
Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones


1
School of Pharmaceutical and Materials Engineering, Taizhou University, Jiaojiang 318000, China
2
School of Environmental Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(3), 673; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25030673
Submission received: 18 January 2020
/
Revised: 1 February 2020
/
Accepted: 3 February 2020
/
Published: 5 February 2020
(This article belongs to the Special Issue Nitro Compounds and Their Derivatives in Organic Synthesis)
Abstract
:Nitro group is one of the most important functional groups in organic syntheses because its strongly electron-withdrawing ability activates the scaffold, facilitating the reaction with nucleophilic reagents or the Diels–Alder reaction. In this review, recent progress in the nitro-promoted direct functionalization of pyridones and quinolones is highlighted to complement previous reviews.
1. Introduction
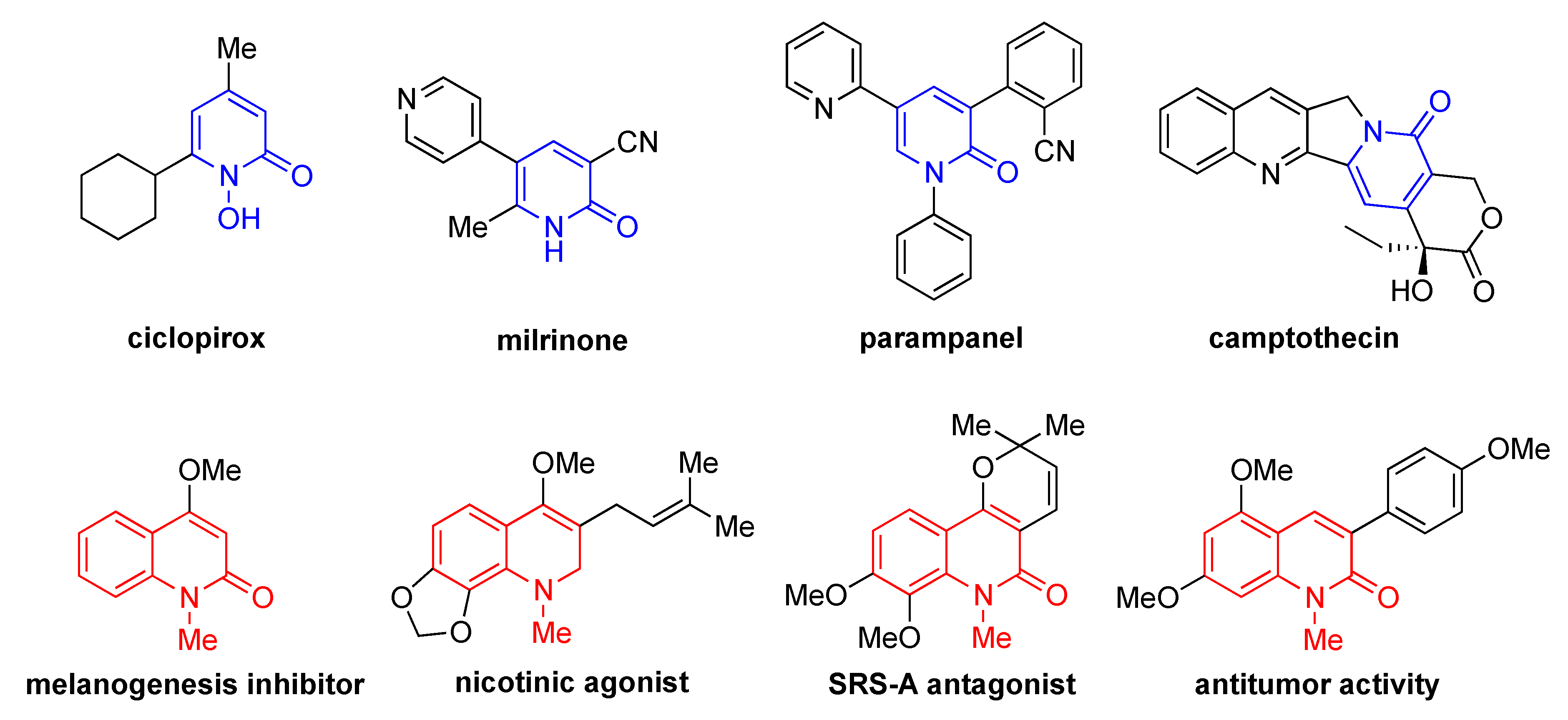
Natural and synthetic aza-heterocycles represent an important class of organic compounds [1,2,3,4,5]. Among the large number of aza-heterocycles available, pyridones and quinolones, both of which have a common six-membered aza-framework, exhibit a wide range of pharmacologically important activities (Figure 1) [6,7,8,9,10]. Therefore, various methods for the preparation of structurally diverse pyridones and quinolones have been studied in detail [6,11,12,13,14,15,16,17,18,19,20,21].
