
Local Structures of Two-Dimensional Zeolites—Mordenite and ZSM-5—Probed by Multinuclear NMR

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
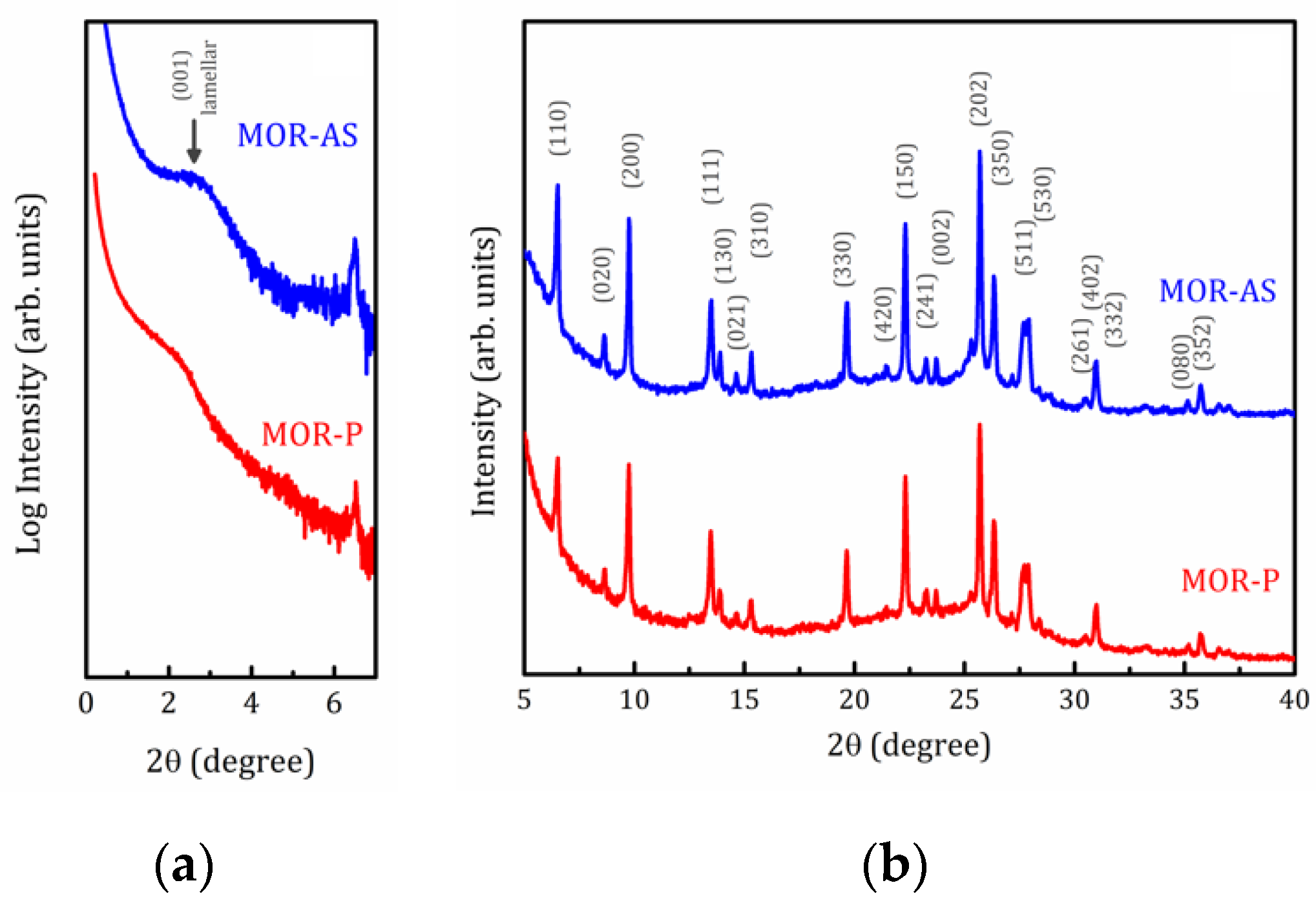
2.1. X-ray Analysis
2.2. SEM-EDX Studies
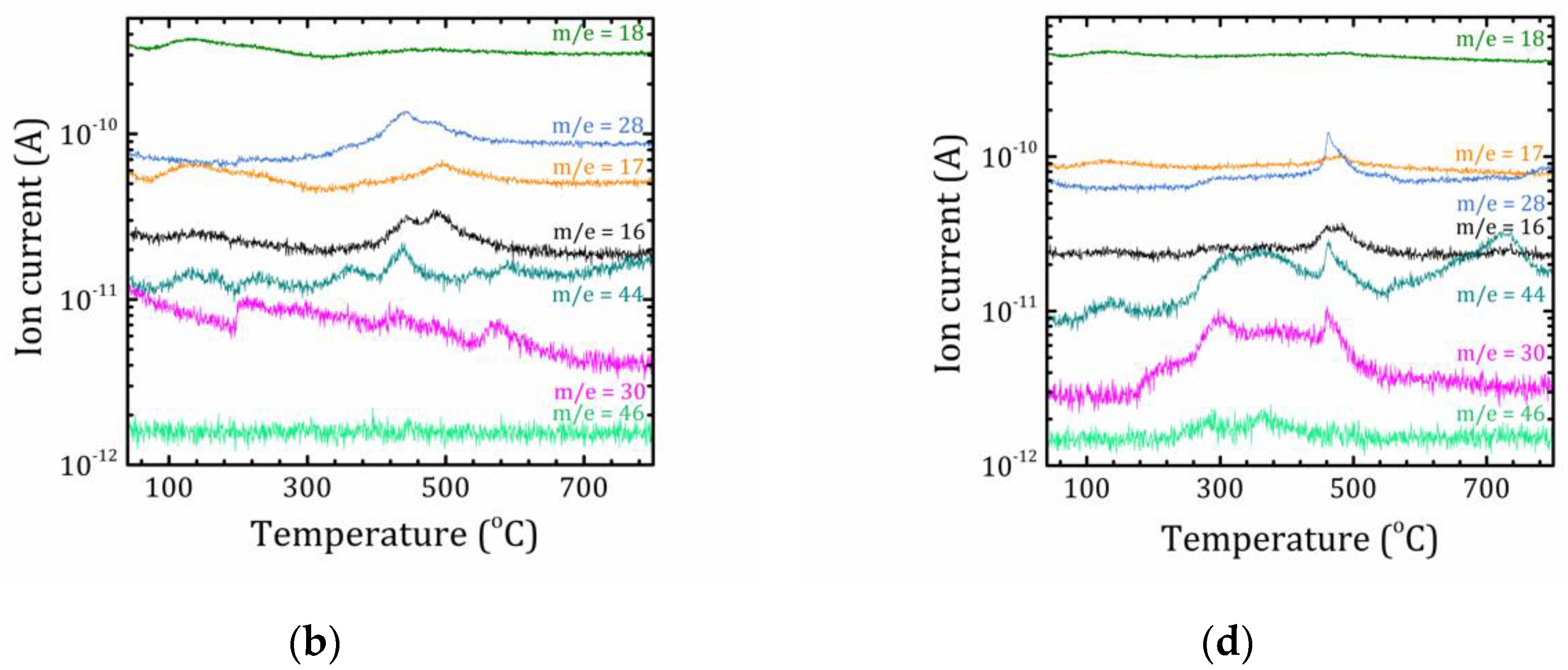
2.3. Thermal Analysis
2.4. NMR Study
2.4.1. 1H MAS NMR and 13C CP-MAS NMR
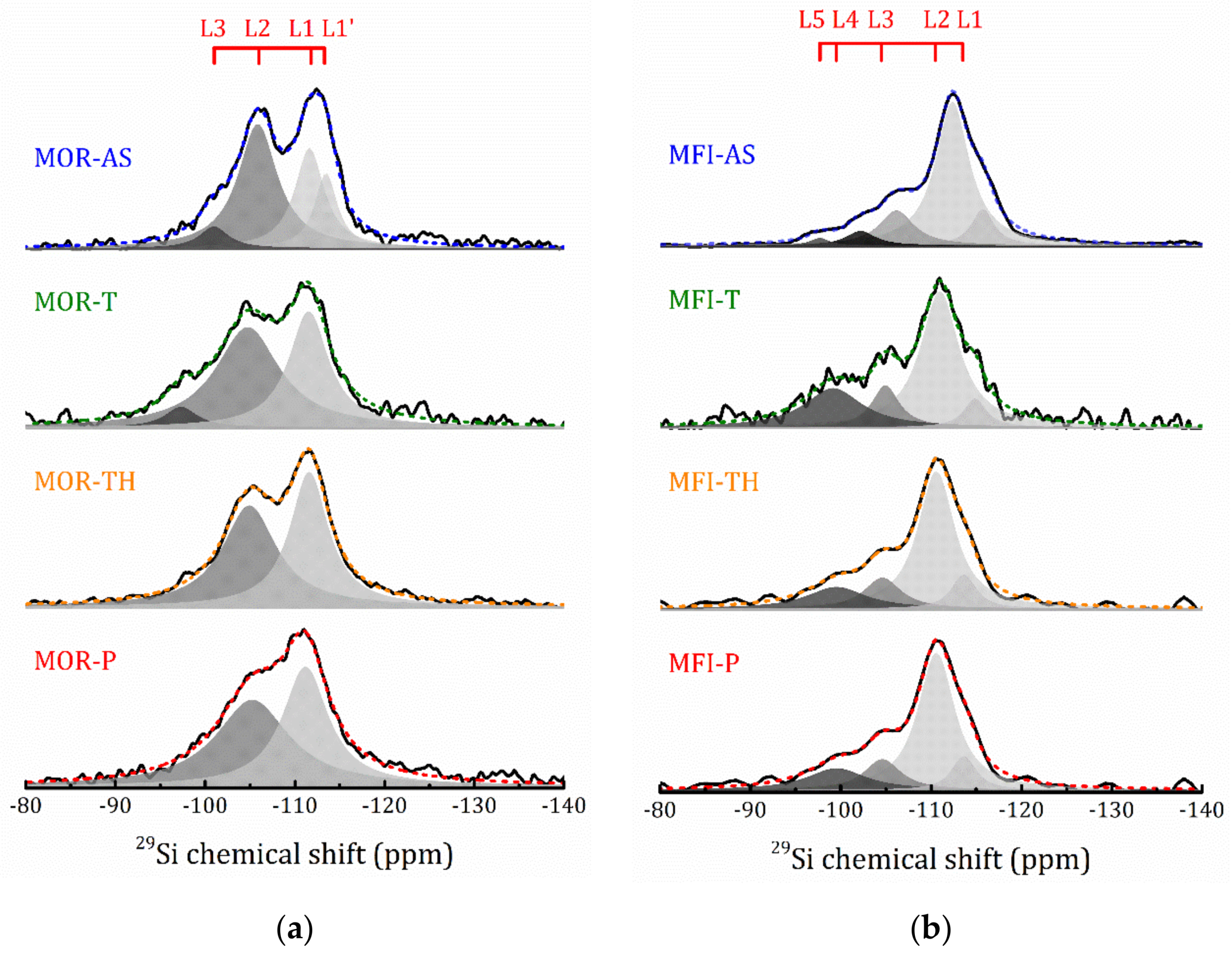
2.4.2. 27Al and 29Si MAS NMR
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wight, A.P.; Davis, M.E. Design and Preparation of Organic−Inorganic Hybrid Catalysts. Chem. Rev. 2002, 102, 3589–3614. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, Z.; Zhou, D.; Xia, Y.; Lu, X.; He, H.; Xia, Q. One-step synthesis of hybrid zeolite with exceptional hydrophobicity to accelerate the interfacial reaction at low temperature. Microporous Mesoporous Mater. 2019, 280, 195–202. [Google Scholar] [CrossRef]
- Čejka, J.; Millini, R.; Opanasenko, M.; Serrano, D.P.; Roth, W.J. Advances and challenges in zeolite synthesis and catalysis. Catal. Today 2020, 345, 2–13. [Google Scholar] [CrossRef]
- Roth, W.J.; Nachtigall, P.; Morris, R.E.; Čejka, J. Two-Dimensional Zeolites: Current Status and Perspectives. Chem. Rev. 2014, 114, 4807–4837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Ostraat, M.L. Innovations in hierarchical zeolite synthesis. Catal. Today 2016, 264, 3–15. [Google Scholar] [CrossRef]
- Ge, T.; Hua, Z.; He, X.; Zhu, Y.; Ren, W.; Chen, L.; Zhang, L.; Chen, H.; Lin, C.; Yao, H.; et al. One-pot synthesis of hierarchically structured ZSM-5 zeolites using single micropore-template. Chin. J. Catal. 2015, 36, 866–873. [Google Scholar] [CrossRef]
- Jia, X.; Khan, W.; Wu, Z.; Choi, J.; Yip, A.C. Modern synthesis strategies for hierarchical zeolites: Bottom-up versus top-down strategies. Adv. Powder Technol. 2019, 30, 467–484. [Google Scholar] [CrossRef]
- Koohsaryan, E.; Anbia, M. Nanosized and hierarchical zeolites: A short review. Chin. J. Catal. 2016, 37, 447–467. [Google Scholar] [CrossRef]
