Process Development and Synthesis of Process-Related Impurities of an Efficient Scale-Up Preparation of 5,2′-Dibromo-2,4′,5′-Trihydroxy Diphenylmethanone as a New Acute Pyelonephritis Candidate Drug

Abstract
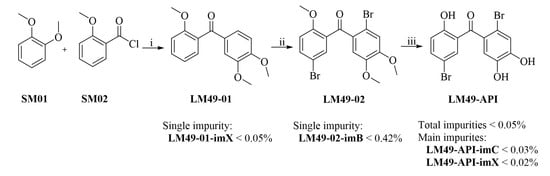
:
1. Introduction
2. Results and Discussion
2.1. Process Optimization
2.1.1. Friedel–Crafts Acylation Step
2.1.2. Bromination Reaction Step
2.1.3. Demethylation Reaction Step
2.2. Process Verification
2.2.1. Process Verification of Friedel–Crafts Acylation Step
2.2.2. Process Verification of Bromination Step
2.2.3. Process Verification of Demethylation Step
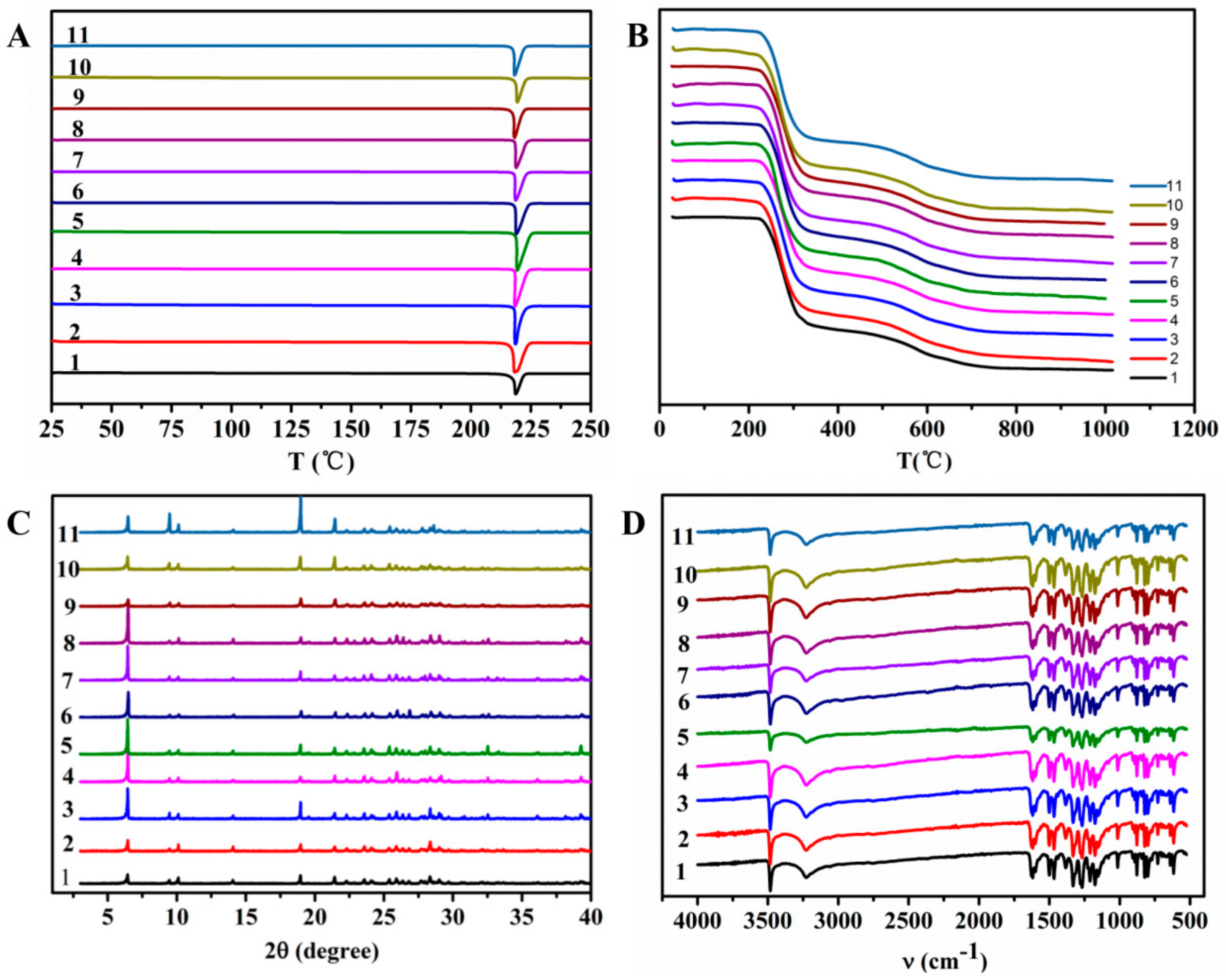
2.3. Polymorph Investigation of LM49-API
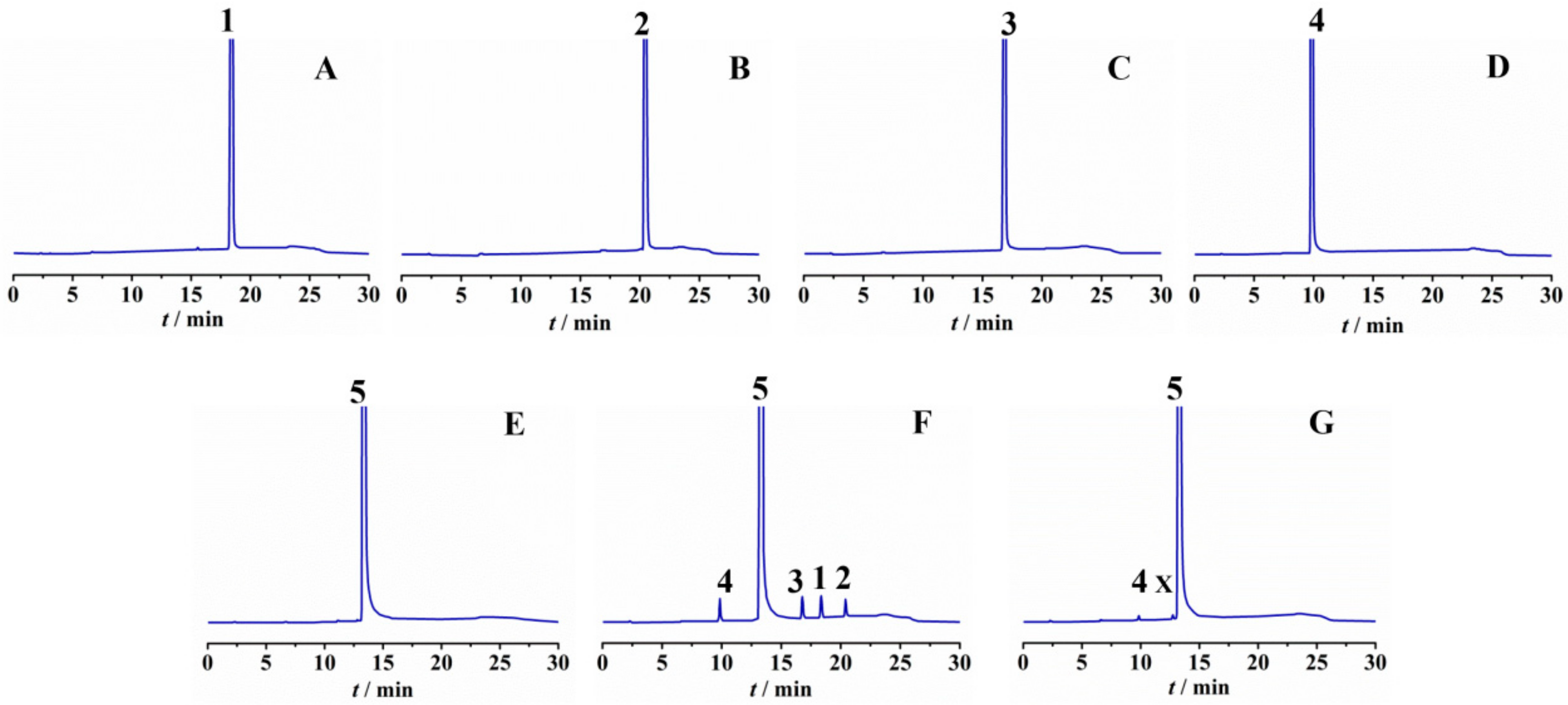
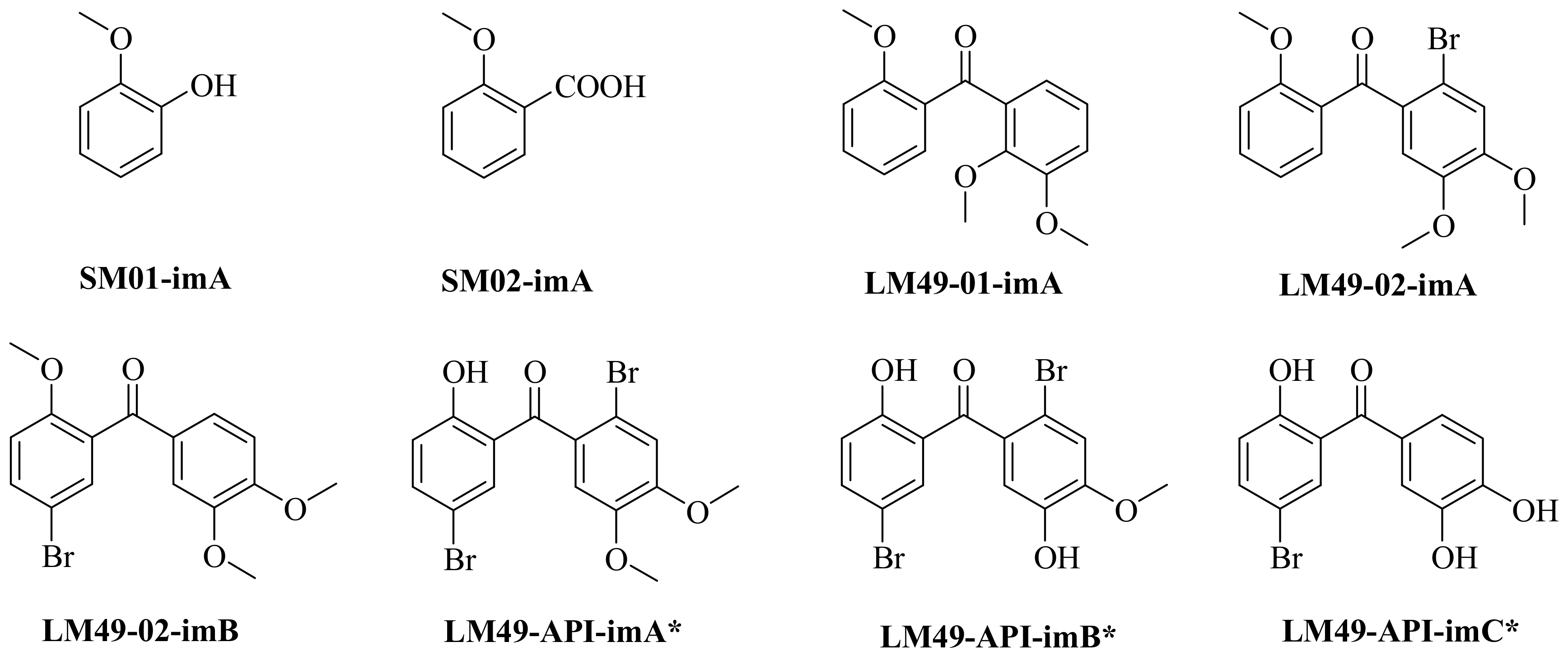
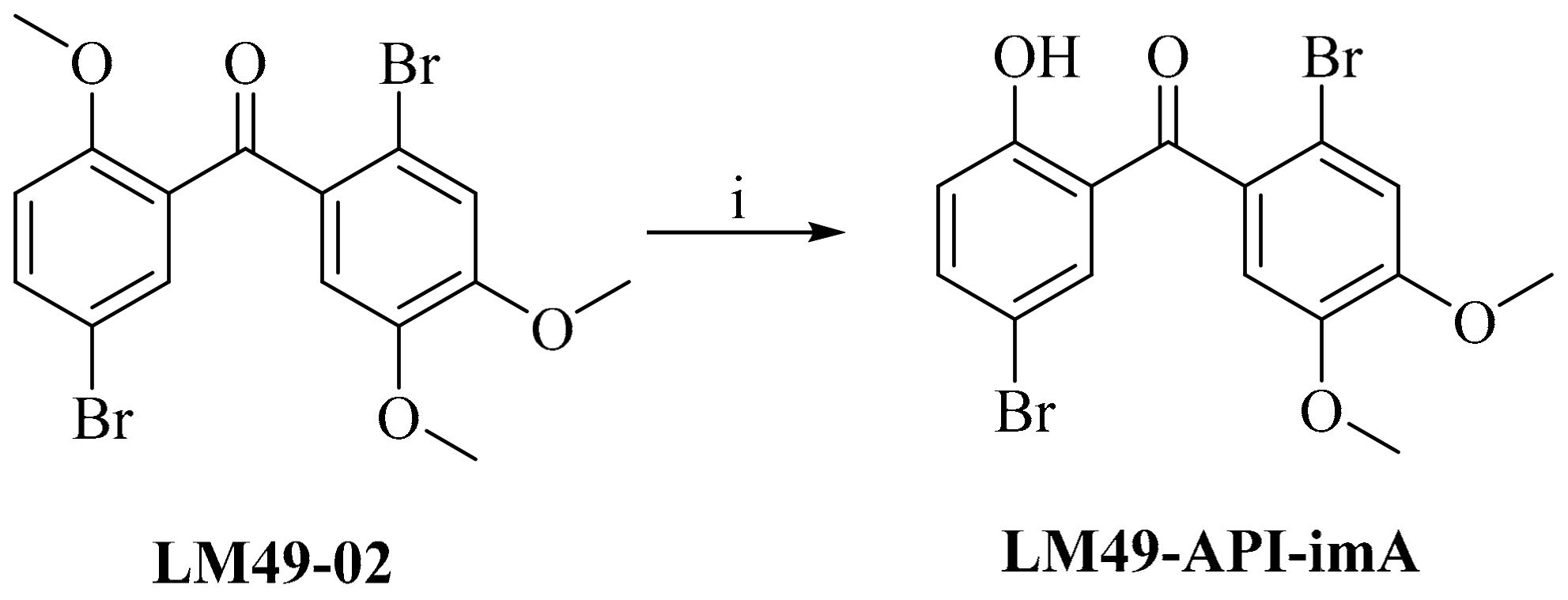
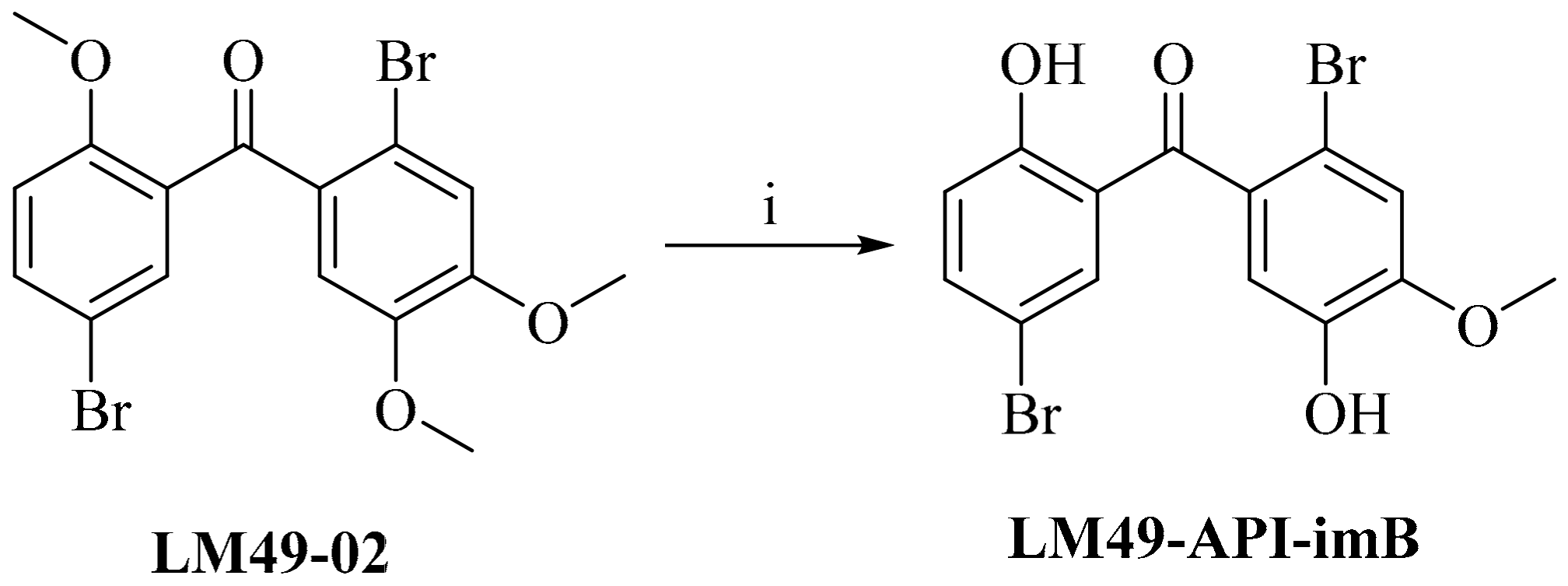
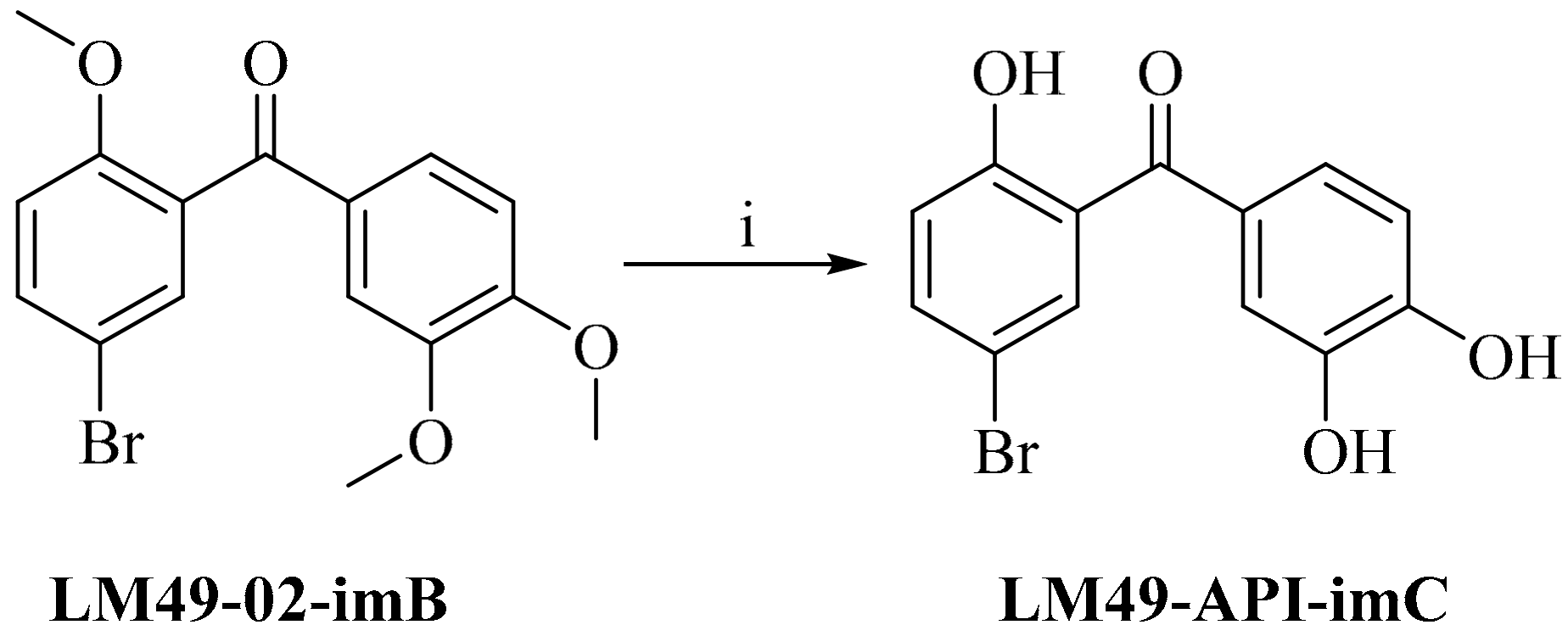
2.4. Impurity Synthesis
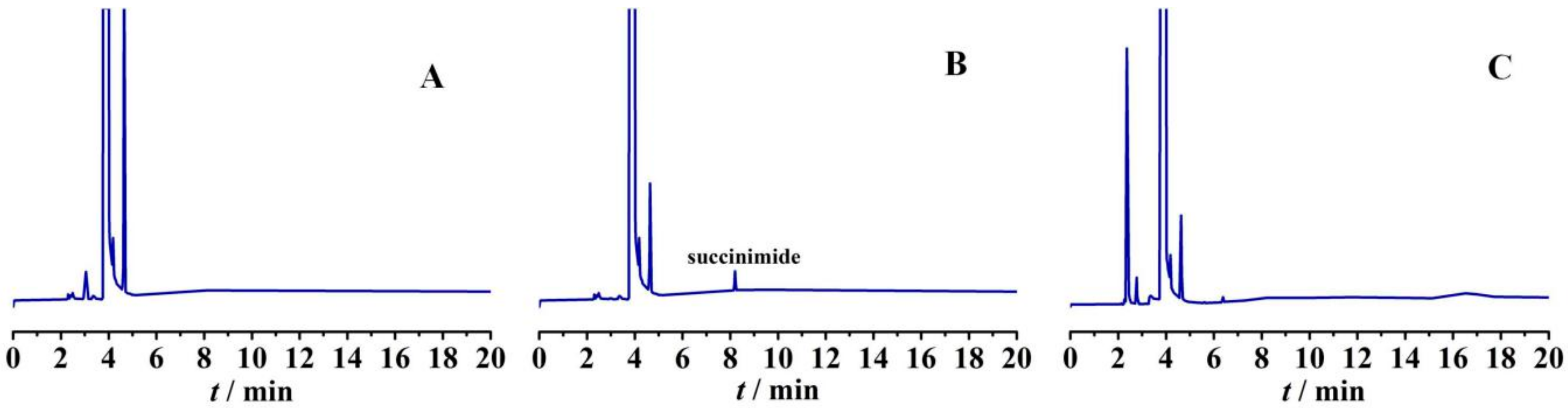
2.4.1. Impurity Analysis
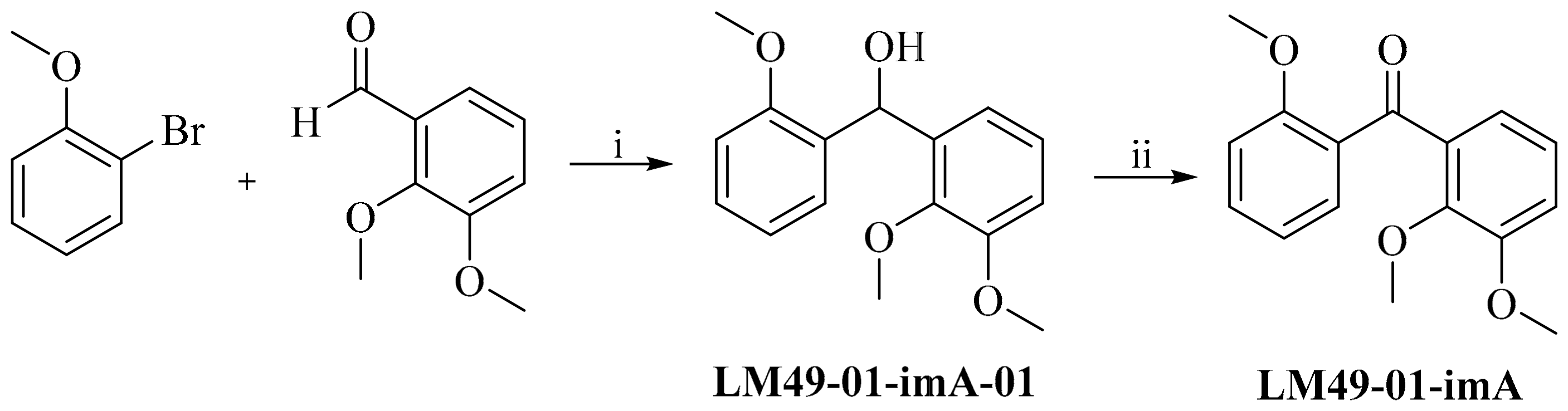
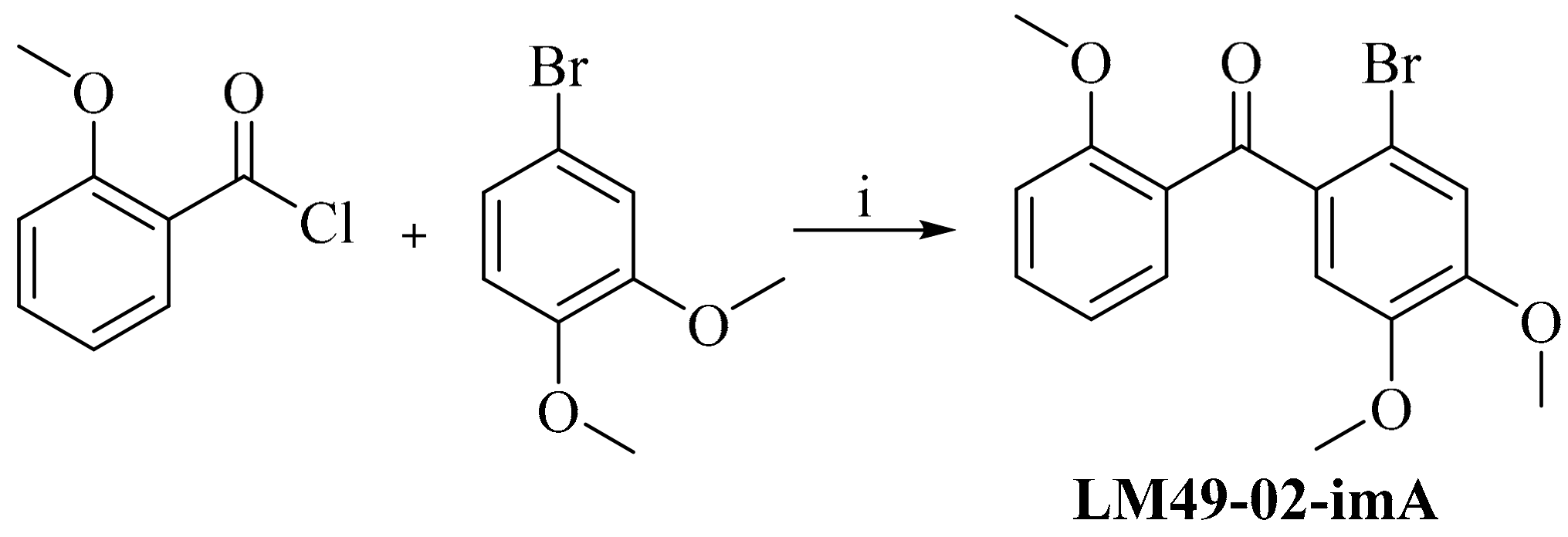
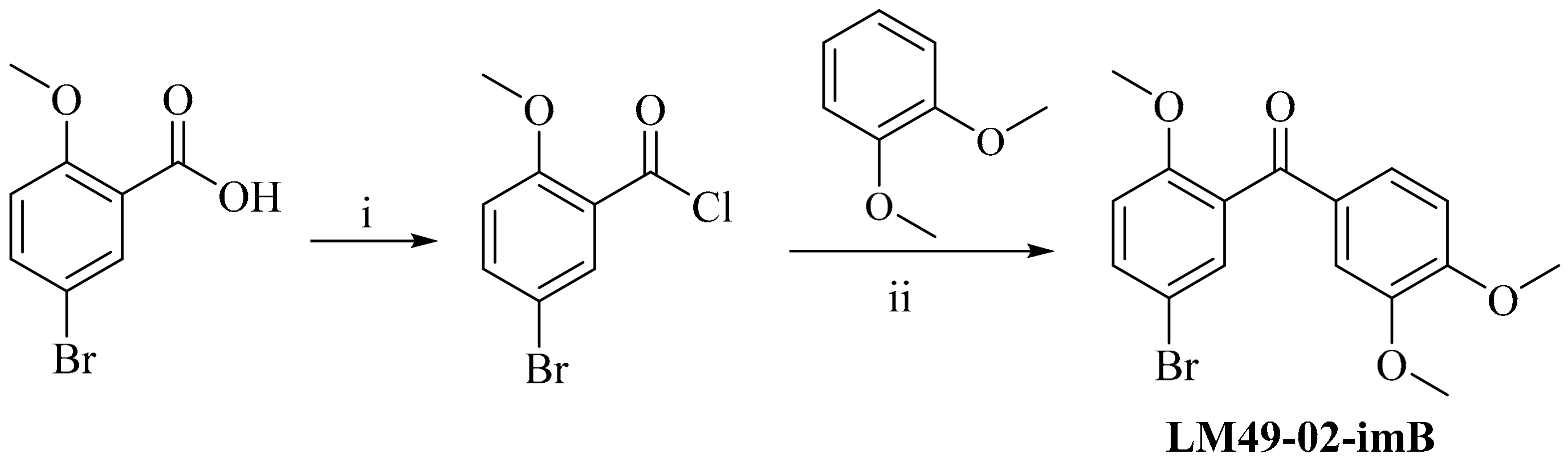
2.4.2. Preparation of Impurities
3. Experimental Section
3.1. Instruments and Reagents
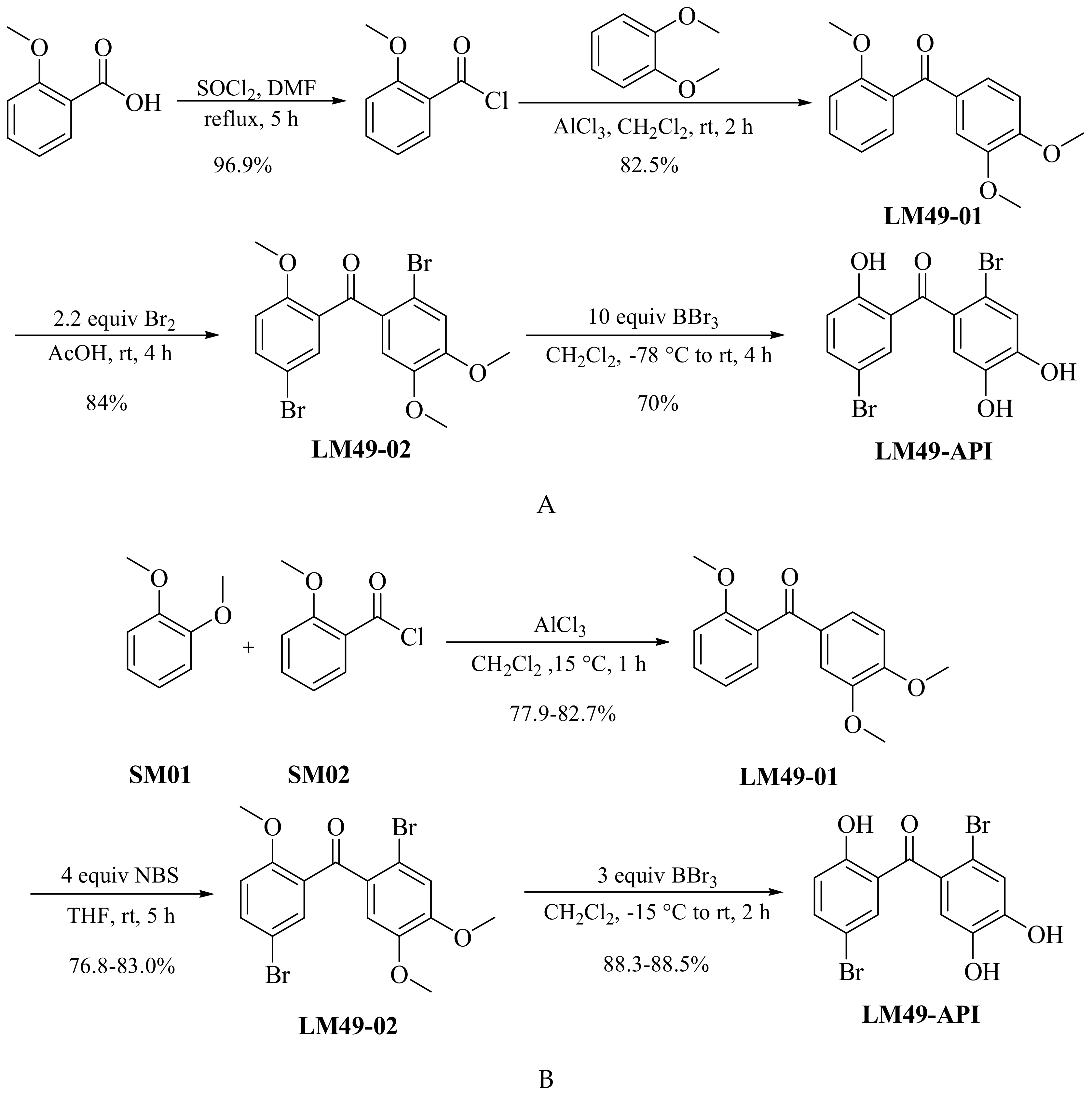
3.2. Improved Prepared Process of LM49-API
3.2.1. Preparation of Intermediate 2,4′5′-Trimethoxybenzophenone (LM49-01)
3.2.2. Preparation of Key Intermediate 5,2′-Dibromo-2,4′5′-Trimethoxybenzophenone (LM49-02)
3.2.3. Preparation of the Target Compound (LM49-API)
3.3. Polymorphs Investigation of LM49-API
3.4. Preparation of Process-Related Impurities
3.4.1. 2,2′3′-Trimethoxylbenzophenone (LM49-01-imA)
3.4.2. 2′-Bromo-2,4′,5′-Trimethoxylbenzophenone (LM49-02-imA)
3.4.3. 5-Bromo-2,3′,4′-Trimethoxylbenzophenone (LM49-02-imB)
3.4.4. 5,2′-Dibromo-2-Hydroxyl-4′,5′-Dimethoxylbenzophenone (LM49-API-imA)
3.4.5. 5,2′-Dibromo-2,5′-Dihydroxyl-4′-Methoxylbenzophenone (LM49-API-imB)
3.4.6. 5-Bromo-2,3′,4′-Trihydroxylbenzophenone (LM49-API-imC)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Czaja, C.A.; Scholes, D.; Hooton, T.M.; Stamm, W.E. Population-Based Epidemiologic Analysis of Acute Pyelonephritis. Clin. Infect. Dis. 2007, 45, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Cattrall, J.W.S.; Asín-Prieto, E.; Freeman, J.; Trocóniz, I.F.; Kirby, A. A Pharmacokinetic-pharmacodynamic Assessment of Oral Antibiotics for Pyelonephritis. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 2311–2321. [Google Scholar] [CrossRef] [Green Version]