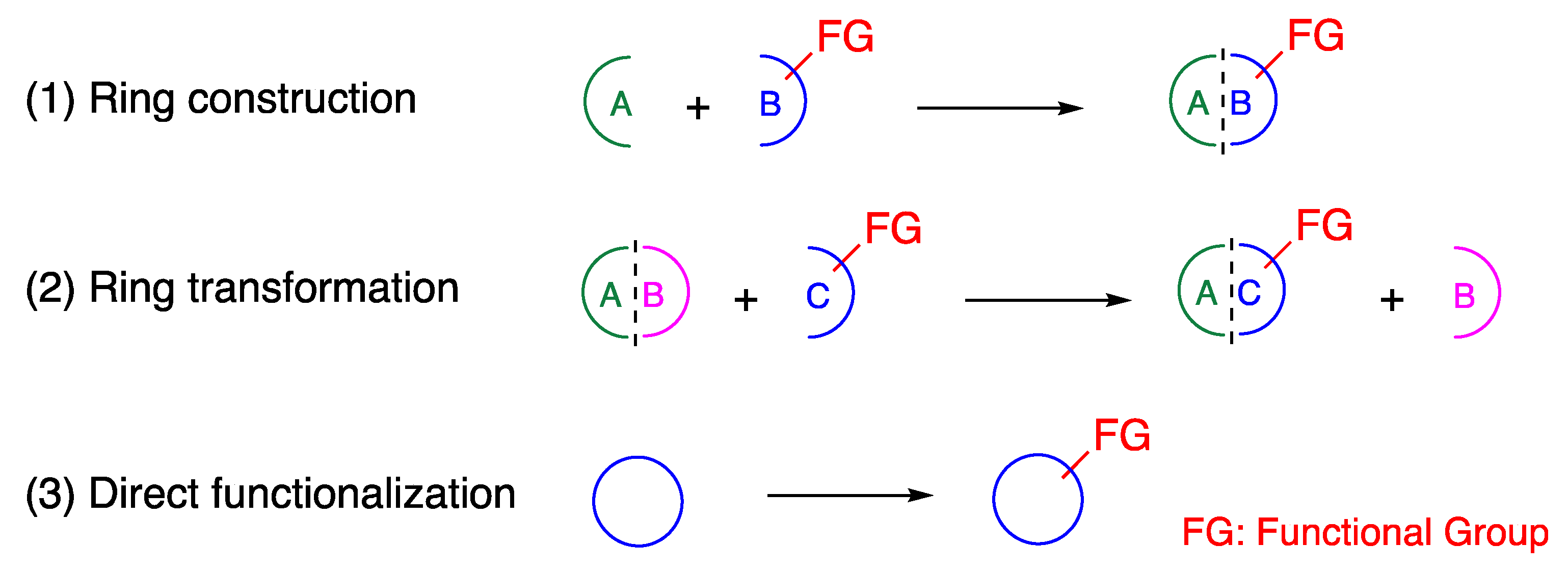
Conventional strategies for the synthesis of aza-heterocycles involve (1) construction of aza-heterocycle frameworks from prefunctionalized starting materials, (2) ring transformation leading to aza-heterocycle frameworks, and (3) direct functionalization of aza-heterocycle frameworks, which are supplementary to each other (Figure 2) [22].
