
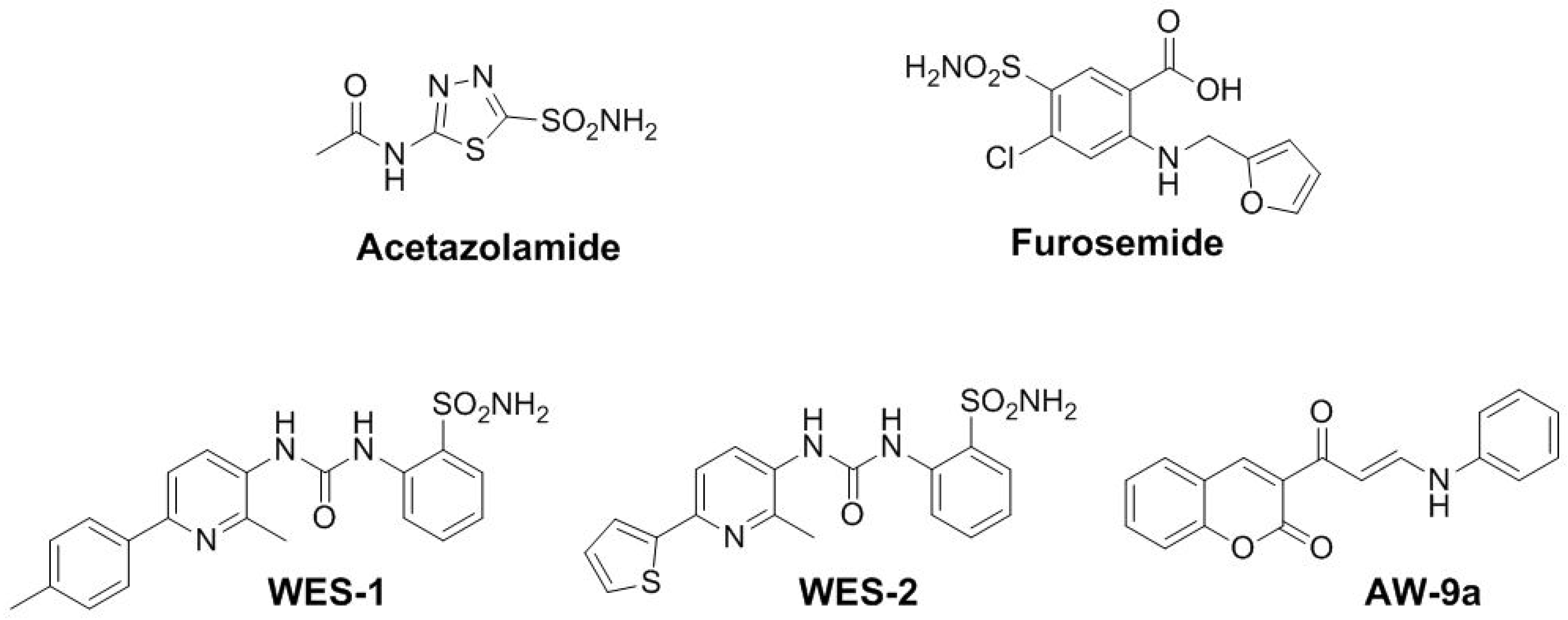
Development and Validation of High-Throughput Bioanalytical Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Method for the Quantification of Newly Synthesized Antitumor Carbonic Anhydrase Inhibitors in Human Plasma


 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Development and Optimization
2.2. Method Validation
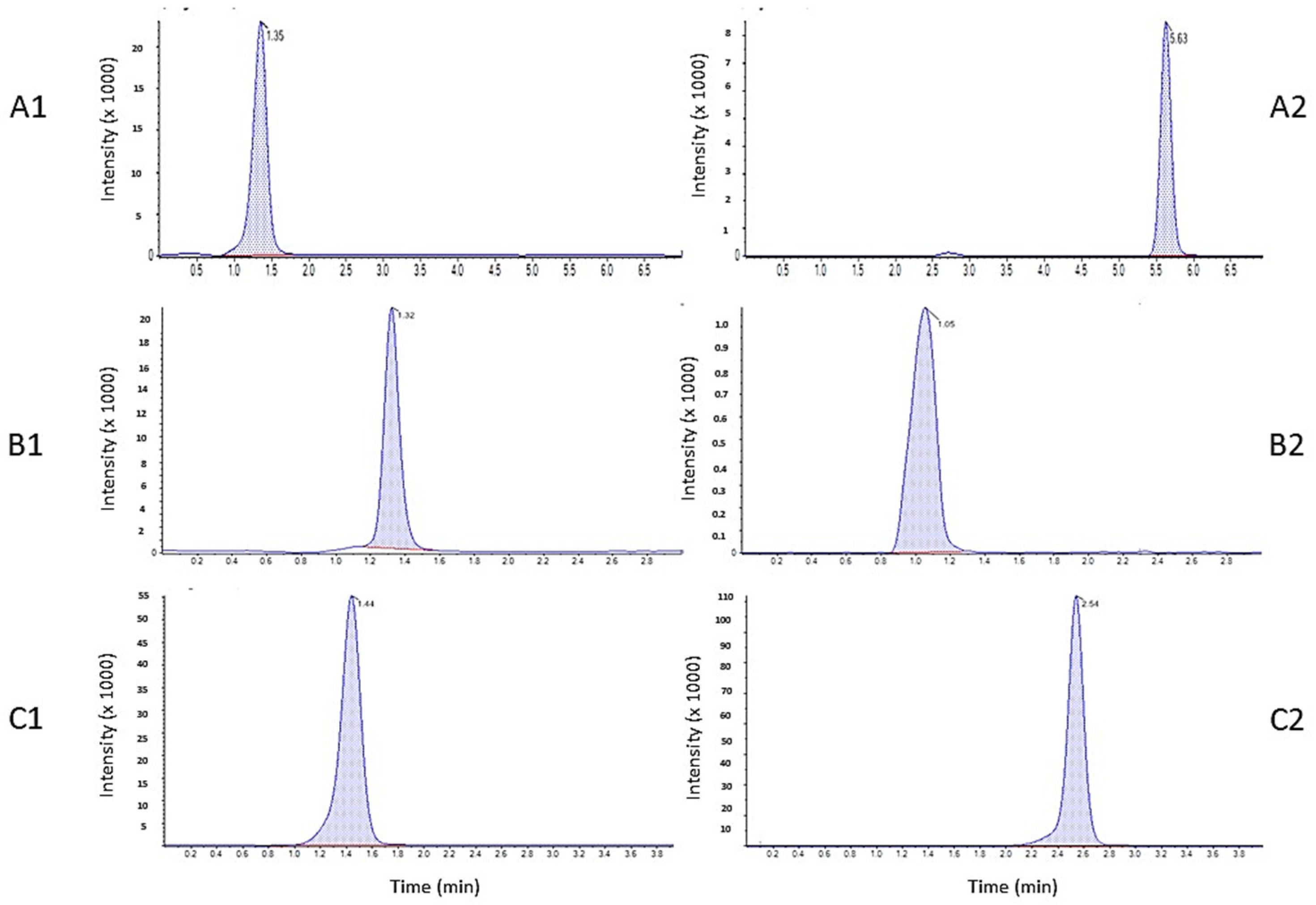
2.2.1. Selectivity
2.2.2. Calibration Curve and Quantitation Range
2.2.3. Accuracy and Precision
2.2.4. Extraction Recovery and Matrix Effect
2.2.5. Carry-Over
2.2.6. Stability
2.2.7. Dilution Integrity
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instruments
3.3. Mass Spectrometric Conditions
3.4. Preparation of Calibration Standards and Quality Control Samples
3.5. Preparation of Fortified Samples
3.6. Assay Validation
3.6.1. Specificity
3.6.2. Linearity
3.6.3. Accuracy and Precision
3.6.4. Matrix Effect and Extraction Recovery
3.6.5. Dilution Integrity
3.6.6. Stability Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Winum, J.-Y.; Scozzafava, A.; Montero, J.-L.; Supuran, C.T. Sulfamates and their therapeutic potential. Med. Res. Rev. 2004, 25, 186–228. [Google Scholar] [CrossRef] [PubMed]
- Temperini, C.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Activation and the Drug Design. Curr. Pharm. Des. 2008, 14, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; Baranauskiene, L.; Salzano, A.M.; Scaloni, A.; Matulis, D.; Innocenti, A.; Scozzafava, A.; Monti, S.M.; Di Fiore, A.; De Simone, G.; et al. Biochemical Characterization of CA IX, One of the Most Active Carbonic Anhydrase Isozymes. J. Biol. Chem. 2008, 283, 27799–27809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3674. [Google Scholar] [CrossRef] [PubMed]
- Le Duc, Y.; Licsandru, E.; Vullo, D.; Barboiu, M.; Supuran, C.T. Carbonic anhydrases activation with 3-amino-1H-1,2,4-triazole-1-carboxamides: Discovery of subnanomolar isoform II activators. Bioorganic Med. Chem. 2017, 25, 1681–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T.; Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Carta, F.; Monti, S.M.; De Simone, G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med. Res. Rev. 2018, 38, 1799–1836. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.A.; Capasso, C.; Supuran, C.T. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum—The eta-carbonic anhydrases. Bioorg. Med. Chem. Lett. 2014, 24, 4389–4396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J. Enzyme Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A.; Conway, J. Carbonic Anhydrase: Its Inhibitors and Activators; CRC Enzyme Inhibitors Series; CRC Press: Boca Raton, FL, USA, 2004; Volume 1. [Google Scholar]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Correction to Deciphering the Mechanism of Carbonic Anhydrase Inhibition with Coumarins and Thiocoumarins. J. Med. Chem. 2015, 58, 5689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomelino, C.L.; Supuran, C.T.; McKenna, R. Non-Classical Inhibition of Carbonic Anhydrase. Int. J. Mol. Sci. 2016, 17, 1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Al-Warhi, T.; Altyar, A.E.; Alkahtani, H.M.; Almehizia, A.A.; Abdel-Aziz, H.A. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J. Enzym. Inhib. Med. Chem. 2019, 34, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, N.; Wang, C.; Deng, S.; Sun, X.; Zhang, W.; Gao, W.; Zhao, D.; Lu, Y.; Chen, X. Development and Validation of a Simple and Reliable LC-MS/MS Method for the Determination of Acetazolamide, an Effective Carbonic Anhydrase Inhibitor, in Plasma and its Application to a Pharmacokinetic Study. Drug Res. 2014, 64, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narapusetti, A.; Bethanabhatla, S.S.; Sockalingam, A.; Pilli, N.R. LC–MS/MS assay for Acetazolamide, A Carbonic Anhydrase Inhibitor in Human Plasma and its Clinical Application. J. Young Pharm. 2015, 7, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Imaduwage, K.P.; Lakbub, J.; Go, E.P.; Desaire, H. Rapid LC-MS Based High-Throughput Screening Method, Affording No False Positives or False Negatives, Identifies a New Inhibitor for Carbonic Anhydrase. Sci. Rep. 2017, 7, 10324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menicatti, M.; Pallecchi, M.; Bua, S.; Vullo, D.; Mannelli, L.D.C.; Ghelardini, C.; Carta, F.; Supuran, C.T.; Bartolucci, G. Resolution of co-eluting isomers of anti-inflammatory drugs conjugated to carbonic anhydrase inhibitors from plasma in liquid chromatography by energy-resolved tandem mass spectrometry. J. Enzym. Inhib. Med. Chem. 2018, 33, 671–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constanzer, M.; Chavez, C.M.; Matuszewski, B.K. Low level determination of dorzolamide and its de-ethylated metabolite in human plasma by liquid chromatography with atmospheric pressure chemical ionization tandem mass spectrometry. J. Pharm. Biomed. Anal. 1997, 15, 1001–1008. [Google Scholar] [CrossRef]