- Wagenlehner, F.M.; Sobel, J.D.; Newell, P.; Armstrong, J.; Huang, X.G.; Stone, G.G.; Yates, K.; Gasink, L.B. Ceftazidime-avibactam Versus Doripenem for the Treatment of Complicated Urinary Tract Infections, Including Acute Pyelonephritis: RECAPTURE, a Phase 3 Randomized Trial Program. Clin. Infect. Dis. 2016, 63, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Hooton, T.M.; Naber, K.G.; Wullt, B.; Colgan, R.; Miller, L.G.; Moran, G.J.; Nicolle, L.E.; Raz, R.; Schaeffer, A.J.; et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin. Infect. Dis. 2011, 52, 103–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Y.; Feng, X.E.; Ban, S.R.; Lin, W.H.; Li, Q.S. Synthesis and Biological Activity of Halophenols as Potent Antioxidant and Cytoprotective Agents. Bioorg. Med. Chem. Lett. 2010, 20, 4132–4134. [Google Scholar] [CrossRef]
- Feng, X.E.; Liang, T.G.; Gao, J.; Kong, D.P.; Ge, R.; Li, Q.S. Heme Oxygenase-1, a Key Enzyme for the Cytoprotective Actions of Halophenols by Upregulating Nrf2 Expression via Activating Erk1/2 and PI3K/Akt in EA.hy926 Cells. Oxid. Med. Cell. Longev. 2017, 7028478. [Google Scholar] [CrossRef] [PubMed]
- Li, J.G.; Feng, X.E.; Ge, R.; Li, J.K.; Li, Q.S. Protective Effect of 2,4′,5′-Trihydroxyl-5,2′-dibromo diphenylmethanone, a New Halophenol, Against Hydrogen Peroxide-Induced EA.hy926 Cells Injury. Molecules 2015, 20, 14254–14264. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.X.; Feng, X.E.; Liu, E.L.; Ge, R.; Zhang, Y.L.; Xiao, B.G.; Li, Q.S. 5,2′-Dibromo-2,4′,5′-trihydroxydiphenylmethanone Attenuates LPS-induced Inflammation and ROS Production in EA.hy926 Cells via HMBOX1 Induction. J. Cell. Mol. Med. 2019, 23, 453–463. [Google Scholar] [CrossRef]
- Yang, F.; Cai, H.H.; Feng, X.E.; Zhang, Y.L.; Ge, R.; Xiao, B.G.; Li, Q.S. 5,2′-Dibromo-2,4,5-trihydroxydiphenylmethanone, a Novel Immunomodulator of T lymphocytes by Regulating the CD4+ T Cell Subset Balance via Activating the Mitogen-activated Protein Kinase Pathway. Int. Immunopharmacol. 2019, 72, 487–495. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Feng, X.E.; Chang, R.R.; Zhang, L.H.; Xiao, B.G.; Li, Q.S.; Hao, X.L. Therapeutic Effects of 5,2′-Dibromo-2,4′,5′-trihydroxydiphenylmethanone (LM49) in an Experimental Rat Model of Acute Pyelonephritis by Immunomodulation and Anti-inflammation. Int. Immunopharmacol. 2018, 62, 155–164. [Google Scholar] [CrossRef]