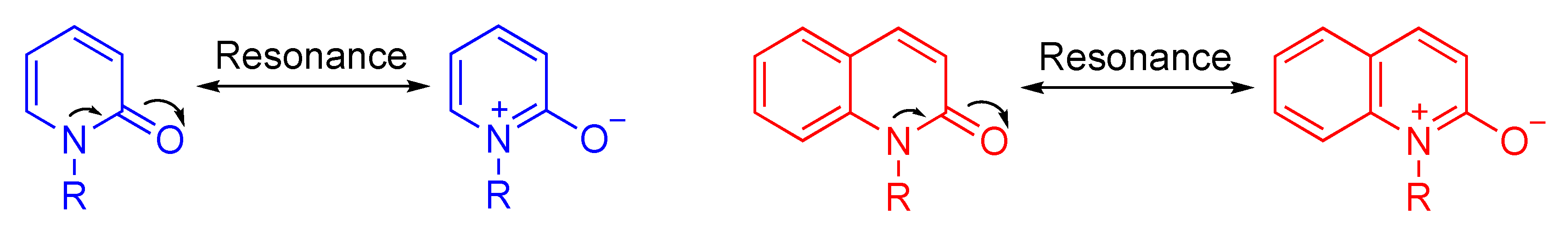
Among these three protocols, direct functionalization of aza-heterocycles, for preparing new diversely functionalized heterocycles, is the most efficient approach from a practical viewpoint, because it requires only simple experimental manipulations. Accordingly, the development of easy and efficient methods for the direct functionalization of quinolone and pyridone frameworks is highly demanded. However, only a few such methods are currently available because these scaffolds are inert due to the aromaticity (Figure 3) [22].
To the best of our knowledge, the currently used methods for direct functionalization of the quinolone and pyridone scaffolds are mainly focused on transition-metal-catalyzed cross-coupling and C–H activation reactions [6,11,12,13,14,15,16,17,18,19,20,21]. However, most of these methods suffer from some limitations, such as the use of potentially poisonous and expensive noble metals, along with harsh reaction conditions.
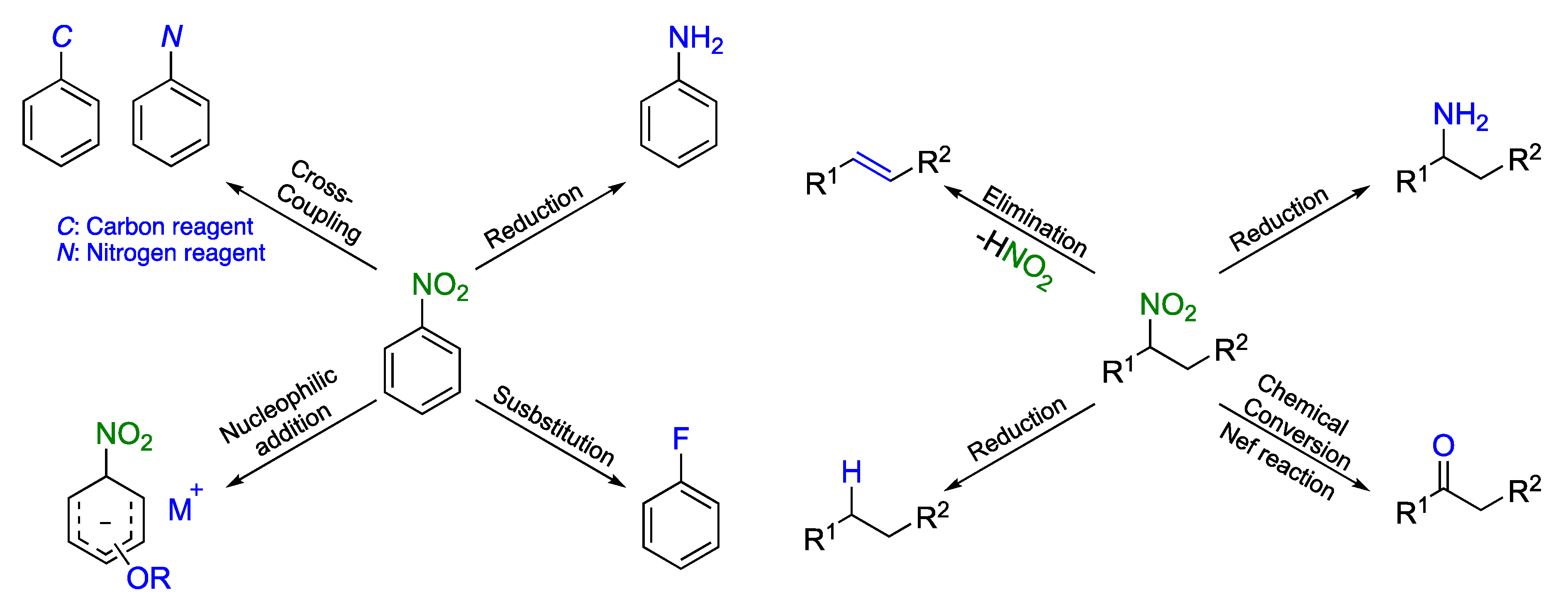
However, the nitro group, which is often described as a “synthetic chameleon [23],” serves as a precursor for versatile functionalities, such as formyl, acyl, cyano, and amino groups (Scheme 1) [24,25,26,27,28]. Moreover, the nitro group has been proved to activate many different scaffolds because of its strong electron-withdrawing ability, facilitating the reaction with nucleophilic reagents [29,30]. The nitro group is also a good leaving group, which is often involved in addition–elimination reactions [31,32].