- Mintova, S.; Grand, J.; Valtchev, V. Nanosized zeolites: Quo Vadis? Comptes Rendus Chim. 2016, 19, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Margarit, V.J.; Portilla, M.T.; Navarro, M.T.; Abudawoud, R.; Al-Zahrani, I.M.; Shaikh, S.; Martínez, C.; Corma, A. One-pot co-crystallization of beta and pentasil nanozeolites for the direct conversion of a heavy reformate fraction into xylenes. Appl. Catal. A Gen. 2019, 581, 11–22. [Google Scholar] [CrossRef]
- Moliner, M. Direct Synthesis of Functional Zeolitic Materials. ISRN Mater. Sci. 2012, 2012, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Moliner, M.; Rey, F.; Corma, A. Towards the Rational Design of Efficient Organic Structure-Directing Agents for Zeolite Synthesis. Angew. Chem. Int. Ed. 2013, 52, 13880–13889. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.-Q.; Long, Y.-C.; Sun, Y.-J. Synthesis of FER type zeolite with tetrahydrofuran as the template. Chem. Commun. 2000, 1893–1894. [Google Scholar] [CrossRef]
- Migliori, M.; Aloise, A.; Giordano, G. Methanol to dimethylether on H-MFI catalyst: The influence of the Si/Al ratio on kinetic parameters. Catal. Today 2014, 227, 138–143. [Google Scholar] [CrossRef]
- Li, H.-J.; Zhou, X.-D.; Di, Y.H.; Zhang, J.-M.; Zhang, Y. Effect of Si-ATP/CTAB ratio on crystal morphology, pore structure and adsorption performance of hierarchical (H) ZSM-11 zeolite. Microporous Mesoporous Mater. 2018, 271, 146–155. [Google Scholar] [CrossRef]
- Catizzone, E.; Migliori, M.; Mineva, T.; Van Daele, S.; Valtchev, V.; Giordano, G. New synthesis routes and catalytic applications of ferrierite crystals. Part 2: The effect of OSDA type on zeolite properties and catalysis. Microporous Mesoporous Mater. 2020, 296, 109988. [Google Scholar] [CrossRef]
- Che, S.; Feng, J.; Che, S. An insight into the role of the surfactant CTAB in the formation of microporous molecular sieves. Dalton Trans. 2014, 43, 3612–3617. [Google Scholar] [CrossRef]