- Zhou, A.N.; Chen, X.W.; Qi, Y.L.; Duan, G.L.; Li, J.Q. Process Development of an Efficient Kilogram-Scale Preparation of a Preferential Dopamine D3 Versus D2 Receptor Antagonist SIPI 6398 as a New Antipsychotic Candidate. Org. Process. Res. Dev. 2019, 23, 1442–1449. [Google Scholar] [CrossRef]
- Wada, Y.; Matsumoto, A.; Asano, K.; Matsubara, S. Enantioselective Bromination of Axially Chiral Cyanoarenes in the Presence of Bifunctional Organocatalysts. RSC Adv. 2019, 9, 31654–31658. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.G.; Du, M.F.; Xia, W.; Hu, J.L. One-pot Synthesis of α,β-Dehydroamino Derivatives From β, β-Dicyanostyrene With 1,3-Dibromo-5,5-dimethylhydantoin Promoted by Mild Base. Chem. Res. Chin. Univ. 2016, 32, 68–75. [Google Scholar] [CrossRef]
- Tan, X.Y.; Pan, H.J.; Tian, H.; Shi, Y. Phosphine oxide-Sc(OTf)3 Catalyzed Enantioselective Bromoaminocyclization of Tri-substituted Allyl N-tosylcarbamates. Sci. China Chem. 2018, 61, 656–659. [Google Scholar] [CrossRef]
- Bogacheva, A.M.; Yarovenko, V.N.; Levchenko, K.S.; Kobeleva, O.I.; Valova, T.M.; Barachevsky, V.A.; Struchkova, M.I.; Shmelin, P.S.; Krayushkin, M.M.; Charushin, V.N. A Convenient Method for the Preparation of Mono- and Bis-substituted Photochromic Bis(benzothienyl)perfluorocyclopentenes via Regioselective Friedel–Crafts Acylation. Tetrahedron. Lett. 2012, 53, 5948–5951. [Google Scholar] [CrossRef]
- Cano, M.; Rojas, C.; Hidalgo, W.; Sáez, J.; Gil, J.; Schneider, B. Improved synthesis of 4-phenylphenalenones: The case of isoanigorufone and structural analogs. Tetrahedron. Lett. 2013, 54, 351–354. [Google Scholar] [CrossRef]
- Ruffell, J.E.; Farmer, T.J.; Macquarrie, D.J.; Stark, M.S. The Autoxidation of Alkenyl Succinimides-Mimics for Polyisobutenyl Succinimide Dispersants. Ind. Eng. Chem. Res. 2019, 58, 19649–19660. [Google Scholar] [CrossRef]
