 98, 129–135. [Google Scholar] [CrossRef]
- Nensala, T.; Nyokong, T. Photosenassisted reduction of thionyl chloride by neodymium, europium, thulium and lutetium diphthalocyanines. Polyhedron 1998, 06, 2356–2364. [Google Scholar] [CrossRef]
- Dyrda, G.; Słota, R.; Broda, M.A.; Mele, G. Meso-Aryl-substituted free-base porphyrins: Formation, structure and photostability of diprotonated species. Res. Chem. Intermed. 2016, 42, 3789–3804. [Google Scholar] [CrossRef] [Green Version]
- Dyrda, G.; Broda, M.A.; Hnatejko, Z.; Pędziński, T.; Słota, R. Adducts of free-base meso-tetraarylporphyrins with trihaloacetic acids: Structure and photostability. J. Photochem. Photobiol. A Chem. 2020, 393, 112445. [Google Scholar] [CrossRef]
- Dyrda, G.; Kocot, K.; Poliwoda, A.; Mele, G.; Pal, S.; Słota, R. Hybrid TiO2 @ phthalocyanine catalysts in photooxidation of 4-nitrophenol: Effect of the matrix and sensitizer type. J. Photochem. Photobiol. A Chem. 2020, 387, 112–124. [Google Scholar] [CrossRef]
- Słota, R.; Dyrda, G.; Szczego, K.; Mele, G.; Pio, I. Photocatalytic activity of nano and microcrystalline TiO2 hybrid systems involving phthalocyanine or porphyrin sensitizers. Photochem. Photobiol. Sci. 2011, 10, 361–366. [Google Scholar] [CrossRef]
- Ghammamy, S.; Azimi, M.; Sedaghat, S. Preparation and identification of two new phthalocyanines and study of their anti-cancer activity and anti-bacterial properties. Sci. Res. Essays 2012, 74, 3751–3757. [Google Scholar] [CrossRef]
- Soncin, M.; Fabris, C.; Busetti, A.; Dei, D.; Nistri, D.; Roncucci, G.; Jori, G. Approaches to selectivity in the Zn(II)–phthalocyaninephotosensitized inactivation of wild-type and antibiotic-resistant Staphylococcus aureus. Photochem. Photobiol. Sci. 2002, 1, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Seven, O.; Bircan, D.; Sohret, A.; Feriha, C. Synthesis, properties and photodynamic activities of some zinc(II) phthalocyanines against Escherichia coli and Staphylococcus aureus. J. Porphyr. Phthalocyanines 2008, 12, 953–963. [Google Scholar] [CrossRef]
- D’Alessandro, N.; Toniucci, L.; Morvillo, A.; Dragani, L.K.; Di Deo, M.; Bressan, M. Thermal stability and photostability of water solutions of sulfophthalocyanines of Ru(II), Cu(II), Ni(II), Fe(III) and Co(II). J. Organomet. Chem. 2005, 690, 2133–2141. [Google Scholar] [CrossRef]
- Caronna, T.; Colleon, C.; Dotti, S.; Fontana, F.; Rosace, G. Decomposition of phthalocyanine dye in various conditions under UV or visible light irradiation. J. Photochem. Photobiol. A Chem. 2006, 184, 135–140. [Google Scholar] [CrossRef]
- Özdemir, M.; Karapınar, B.; Yalçın, B.; Salan, Ü.; Durmuş, M.; Bulut, M. Synthesis and characterization of novel 7-oxy- 3-ethyl-6-hexyl-4-methylcoumarin substitute metallo phthalocyanines and investigation of their photophysical and photochemical properties. Dalton Trans. 2019, 48, 13046–13056. [Google Scholar] [CrossRef]
- Singh-Rachford, T.N.; Castellano, F.N. Photon upconversion based on sensitized triplet–triplet annihilation. Coord. Chem. Rev. 2010, 254, 2560–2573. [Google Scholar] [CrossRef]
- Mele, G.; García-López, E.; Palmisano, L.; Dyrda, G.; Słota, R. Photocatalytic degradation of 4- nitrophenol in aqueous suspension by using polycrystalline TiO2 impregnated with lanthanide double-decker phthalocyanine complexes. J. Phys. Chem. C 2007, 111, 6581–6588. [Google Scholar] [CrossRef]
- Obłoza, M.; Łapok, Ł.; Solarski, J.; Pędziński, T.; Nowakowska, M. Facile synthesis, triplet-state properties, and electrochemistry of Hexaiodo-Subphthalocyanine. Chem. Eur. J. 2018, 24, 17080–17090. [Google Scholar] [CrossRef]