- Wang, X.; Chen, H.; Meng, F.; Gao, F.; Sun, C.; Sun, L.; Wang, S.; Wang, L.; Wang, Y. CTAB resulted direct synthesis and properties of hierarchical ZSM-11/5 composite zeolite in the absence of template. Microporous Mesoporous Mater. 2017, 243, 271–280. [Google Scholar] [CrossRef]
- Xu, L.; Ji, X.; Li, S.; Zhou, Z.; Du, X.; Sun, J.; Deng, F.; Che, S.; Wu, P. Self-Assembly of Cetyltrimethylammonium Bromide and Lamellar Zeolite Precursor for the Preparation of Hierarchical MWW Zeolite. Chem. Mater. 2016, 28, 4512–4521. [Google Scholar] [CrossRef]
- Chizhik, V.I.; Chernyshev, Y.S.; Donets, A.V.; Frolov, V.V.; Komolkin, A.V.; Shelyapina, M.G. Magnetic Resonance and Its Applications; Springer International Publishing: Cham, Switzerland, 2014. [Google Scholar] [CrossRef]
- Shelyapina, M.; Nefedov, D.Y.; Kostromin, A.V.; Siluykov, O.; Zvereva, I. Proton mobility in Ruddlesden-Popper phase H2La2Ti3O10 studied by 1H-NMR. Ceram. Int. 2019, 45, 5788–5795. [Google Scholar] [CrossRef]
- Shelyapina, M.; Lushpinskaya, I.P.; Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Identification of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10 by Multinuclear NMR. Russ. J. Gen. Chem. 2020, 90, 760–761. [Google Scholar] [CrossRef]
- Kharkov, B.B.; Dvinskikh, S.V. Chain dynamics of surfactants in mesoporous silica. Phys. Chem. Chem. Phys. 2013, 15, 18620–18626. [Google Scholar] [CrossRef] [PubMed]
- Kharkov, B.B.; Corkery, R.W.; Dvinskikh, S.V. Phase Transitions and Chain Dynamics of Surfactants Intercalated into the Galleries of Naturally Occurring Clay Mineral Magadiite. Langmuir 2014, 30, 7859–7866. [Google Scholar] [CrossRef]
- Khimyak, Y.Z.; Klinowski, J. Solid-state NMR studies of the organic template in mesostructured aluminophosphates. Phys. Chem. Chem. Phys. 2001, 3, 616–626. [Google Scholar] [CrossRef]