- Hu, Y.; Erxleben, A.; Ryder, A.G.; McArdle, P. Quantitative Analysis of Sulfathiazole Polymorphs in Ternary Mixtures by Attenuated Total Reflectance Infrared, Near-infrared and Raman spectroscopy. J. Pharm. Biomed. Anal. 2010, 53, 412–420. [Google Scholar] [CrossRef]
- Deng, J.; Staufenbiel, S.; Bodmeier, R. Evaluation of a Biphasic in Vitro Dissolution Test for Estimating the Bioavailability of Carbamazepine Polymorphic Forms. Eur. J. Pharm. Sci. 2017, 105, 64–70. [Google Scholar] [CrossRef]
- Wardhana, Y.W.; Soewandhi, S.N.; Wikarsa, S.; Suendo, V. Polymorphic Properties and Dissolution Profile of Efavirenz due to Solvents Recrystallization. Pak. J. Pharm. Sci. 2019, 32, 981–986. [Google Scholar]
- Antony, P.; Sundaram, S.J.; Ramaclus, J.V.; Inglebert, S.A.; Raj, A.A.; Dominique, S.; Hegde, T.A.; Vinitha, G.; Sagayaraj, P. Synthesis, Growth, Crystal Structure, Thermal, Linear and Nonlinear Opticalanalysis of New Extended p-conjugated Organic Material Based on Methyl Pyridinium Compound of 4-(4-(4-(dimethylamino) phenyl) buta-1,3-dienyl)-1-methylpyridinium p-styrenesulfonate Hydrate. J. Mol. Struct. 2019, 1196, 699–706. [Google Scholar]
- De la Fuente, J.L.; Ruiz-Bermejo, M.; Menor-Salván, C.; Osuna-Esteban, S. Thermal Characterization of HCN Polymers by TG–MS, TG, DTA and DSC Methods. Polym. Degrad. Stabil. 2011, 96, 943–948. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, H.W.; Wu, J.M.; Shi, Y.Q.; Yang, L.H. Polymorphs of Clopidogrel Bisulfate. Acta Pharm. Sin. 2013, 48, 1358–1360. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry # | SM01 (g) | Solvent Amount (mL) | SM01/SM02 * | SM01/AlCl3 * | Temp. (°C) | Yield (%) |
|---|---|---|---|---|---|---|
| Solvent Amount | 5.0 | 25 | 1:1.05 | 1:1 | 15 ± 5 | 98.5 a |
| 5.0 | 50 | 1:1.05 | 1:1 | 15 ± 5 | 98.8 a | |
| 5.0 | 75 | 1:1.05 | 1:1 | 15 ± 5 | 98.3 a | |
| SM01/SM02 | 5.0 | 50 | 1:1 | 1:1 | 15 ± 5 | 96.7 a |
| 5.0 | 50 | 1:1.05 | 1:1 | 15 ± 5 | 98.8 a | |
| 5.0 | 50 | 1:1.1 | 1:1 | 15 ± 5 | 98.7 a | |
| 5.0 | 50 | 1:1.2 | 1:1 | 15 ± 5 | 98.5 a | |
| SM01/AlCl3 | 5.0 | 50 | 1:1.05 | 1:0.75 | 15 ± 5 | 74.7 a |
| 5.0 | 50 | 1:1.05 | 1:1 | 15 ± 5 | 98.8 a | |
| 5.0 | 50 | 1:1.05 | 1:1.5 | 15 ± 5 | 98.5 a | |
| Temp. | 5.0 | 50 | 1:1.05 | 1:1 | 0 ± 5 | 97.9 a |
| 5.0 | 50 | 1:1.05 | 1:1 | 15 ± 5 | 98.8 a | |
| 5.0 | 50 | 1:1.05 | 1:1 | 30 ± 5 | 97.3 a | |
| Time | 5.0 | 50 | 1:1.05 | 1:1 | 15 ± 5 | 98.8 a |