Based on these significant properties of the nitro group, the synthetic utility of nitrated aza-heterocycles in the preparation of functionalized aza-heterocycles has been widely investigated [33]. However, electrophilic nitration of pyridines and quinolines is difficult because of the electron deficiency of the aromatic cores. On the contrary, it is possible to nitrate pyridones and quinolones because the dearomatization of these scaffolds is easier than that of pyridines and quinolines. Indeed, the introduced nitro groups activate the scaffolds to facilitate direct functionalization, which affords structurally diverse aza-heterocycles. Herein, recent progress in the nitro-promoted direct functionalization of pyridones and quinolones in the past couple decades is highlighted.
2. Cycloaddition of Nitropyridones
The nitro group is a strongly electron-withdrawing group that reduces the electron density on the scaffold. Further, 2-pyridones possessing a nitro group are highly electron-deficient, and they serve as dienophiles that undergo Diels–Alder (D–A) cycloaddition with electron-rich dienes, forming fused aza-heterocycles [34].
When 5-nitro-2-pyridones 1 are reacted with 2,3-dimethyl-1,3-butadiene 2, quinolones 3 are formed via regioselective D–A cycloaddition at the 5- and 6-positions and subsequent aromatization accompanied by elimination of nitrous acid (Table 1). For 5-nitropyridone bearing a methoxycarbonyl group at the 3-position, the D–A reaction occurs chemoselectively to yield the corresponding 3-functionalized quinolone 3c.
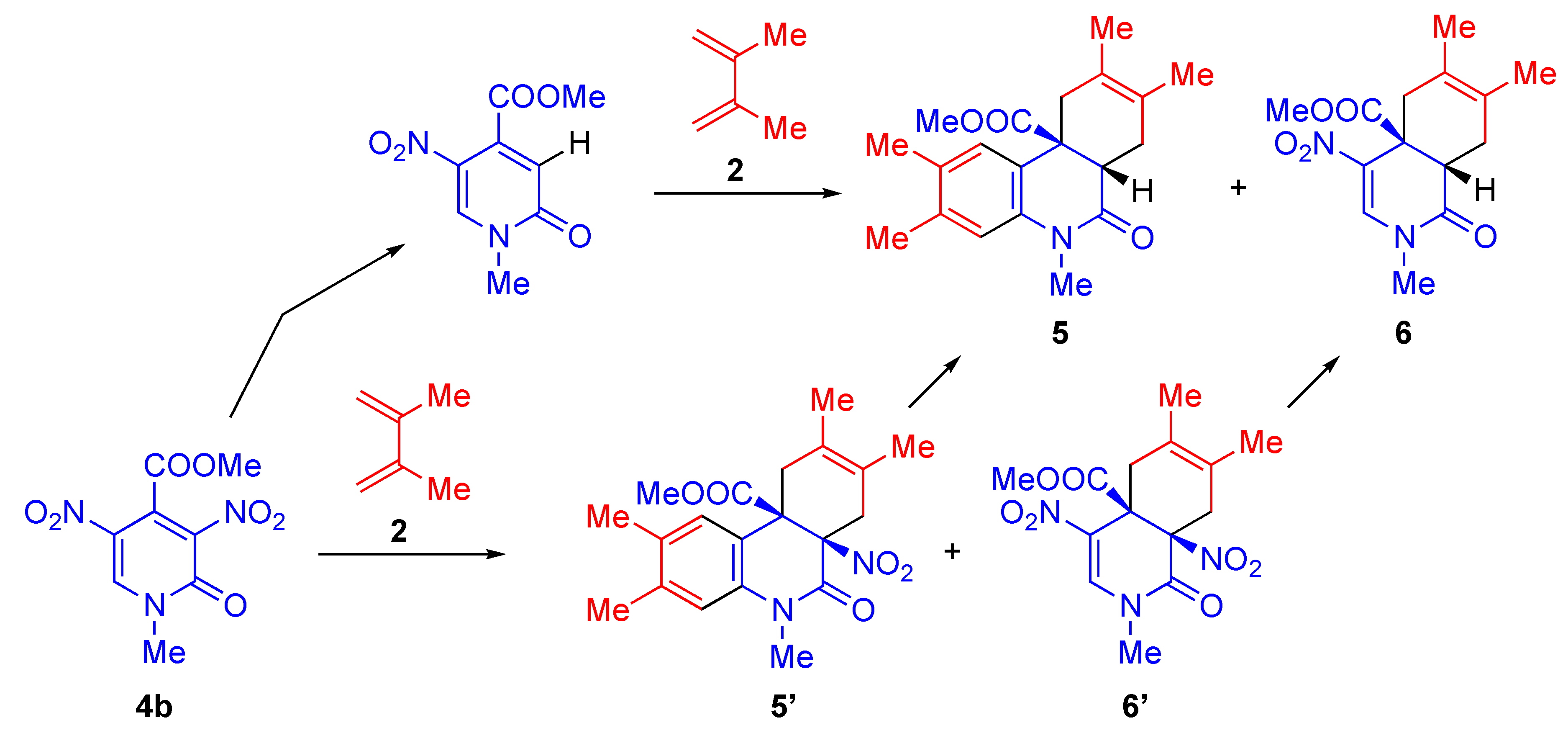
It is known that 5-nitropyridones 4 possessing electron-withdrawing groups at the 3- and/or 4-positions have two electron-deficient sites on the ring. When these substrates are subjected to D–A reactions with diene 2, the reaction proceeds stereoselectively to produce the functionalized cis-adducts 5 and 6, accompanied by denitration (Table 2). Since the reaction is conducted under harsh conditions, the denitration of either pyridone 4b or the cycloadducts 5′ and 6′ might occur (Scheme 2), however a detailed explanation has not been reported in the literature [34].