- Yocupicio-Gaxiola, R.I.; Petranovskii, V.; Antúnez-García, J.; Moyado, S.F. One-pot synthesis of lamellar mordenite and ZSM-5 zeolites and subsequent pillaring by amorphous SiO2. Appl. Nanosci. 2019, 9, 557–565. [Google Scholar] [CrossRef]
- Baerlocher, C.; McCusker, L.B.; Olson, D.H. Atlas of Zeolite Framework Types, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Na, K.; Choi, M.; Park, W.; Sakamoto, Y.; Terasaki, O.; Ryoo, R. Pillared MFI Zeolite Nanosheets of a Single-Unit-Cell Thickness. J. Am. Chem. Soc. 2010, 132, 4169–4177. [Google Scholar] [CrossRef]
- Na, K.; Park, W.; Seo, Y.; Ryoo, R. Disordered Assembly of MFI Zeolite Nanosheets with a Large Volume of Intersheet Mesopores. Chem. Mater. 2011, 23, 1273–1279. [Google Scholar] [CrossRef]
- Banfi, D.; Patiny, L. www.nmrdb.org: Resurrecting and Processing NMR Spectra On-line. Chim. Int. J. Chem. 2008, 62, 280–281. [Google Scholar] [CrossRef]
- Ishikawa, S.; Kurosu, H.; Ando, I. Structural studies of n-alkanes by variable-temperature solid-state high-resolution 13C-NMR spectroscopy. J. Mol. Struct. 1991, 248, 361–372. [Google Scholar] [CrossRef]
- Janicke, M.; Landry, C.C.; Christiansen, S.C.; Kumar, D.; Stucky, G.D.; Chmelka, B.F. Aluminum Incorporation and Interfacial Structures in MCM-41 Mesoporous Molecular Sieves. J. Am. Chem. Soc. 1998, 120, 6940–6951. [Google Scholar] [CrossRef]
- Baccile, N.; Babonneau, F. Organo-modified mesoporous silicas for organic pollutant removal in water: Solid-state NMR study of the organic/silica interactions. Microporous Mesoporous Mater. 2008, 110, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Voelkel, R. High-Resolution Solid-State 13C-NMR Spectroscopy of Polymers [New Analytical Methods(37)]. Angew. Chem. Int. Ed. 1988, 27, 1468–1483. [Google Scholar] [CrossRef]
- Deng, F.; Yue, Y.; Ye, C. Observation of Nonframework Al Species in Zeolite β by Solid-State NMR Spectroscopy. J. Phys. Chem. B 1998, 102, 5252–5256. [Google Scholar] [CrossRef]