| 5.0 | 50 | 1:1.05 | 1:1 | 0 ± 5 | 98.9 b | |
| 5.0 | 50 | 1:1.05 | 1:1 | 15 ± 5 | 98.3 b |
| Entry # | LM49-01 (g) | Amount (V(mL)/V(mL)) | Yield (%) | Appearance | Purity (%) * | Maximal Impurity LM49-01-imX (%) * |
|---|---|---|---|---|---|---|
| a | 97.66 | 1.45 | ||||
| CH2Cl2/n-hexane | 10.0 | 10/100 | - | - | - | - |
| 10.0 | 10/200 | 80 | white crystal powder | 99.90 | 0.10 | |
| 10.0 | 10/300 | 78 | white block solid | 99.47 | 0.53 | |
| 10.0 | 20/200 | - | - | - | - | |
| 10.0 | 20/300 | 69 | white crystal powder | 99.89 | 0.11 | |
| 10.0 | 20/400 | 52 | white crystal powder | 99.78 | 0.22 | |
| ethyl acetate/n-hexane | 10.0 | 50/50 | - | - | - | - |
| 10.0 | 50/100 | - | - | - | - | |
| 10.0 | 50/150 | 25 | white crystal powder | 98.82 | 1.01 | |
| 10.0 | 50/200 | 31 | white crystal powder | 99.84 | 0.06 | |
| 10.0 | 50/300 | 40 | white crystal powder | 99.85 | 0.09 | |
| 10.0 | 50/500 | 48 | white block solid | 99.89 | 0.11 | |
| 10.0 | 30/90 | 44 | white crystal powder | 99.80 | 0.09 | |
| 10.0 | 40/120 | 49 | white crystal powder | 99.79 | 0.10 |
| Entry # | LM49-01 (g) | Reagent | Solvent/Amount (mL) | LM49-01/Reagent * | Temp. (°C) | Yield (%) |
|---|---|---|---|---|---|---|
| Reagent | 1.0 | NBS | THF/20 | 1:12 | 20 ± 5 | 90.6 a |
| 1.0 | DBDMH | THF/20 | 1:5 | 20 ± 5 | 87.6 b | |
| 1.0 | PyBr3 | MeOH/20 | 1:10 | reflux | 86.3 b | |
| Solvent | 1.0 | NBS | MeOH/20 | 1:12 | 20 ± 5 | 69.5 b |
| 1.0 | NBS | DMF/20 | 1:12 | 20 ± 5 | 47.4 b | |
| 1.0 | NBS | CH2Cl2/20 | 1:12 | 20 ± 5 | 42.9 b | |
| 1.0 | NBS | HOAc/20 | 1:12 | 20 ± 5 | 67.6 b | |
| 1.0 | NBS | THF/20 | 1:12 | 20 ± 5 | 90.6 a | |
| LM49-01/Reagent | 5.0 | NBS | THF/25 | 1:2.5 | 20 ± 5 | 49.7 b |
| 5.0 | NBS | THF/25 | 1:3 | 20 ± 5 | 64.2 b | |
| 5.0 | NBS | THF/25 | 1:3.5 | 20 ± 5 | 78.4 b | |
| 5.0 | NBS | THF/25 | 1:4 | 20 ± 5 | 91.3 a | |
| Temp. | 5.0 | NBS | THF/25 | 1:4 | 0 ± 5 | 45.3 b |
| 5.0 | NBS | THF/25 | 1:4 | 20 ± 5 | 91.3 a | |
| 5.0 | NBS | THF/25 | 1:4 | reflux | 75.5 b | |
| Time | 5.0 | NBS | THF/25 | 1:4 | 20 ± 5 | 69.5 c |
| 5.0 | NBS | THF/25 | 1:4 | 20 ± 5 | 91.3 a | |
| 5.0 | NBS | THF/25 | 1:4 | 20 ± 5 | 91.5 b |
| LM49-02 (g) | Crystallization Temp. (°C) | Method | Amount (mL) | Yield (%) | Purity (%) * | Maximal Impurity LM49-02-imB (%) * |