- Gomha, S.M.; Zaki, Y.H.; Abdelhamid, A.O. Utility of 3-Acetyl-6-bromo-2H-chromen-2-one for the Synthesis of New Heterocycles as Potential Antiproliferative Agents. Molecules 2015, 20, 21826–21839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research and Center for Veterinary Medicine. Guidance for industry: Bioanalytical method validation. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf (accessed on 1 October 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intercept | Slope | R2 | LLOQ | LLOD | Linearity Range | ||||
|---|---|---|---|---|---|---|---|---|---|
| Average * | SD | Average * | SD | Average * | SD | (ng/mL) | (ng/mL) | ||
| AW-9a | 0.018 | 0.0116 | 0.011 | 0.006 | 0.9991 | 0.0002 | 1 | 0.03 | 1–1000 |
| WES-1 | 0.0014 | 0.0004 | 0.0015 | 0.001 | 0.9991 | 0.0005 | 2.5 | 1 | 2.5–800 |
| WES-2 | 0.053 | 0.0270 | 0.006 | 0.0003 | 0.992 | 0.006 | 5 | 1.5 | 5–500 |
| Analyte | Concentration | Intra-Day | Inter-Day | |||
|---|---|---|---|---|---|---|
| (ng/mL) | Recovery (%) | CV (%) | Recovery (%) | CV (%) | ||
| AW-9a | LLOQ | 1 | 103.5 | 5.35 | 99.20 | 4.55 |
| QCL | 3 | 93.3 | 6.40 | 94.44 | 5.17 | |
| QCM | 50 | 99.3 | 5.69 | 98.97 | 5.19 | |
| QCH | 800 | 100.4 | 8.44 | 99.71 | 7.61 | |
| WES-1 | LLOQ | 2.5 | 86.05 | 7.86 | 90.15 | 4.91 |
| QCL | 15 | 104.2 | 2.01 | 102.3 | 5.01 | |
| QCM | 200 | 98.40 | 1.24 | 99.72 | 2.31 | |
| QCH | 650 | 94.82 | 8.30 | 91.24 | 10.8 | |
| WES-2 | LLOQ | 5 | 94.32 | 4.35 | 93.87 | 4.1 |
| QCL | 15 | 99.38 | 2.66 | 97.90 | 5.18 | |
| QCM | 120 | 97.34 | 1.77 | 98.38 | 1.63 | |
| QCH | 400 | 96.81 | 1.15 | 97.04 | 2.91 | |
| N | 6 | 18 | ||||
| Analyte | Concentration | Extraction Recover (%) | Matrix Effect | |||
|---|---|---|---|---|---|---|
| (ng/mL) | Mean | RSD (%) * | CV (%) | RSD (%) | ||
| AW-9a | QCL | 3 | 82.31 | 12.2 | 7.65 | 5.13 |
| QCM | 50 | 89.91 | 4.20 | 7.17 | 7.11 | |
| QCH | 800 | 96.13 | 9.13 | 8.20 | 7.05 | |
| WES-1 | QCL | 15 | 98.74 | 5.60 | 6.31 | 6.10 |
| QCM | 200 | 96.70 | 3.50 | 5.22 | 6.15 | |
| QCH | 650 | 87.59 | 5.74 | 5.39 | 6.32 | |
| WES-2 | QCL | 15 | 92.77 | 6.29 | 2.66 | 6.15 |
| QCM | 120 | 83.61 | 8.00 | 0.71 | 5.57 | |
| QCH | 400 | 83.60 | 4.64 | 2.91 | 6.86 | |
| Analyte | Concentration (ng/mL) | Short Term Stability at Room Temperature (24 h) | Freeze and Thaw Stability at −80 °C (3 cycles) | Processed Sample Stability at 4 °C (72 h) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy (%) | CV (%) | Stability (%) | Accuracy (%) | CV (%) | Stability (%) | Accuracy (%) | CV (%) | Stability (%) | |||