D–A cycloaddition of 1-unsubstituted 3-nitro-2-pyridones 7a with diene 2 yields the cis-condensed tetrahydroisoquinolone 8a stereoselectively. For 1-methyl-3-nitro-2-pyridone 7b, cis-tetrahydroisoquinolone 8b as well as aromatized isoquinolone 9b is formed via dehydrogenation and release of a nitrous acid. The use of a substrate with 4-methoxycarbonyl substitution affords cis-tetrahydroisoquinolone 8c as the sole product (Table 3).
The reaction of 1-unsubstituted 3,5-dinitropyridone 10a gives an aromatized isoquinolone 11a via cycloaddition at the 3- and 4-positions, followed by dehydrogenation and elimination of nitrous acid; an aromatized phenanthridone 12a is also obtained via double D–A adduct formation (Table 4). However, the reaction of 1-methyl-3,5-dinitro-2-pyridone 10b furnishes not only 4-nitroisoquinolone 11b and phenanthridone 12b, but also cis-tetrahydroisoquinolone 8b, via cycloaddition at the 3- and 4-positions accompanied by heating-promoted elimination of the nitro group at the 5-position. D–A reactions of 3-nitro-2-pyridones 10c and 10d with 5-methoxycarbonyl substitution mainly yield the aromatized isoquinolones 11c and 11d, respectively, in addition to the incompletely aromatized cis-phenanthridone adducts 13c and 13d, respectively.
3. Cycloaddition of Nitroquinolones
The D–A reactions at the nitroalkene moiety of 3-nitrated 1-methyl-2-quinolones 14 with electron-rich dienes yield aromatized phenanthridone derivatives 15 (Table 5). Although this method enables simultaneous C–C bond formation at the 3- and 4-positions of the quinolone framework, harsh reaction conditions must be employed [35,36].