|---|---|---|---|---|---|---|
| a | 98.39 | 1.61 | ||||
| b | 96.16 | 3.84 | ||||
| 5.0 a | 25 | standing | 150 c | 78.0 | 99.71 | 0.29 |
| 5.0 a | 15 | standing | 150 c | 86.0 | 99.73 | 0.27 |
| 5.0 a | 25 | stirring | 150 c | 84.0 | 99.93 | 0.07 |
| 5.0 a | 15 | stirring | 150 c | 88.0 | 99.87 | 0.13 |
| 5.0 a | 25 | standing | 150 d | 66.0 | 99.74 | 0.26 |
| 5.0 a | 15 | standing | 150 d | 86.0 | 99.57 | 0.43 |
| 5.0 a | 25 | stirring | 150 d | 82.0 | 99.94 | 0.06 |
| 5.0 a | 15 | stirring | 150 d | 88.0 | 99.69 | 0.31 |
| 5.0 b | 25 | stirring | 50 d | 91.0 | 99.84 | 0.16 |
| 5.0 b | 25 | stirring | 75 d | 89.6 | 99.69 | 0.31 |
| 5.0 b | 25 | stirring | 100 d | 87.6 | 99.78 | 0.22 |
| 5.0 b | 25 | stirring | 125 d | 84.8 | 99.79 | 0.21 |
| 5.0 b | 25 | stirring | 150 d | 82.5 | 99.80 | 0.20 |
| Entry # | LM49-02 (g) | LM49-02/BBr3 * | Temp. (°C) | Yield (%) |
|---|---|---|---|---|
| LM49-02/BBr3 | 4.0 | 1:2 | −15 ± 5 | 95.8 a |
| 4.0 | 1:3 | −15 ± 5 | 93.9 a | |
| 4.0 | 1:4 | −15 ± 5 | 93.9 a | |
| 4.0 | 1:5 | −15 ± 5 | 93.4 a | |
| Temp. | 5.0 | 1:3 | 15 ± 5 | 94.7 a |
| 5.0 | 1:3 | 0 ± 5 | 94.5 a | |
| 5.0 | 1:3 | −15 ± 5 | 95.1 a | |
| 5.0 | 1:3 | −25 ± 5 | 95.8 a | |
| Time | 5.0 | 1:3 | −15 ± 5 | 96.7 b |
| 5.0 | 1:3 | −15 ± 5 | 97.6 a | |
| 5.0 | 1:3 | −15 ± 5 | 98.2 c |
| LM49-API (g) | Solvent/Amount (mL) | Yield (%) | Appearance | Purity (%) * | Maximal Impurity LM49-API-imC (%) * |
|---|---|---|---|---|---|
| a | 99.72 | 0.22 | |||
| 3.0 a | MeOH/6.0 | 42.0 | light yellow crystal powder | 99.93 | 0.05 |
| 3.0 a | Ethyl Acetate/12 | 8.7 | Saffron crystal | 99.79 | 0.15 |
| 3.0 a | EtOH/4.8 | 59.3 | Light yellow crystal powder | 99.92 | 0.06 |
| 3.0 a | Isopropanol/6.5 | 66.7 | Light yellow crystal powder | 99.86 | 0.09 |
| 3.0 a | Isopropyl Acetate/10.0 | 56.0 | Saffron crystal | 99.84 | 0.05 |
| 3.0 a | MeOH + H2O/12 + 6 | 80.7 | Light yellow crystal powder | 99.85 | 0.11 |
| 3.0 a | EtOH + H2O/12 + 6 | 81.5 | Light yellow crystal powder | 99.90 | 0.06 |
| 3.0 a | EtOH + H2O/12 + 12 | 88.7 | Light yellow crystal powder | 99.91 | 0.08 |
| 3.0 a | EtOH + H2O/15 + 15 | 82.6 | Light yellow crystal powder | 99.85 | 0.12 |
| 3.0 a | EtOH + H2O/18 + 18 | 76.0 | Light yellow crystal powder | 99.90 | 0.08 |