| AW-9a | QCL | 3 | 96.76 | 4.67 | 89.28 | 90.92 | 2.89 | 86.28 | 102.79 | 13.99 | 98.85 |
| QCH | 800 | 100.81 | 7.06 | 94.15 | 105.94 | 6.71 | 101.32 | 100.20 | 4.73 | 95.81 | |
| WES-1 | QCL | 15 | 86.02 | 1.05 | 97.37 | 89.30 | 3.97 | 96.86 | 93.38 | 6.91 | 95.77 |
| QCH | 650 | 88.69 | 0.86 | 94.51 | 91.93 | 5.11 | 111.37 | 90.00 | 3.35 | 95.77 | |
| WES-2 | QCL | 15 | 89.63 | 4.10 | 91.22 | 87.92 | 4.09 | 91.95 | 84.58 | 3.87 | 97.29 |
| QCH | 400 | 94.12 | 1.32 | 99.96 | 95.33 | 2.08 | 98.69 | 92.27 | 6.48 | 101.13 | |
| N | 3 | 3 | 3 | ||||||||
| Analyte | Spiked Concentration (ng/mL) | Dilution Fold | Accuracy (%) | CV (%) |
|---|---|---|---|---|
| AW-9a | 800 | 1:2 | 98.16 | 8.75 |
| 1:4 | 95.17 | 7.36 | ||
| WES 1 | 650 | 1:2 | 89.54 | 2.50 |
| 1:4 | 89.67 | 1.90 | ||
| WES 2 | 400 | 1:2 | 95.65 | 2.83 |
| 1:4 | 99.66 | 6.44 |
| Analyte | Q1 a (m/z) | Q3 b (m/z) | DP c (V) | EP d (V) | CE e (V) | CXP f (V) |
|---|---|---|---|---|---|---|
| AW-9a | 291.978 | 173.037 | 81.0 | 10 | 40 | 16 |
| WES-1 | 396.979 | 225.140 | 80 | 10 | 90 | 8 |
| WES 2 | 388.930 | 217.056 | 111 | 10 | 35 | 8.0 |
| IS | 146.941 | 91.030 | 70.0 | 10 | 35 | 12 |
Sample Availability: not available. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Megied, A.M.; Eldehna, W.M.; Abdelrahman, M.A.; Elbarbry, F.A. Development and Validation of High-Throughput Bioanalytical Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Method for the Quantification of Newly Synthesized Antitumor Carbonic Anhydrase Inhibitors in Human Plasma. Molecules 2020, 25, 5753. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235753
Abdel-Megied AM, Eldehna WM, Abdelrahman MA, Elbarbry FA. Development and Validation of High-Throughput Bioanalytical Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Method for the Quantification of Newly Synthesized Antitumor Carbonic Anhydrase Inhibitors in Human Plasma. Molecules. 2020; 25(23):5753. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235753
Chicago/Turabian StyleAbdel-Megied, Ahmed M., Wagdy M. Eldehna, Mohamed A. Abdelrahman, and Fawzy A. Elbarbry. 2020. "Development and Validation of High-Throughput Bioanalytical Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Method for the Quantification of Newly Synthesized Antitumor Carbonic Anhydrase Inhibitors in Human Plasma" Molecules 25, no. 23: 5753. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25235753