- Mihaylov, T.; Trendafilova, N.; Kostova, I.; Georgieva, I.; Bauer, G. DFT modeling and spectroscopic study of metal-ligand bonding in La(III) complex of coumarin-3-carboxylic acid. Chem. Phys. 2006, 327, 209–219. [Google Scholar] [CrossRef]
- Núñez, C.; Bastida, R.; MacÍas, A.; Mato-Iglesias, M.; Platas-Iglesias, C.; Valencia, L.A. Hexaaza macrocyclic ligand containing acetohydrazide pendants for Ln(III) complexation in aqueous solution. Solid-state and solution structures and DFT calculations. Dalton Trans. 2008, 29, 3841–3850. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Nagase, S. Theoretical calculations of vibrational modes in endohedral metallofullerenes: La@C82 and Sc2@C84. Mol. Phys. 2003, 101, 49–254. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, G.; Scalmani, J.R.; Barone, B.; Mennucci, G.; Petersson, A.; et al. Gaussian 09, Revision, A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds studied are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Band | NdPc2 | SmPc2 | EuPc2 | GdPc2 | YbPc2 | LuPc2 | |
|---|---|---|---|---|---|---|---|
| LnPc2 (green form) | B | 324 | 322 | 323 | 322 | 319 | 318 |
| MLCT | 461 | 459 | 458 | 458 | 456 | 456 | |
| Q | 675 | 669 | 668 | 667 | 659 | 658 | |
| LMCT | 900 | 901 | 901 | 904 | 912 | 915 | |
| LnPc2@TFA (orange form) | B | 312 | 311 | 311 | 308 | 311 | 311 |
| MLCT | 514 | 503 | 509 | 498 | 486 | 481 | |
| Q | 710 | 707 | 709 | 708 | 694 | 694 | |
| LMCT | 1042 | 1055 | 1070 | 989 | 906 | 892 | |
| Compound | NdPc2 | SmPc2 | EuPc2 | GdPc2 | YbPc2 | LuPc2 |
|---|---|---|---|---|---|---|
| δ% | 100 | 28 | 27 | 6 | 0 | 0 |
| Compound | ΔE kJ/mol | Lu−Np | Sc | Nµ⸱⸱H | Interplanar Spacing | α (°) | β (°) | γ (°) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 4Np | 4Nµ | 8Cb | ||||||||
| LuPc2 | − | 2.416 | 21.602 | − | 2.735 | 3.328 | 5.004 | 14.0 | − | − |
| LuPc2@TFA | 245 | 2.422 | 21.645 | 1.800 | 2.747 | 3.310 | 4.123 | 8.0 | 32.0 | 21.4 |
| λexc, nm | NdPc2 | SmPc2 | EuPc2 | GdPc2 | YbPc2 | LuPc2 |
|---|---|---|---|---|---|---|
| 350 | 434 | 435 | 482 | 487 | 430 | 429 |
| 633 | 694 | 691 | 688 | 695 | 692 | 693 |
| ΔS (B) | 110 | 113 | 159 | 165 | 111 | 111 |
| ΔS (Q) | 19 | 22 | 20 | 28 | 33 | 35 |
| LnPc2@TFA | ||||||
| 350 | 428 | 471 | 431 | 430 | 430 | 430 |
| 650 | 695 | 694 | 695 | 693 | 693 | 694 |
| ΔS (B) | 116 | 160 | 120 | 122 | 119 | 119 |
| ΔS (Q) | −15 | −13 | −14 | −15 | −1 | 0 |
| Photolysis | NdPc2 | SmPc2 | EuPc2 | GdPc2 | YbPc2 | LuPc2 |
|---|---|---|---|---|---|---|
| I stage | 1.8 × 10−2 | 1.4 × 10−2 | 2.1 × 10−2 | 1.5 × 10−2 | 1.3 × 10−2 | 4.6 × 10−3 |
| II stage | 1.7 × 10−3 | 8.0 × 10−4 | 1.2 × 10−3 | 1.4 × 10−3 | 1.8 × 10−3 | 1.3 × 10−3 |
| LnPc2@TFA | 4.4 × 10−1 | 2.8 × 10−2 | 3.0 × 10−2 | 3.0 × 10−2 | 5.6 × 10−3 | 3.7 × 10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyrda, G.; Zakrzyk, M.; Broda, M.A.; Pędziński, T.; Mele, G.; Słota, R. Hydrogen Bond-Mediated Conjugates Involving Lanthanide Diphthalocyanines and Trifluoroacetic Acid (Lnpc2@TFA): Structure, Photoactivity, and Stability. Molecules 2020, 25, 3638. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25163638
Dyrda G, Zakrzyk M, Broda MA, Pędziński T, Mele G, Słota R. Hydrogen Bond-Mediated Conjugates Involving Lanthanide Diphthalocyanines and Trifluoroacetic Acid (Lnpc2@TFA): Structure, Photoactivity, and Stability. Molecules. 2020; 25(16):3638. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25163638
Chicago/Turabian StyleDyrda, Gabriela, Maja Zakrzyk, Małgorzata A. Broda, Tomasz Pędziński, Giuseppe Mele, and Rudolf Słota. 2020. "Hydrogen Bond-Mediated Conjugates Involving Lanthanide Diphthalocyanines and Trifluoroacetic Acid (Lnpc2@TFA): Structure, Photoactivity, and Stability" Molecules 25, no. 16: 3638. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25163638