| Batch No. a | SM01 (g)/SM02 (g) | Product | Output (g) | Crude Product Yield (%) | Recrystallization Yield (%) | Yield (%) | Purity (%) * | Total Impurities (%) * |
|---|---|---|---|---|---|---|---|---|
| 1st | 300/419 | Crude product | 602 | 101.5 | 76.7 | 77.9 | 96.68 | 3.32 |
| Purified product | 461 | 99.97 | 0.03 | |||||
| 2nd | 300/419 | Crude product | 601 | 101.4 | 77.5 | 78.6 | 96.13 | 3.87 |
| Purified product | 465 | 99.96 | 0.04 | |||||
| 3rd | 300/419 | Crude product | 605 | 102.2 | 80.1 | 82.7 | 96.67 | 3.33 |
| Purified product | 490 | 99.95 | 0.05 |
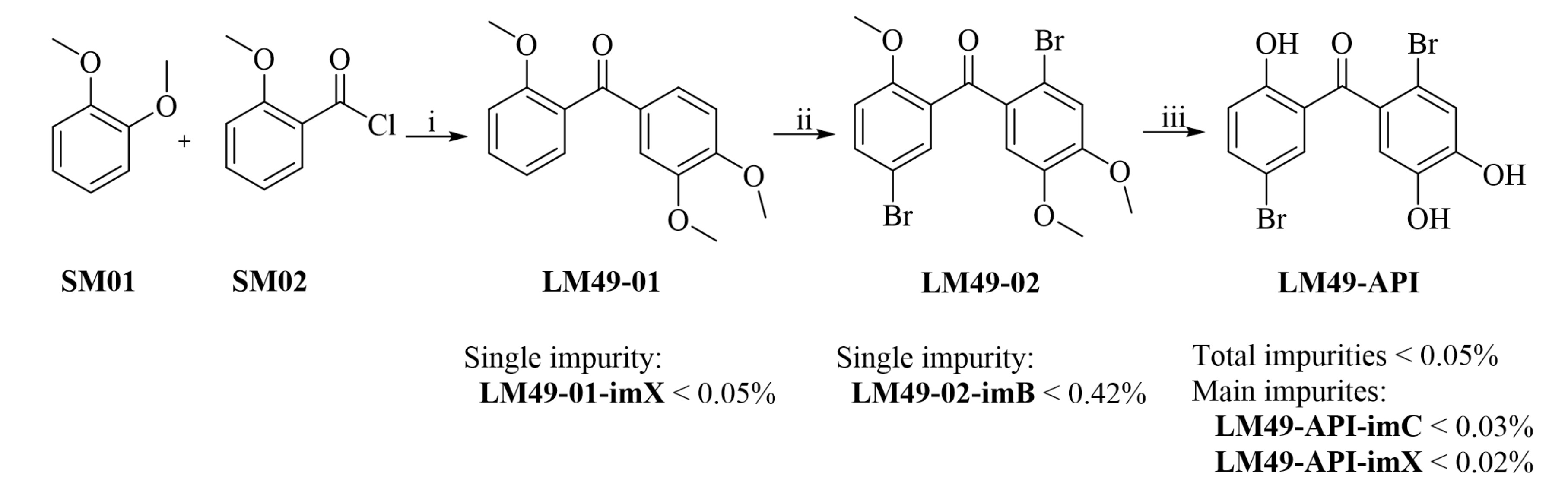
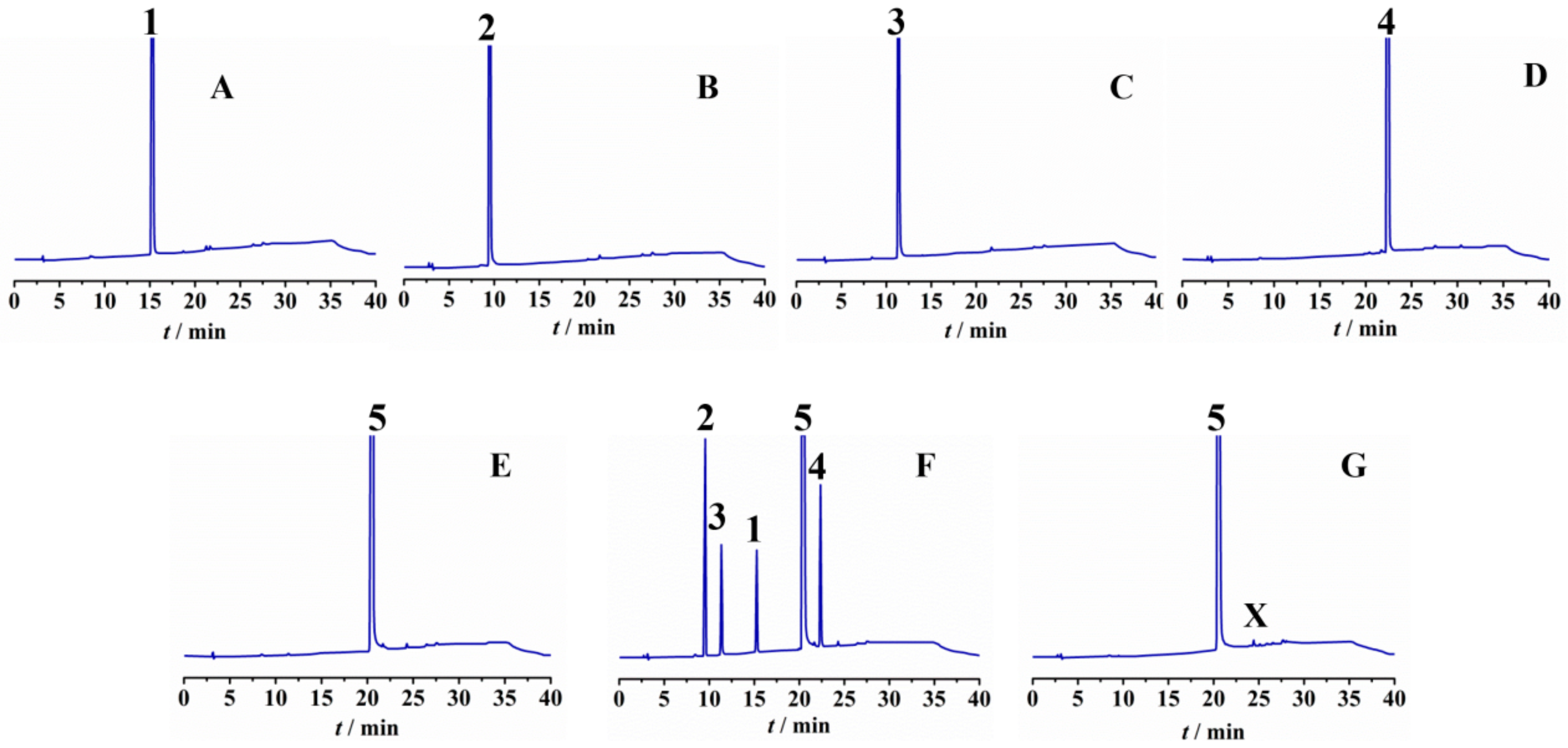
| Batch No. | SM01 (tR = 15.3) (%) * | SM02-imA (tR = 9.5) (%) * | SM01-imA (tR = 11.4) (%) * | LM49-01-imA (tR = 22.4) (%) * | LM49-01-imX (tR = 24.4) (%) * |
|---|---|---|---|---|---|
| 1st | - | - | - | - | 0.03 |
| 2nd | - | - | - | - | 0.04 |
| 3rd | - | - | - | - | 0.05 |
| Batch No. a | LM49-01 (g)/NBS(g) | Product | Output (g) | Crude Product Yield (%) | Recrystallization Yield (%) | Yield (%) | Purity (%) * | Total Impurities (%) * |
|---|---|---|---|---|---|---|---|---|
| 1st | 435/1137 | Crude product | 619 | 90.2 | 92.0 | 83.0 | 97.80 | 2.20 |
| Purified product | 460 | 99.83 | 0.17 | |||||
| 2nd | 450/1174 | Crude product | 604 | 85.0 | 90.4 | 76.8 | 95.77 | 4.23 |
| Purified product | 452 | 99.58 | 0.42 | |||||
| 3rd | 480/1255 | Crude product | 646 | 85.3 | 91.0 | 77.6 | 97.73 | 2.27 |
| Purified product | 490 | 99.62 | 0.38 |
| Batch No. | LM49-01 (tR = 10.7) (%) * | LM49-02-imA (tR = 14.3) (%) * | LM49-02-imB (tR = 16.5) (%) * |
|---|---|---|---|
| 1st | - | - | 0.17 |
| 2nd | - | - | 0.42 |
| 3rd | - | - | 0.38 |
| Batch No. a | LM49-02 (g)/BBr3 (g) | Product | Output (g) | Crude Product Yield (%) | Recrystallization Yield (%) | Yield (%) | Total Yield (%) | Purity (%) * | Total Impurities (%) * |
|---|---|---|---|---|---|---|---|---|---|
| 1st | 450/786 | Crude product | 400 | 98.5 | 89.7 | 88.3 | 57.1 | 99.95 | 0.05 |
| Final API | 350 | 99.98 | 0.02 | ||||||
| 2nd | 440/769 | Crude product | 390 | 98.2 | 86.6 | 88.5 | 53.4 | 99.71 | 0.29 |
| Final API | 329 | 99.95 | 0.05 | ||||||
| 3rd | 530/926 | Crude product | 474 | 99.2 | 89.1 | 88.4 | 56.7 | 99.77 | 0.23 |
| Final API | 410 | 99.96 | 0.04 |