- Li, S.; Zheng, A.; Su, Y.; Fang, H.; Shen, W.; Yu, Z.; Chen, L.; Deng, F. Extra-framework aluminium species in hydrated faujasite zeolite as investigated by two-dimensional solid-state NMR spectroscopy and theoretical calculations. Phys. Chem. Chem. Phys. 2010, 12, 3895. [Google Scholar] [CrossRef]
- Zhukov, Y.; Efimov, A.Y.; Shelyapina, M.; Petranovskii, V.; Zhizhin, E.; Burovikhina, A.; Zvereva, I. Effect of preparation method on the valence state and encirclement of copper exchange ions in mordenites. Microporous Mesoporous Mater. 2016, 224, 415–419. [Google Scholar] [CrossRef]
- Shelyapina, M.; Krylova, E.A.; Zhukov, Y.M.; Zhukov, Y.; Rodríguez-Iznaga, I.; Petranovskii, V.; Fuentes-Moyado, S.; Iznaga, R.; Moyado, F. Comprehensive Analysis of the Copper Exchange Implemented in Ammonia and Protonated Forms of Mordenite Using Microwave and Conventional Methods. Molecules 2019, 24, 4216. [Google Scholar] [CrossRef] [Green Version]
- Freude, D. Quadrupolar nuclei in solid-state muclear magnetic resonance. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Dybowski, C., Eds.; Wiley: New York, NY, USA, 2006; pp. 1–37. [Google Scholar]
- Kasperovich, V.S.; Sodel’, N.E.; Shelyapina, M. Nonempirical cluster calculations of the electric field gradient tensor in yttrium-aluminum garnet Y3Al5O12. Phys. Solid State 2006, 48, 1684–1688. [Google Scholar] [CrossRef]
- Shelyapina, M.; Kasperovich, V.; Wolfers, P. Electronic structure and electric-field-gradients distribution in Y3Al5O12: An ab initio study. J. Phys. Chem. Solids 2006, 67, 720–724. [Google Scholar] [CrossRef]
- Fyfe, C.A.; Feng, Y.; Grondey, H.; Kokotailo, G.T.; Gies, H. One- and two-dimensional high-resolution solid-state NMR studies of zeolite lattice structures. Chem. Rev. 1991, 91, 1525–1543. [Google Scholar] [CrossRef]
- Engelhardt, G.; Michel, D. High Resolution Solid State NMR of Silicates and Zeolites; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Brouwer, D.H.; Brouwer, C.C.; Mesa, S.; Semelhago, C.A.; Steckley, E.E.; Sun, M.P.; Mikolajewski, J.G.; Baerlocher, C. Solid-state 29Si NMR spectra of pure silica zeolites for the International Zeolite Association Database of Zeolite Structures. Microporous Mesoporous Mater. 2020, 297, 110000. [Google Scholar] [CrossRef]
- Kato, M.; Itabashi, K.; Matsumoto, A.; Tsutsumi, K. Characteristics of MOR-Framework Zeolites Synthesized in Fluoride-Containing Media and Related Ordered Distribution of Al Atoms in the Framework. J. Phys. Chem. B 2003, 107, 1788–1797. [Google Scholar] [CrossRef]
- Paixão, V.; Carvalho, A.P.; Rocha, J.; Fernandes, A.; Martins, F. Modification of MOR by desilication treatments: Structural, textural and acidic characterization. Microporous Mesoporous Mater. 2010, 131, 350–357. [Google Scholar] [CrossRef]
- Klinowski, J. Recent Advances in Solid-State NMR of Zeolites. Annu. Rev. Mater. Res. 1988, 18, 189–218. [Google Scholar] [CrossRef]
- Glaser, R.H.; Wilkes, G.L.; Bronnimann, C.E. Solid-state 29Si NMR of TEOS-based multifunctional sol-gel materials. J. Non-Cryst. Solids 1989, 113, 73–87. [Google Scholar] [CrossRef]
- Brinker, C.; Kirkpatrick, R.; Tallant, D.; Bunker, B.; Montez, B. NMR confirmation of strained “defects” in amorphous silica. J. Non-Cryst. Solids 1988, 99, 418–428. [Google Scholar] [CrossRef]
- Rainho, J.P.; Rocha, J.; Carlos, L.D.; Almeida, R.M. 29Si nuclear-magnetic-resonance and vibrational spectroscopy studies of SiO2–TiO2 powders prepared by the sol-gel process. J. Mater. Res. 2001, 16, 2369–2376. [Google Scholar] [CrossRef]
- Klinowski, J.; Carpenter, T.; Gladden, L. High-resolution solid-state NMR studies of temperature-induced phase transitions in silicalite (zeolite ZSM-5). Zeolites 1987, 7, 73–78. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the author (R.I.Y.-G.) upon request. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Na/Al | Si/Al | Br/Al |
|---|---|---|---|
| MOR-AS | 1.14 ± 0.03 | 8.4 ± 0.3 | 0.24 ± 0.05 |