| Batch No. | LM49-02 (tR = 18.3) (%) * | LM49-API-imA (tR = 20.4) (%) * | LM49-API-imB (tR = 16.8) (%) * | LM49-API-imC (tR = 9.8) (%) * | LM49-API-imX (tR = 12.8) (%) * |
|---|---|---|---|---|---|
| 1st | - | - | - | 0.01 | 0.01 |
| 2nd | - | - | - | 0.03 | 0.02 |
| 3rd | - | - | - | 0.02 | 0.02 |
| Entry | LM49-API (g) | Solvent/Amount (mL) | Yield (%) | Appearance | Mp (°C) |
|---|---|---|---|---|---|
| 1 | a | Light yellow powder | 218.0–218.5 | ||
| 2 | 3.0 a | EthylAcetate/12 | 8.7 | Saffron crystal | 218.0–218.5 |
| 3 | 3.0 a | MeOH/6.0 | 42.0 | Light yellow crystal | 218.0–219.0 |
| 4 | 3.0 a | EtOH/4.8 | 59.3 | Light yellow crystal | 218.5–219.5 |
| 5 | 3.0 a | Isopropanol/6.5 | 66.7 | Light yellow crystal | 218.0–219.5 |
| 6 | 3.0 a | IsopropylAcetate/10.0 | 56.0 | Light yellow crystal | 218.0–219.0 |
| 7 | 3.0 a | EtOH + H2O/4 + 0.2 | 51.5 | Light yellow crystal | 218.5–219.5 |
| 8 | 3.0 a | MeOH + H2O/4 + 0.2 | 54.5 | Light yellow crystal | 218.5–219.5 |
| 9 | 3.0 a | EtOH + H2O/12 + 6 | 81.5 | Light yellow crystal | 219.0–220.0 |
| 10 | 3.0 a | EtOH + H2O/12 + 12 | 88.7 | Light yellow crystal | 219.0–220.0 |
| 11 | b | Light yellow crystal | 218.0–219.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, X.E.; Cui, K.M.; Li, Q.S.; Wu, Z.C.; Lei, F. Process Development and Synthesis of Process-Related Impurities of an Efficient Scale-Up Preparation of 5,2′-Dibromo-2,4′,5′-Trihydroxy Diphenylmethanone as a New Acute Pyelonephritis Candidate Drug. Molecules 2020, 25, 468. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25030468
Feng XE, Cui KM, Li QS, Wu ZC, Lei F. Process Development and Synthesis of Process-Related Impurities of an Efficient Scale-Up Preparation of 5,2′-Dibromo-2,4′,5′-Trihydroxy Diphenylmethanone as a New Acute Pyelonephritis Candidate Drug. Molecules. 2020; 25(3):468. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25030468
Chicago/Turabian StyleFeng, Xiu E, Ke Meng Cui, Qing Shan Li, Zi Cheng Wu, and Fei Lei. 2020. "Process Development and Synthesis of Process-Related Impurities of an Efficient Scale-Up Preparation of 5,2′-Dibromo-2,4′,5′-Trihydroxy Diphenylmethanone as a New Acute Pyelonephritis Candidate Drug" Molecules 25, no. 3: 468. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25030468