| MOR-P | 0.17 ± 0.06 | 15.2 ± 1.3 | Not detected |
| MFI-AS | 1.11 ± 0.09 | 8.8 ± 0.4 | Not detected |
| MFI-P | 0.34 ± 0.05 | 14.8 ± 1.0 | Not detected |
| CTAB Carbon Site | δ (ppm) | TCH (ms) | T1ρ (ms) |
|---|---|---|---|
| CN | 54.5 | 2.21 ± 0.04 | 31 ± 13 |
| C1 | 67.3 | 0.37 ± 0.04 | 4.5 ± 0.5 |
| C4–13 | 30.5 | 0.98 ± 0.12 | 70 ± 30 |
| C16 | 14.6 | 2.8 ± 0.3 | - |
| Sample | ν0 (ppm) | Δν1/2 (ppm) | Sample | ν0 (ppm) | Δν1/2 (ppm) |
|---|---|---|---|---|---|
| MOR-AS | 54.6 ± 0.1 | 4.8 ± 0.1 | MFI-AS | 53.7 ± 0.1 | 4.9 ± 0.1 |
| MOR-T | 56.9 ± 0.1 | 5.6 ± 0.1 | MFI-T | 55.9 ± 0.1 | 4.9 ± 0.1 |
| MOR-TH | 56.3 ± 0.1 | 5.0 ± 0.1 | MFI-TH | 56.1 ± 0.1 | 4.7 ± 0.1 |
| MOR-P | 58.1 ± 0.1 2.6 ± 0.1 | 5.7 ± 0.1 4.2 ± 0.2 | MFI-P | 57.2 ± 0.1 1.6 ± 0.1 | 6.2 ± 0.1 8.5 ± 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shelyapina, M.G.; Yocupicio-Gaxiola, R.I.; Zhelezniak, I.V.; Chislov, M.V.; Antúnez-García, J.; Murrieta-Rico, F.N.; Galván, D.H.; Petranovskii, V.; Fuentes-Moyado, S. Local Structures of Two-Dimensional Zeolites—Mordenite and ZSM-5—Probed by Multinuclear NMR. Molecules 2020, 25, 4678. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204678
Shelyapina MG, Yocupicio-Gaxiola RI, Zhelezniak IV, Chislov MV, Antúnez-García J, Murrieta-Rico FN, Galván DH, Petranovskii V, Fuentes-Moyado S. Local Structures of Two-Dimensional Zeolites—Mordenite and ZSM-5—Probed by Multinuclear NMR. Molecules. 2020; 25(20):4678. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204678
Chicago/Turabian StyleShelyapina, Marina G., Rosario I. Yocupicio-Gaxiola, Iuliia V. Zhelezniak, Mikhail V. Chislov, Joel Antúnez-García, Fabian N. Murrieta-Rico, Donald Homero Galván, Vitalii Petranovskii, and Sergio Fuentes-Moyado. 2020. "Local Structures of Two-Dimensional Zeolites—Mordenite and ZSM-5—Probed by Multinuclear NMR" Molecules 25, no. 20: 4678. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25204678