On the contrary, 1-methyl-3,6,8-trinitro-2-quinolone 16 undergoes cycloaddition with dienes easily under mild conditions (Scheme 3). Indeed, the cycloaddition of 16 with cyclopentadiene proceeds smoothly to furnish a tetracyclic compound 17 that aromatizes via elimination of a nitrous acid in the presence of triethylamine to afford compound 18 [37]. Similarly, the cycloaddition using α,β-unsaturated oxime, instead of cyclopentadiene, as a heterodiene affords the polycyclic diazaphenanthrene 19 (Scheme 4) [38].
The high reactivity of trinitroquinolone 16 is due to the steric repulsion between the 1-methyl and 8-nitro groups, disturbing the coplanarity of the pyridone moiety and the benzene ring. Consequently, the pyridone ring of 16 loses its aromaticity and serves as an activated nitroalkene (Figure 4) [39].
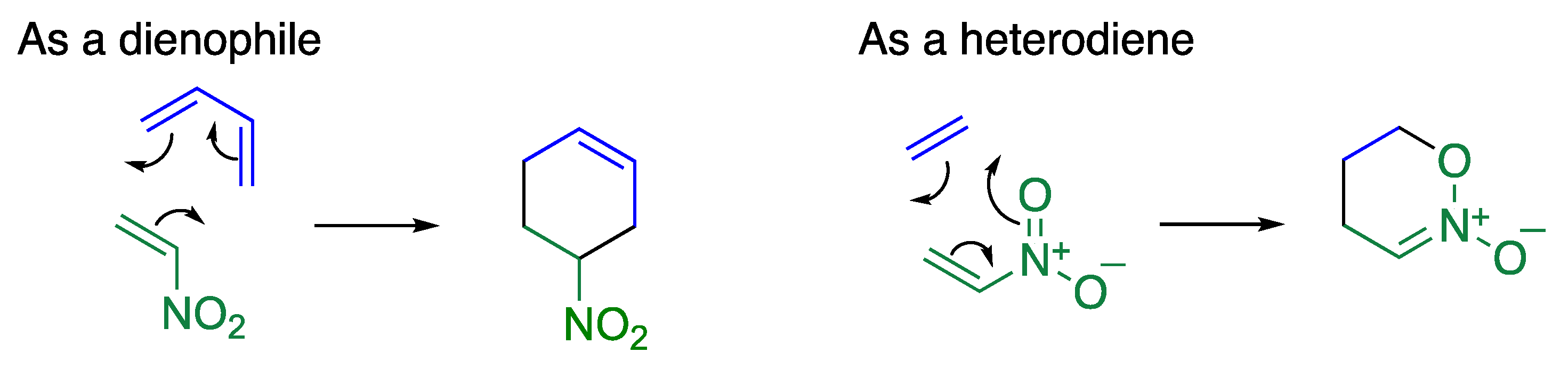
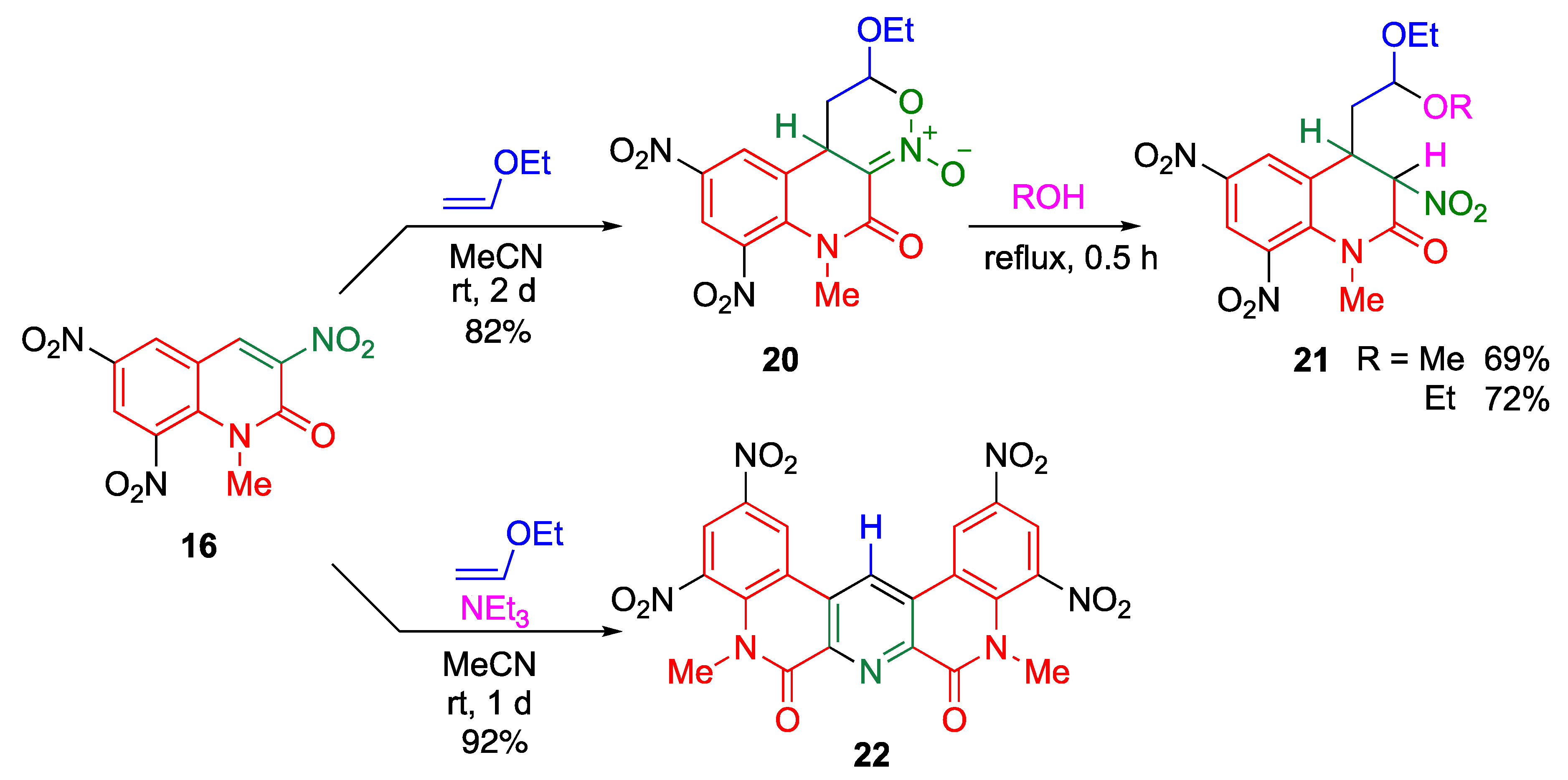
A nitroalkene shows dual behavior in cycloaddition reactions (Figure 5). In reaction with a diene, the nitroalkene serves as a dienophile to form a cyclohexene ring. On the other hand, it serves as a heterodiene in reaction with an electron-rich alkene to construct an oxazine ring. The nitroalkene moiety of trinitroquinolone 16 also serves as a heterodiene in the reaction with ethoxyethene to construct a fused oxazine ring 20 (Scheme 5) [38], which yields an acetal 21 via ring-opening reaction upon treatment with alcohol under reflux conditions.
Interestingly, a quinolino[3, 4-b][1,9]diazaphenanthrene derivative 22 is formed when the same reaction is conducted in the presence of triethylamine (Scheme 5) [38]. A plausible mechanism is shown in Scheme 6. After forming the cyclic nitronate 20, triethylamine assists the proton transfer from the 4-position to the anionic oxygen of the nitronate. The subsequent retro D–A reaction gives the α,β-unsaturated oxime A, accompanied by a loss of ethyl formate. Oxime A serves as an electron-rich heterodiene that undergoes cycloaddition with another molecule of 16 to afford a new pyridine ring, and subsequent aromatization and elimination of nitrous acid and water furnishes the polycyclic product 22. In this reaction, two molecules of trinitroquinolone 16 undergo two kinds of cycloaddition reactions: one molecule serves as a heterodiene and the other serves as a dienophile. This is the first example of a nitroalkene that exhibits dual behavior in the same reaction mixture (Figure 5).
4. Nitro-Promoted Cyclization of Pyridones via Nucleophilic Addition
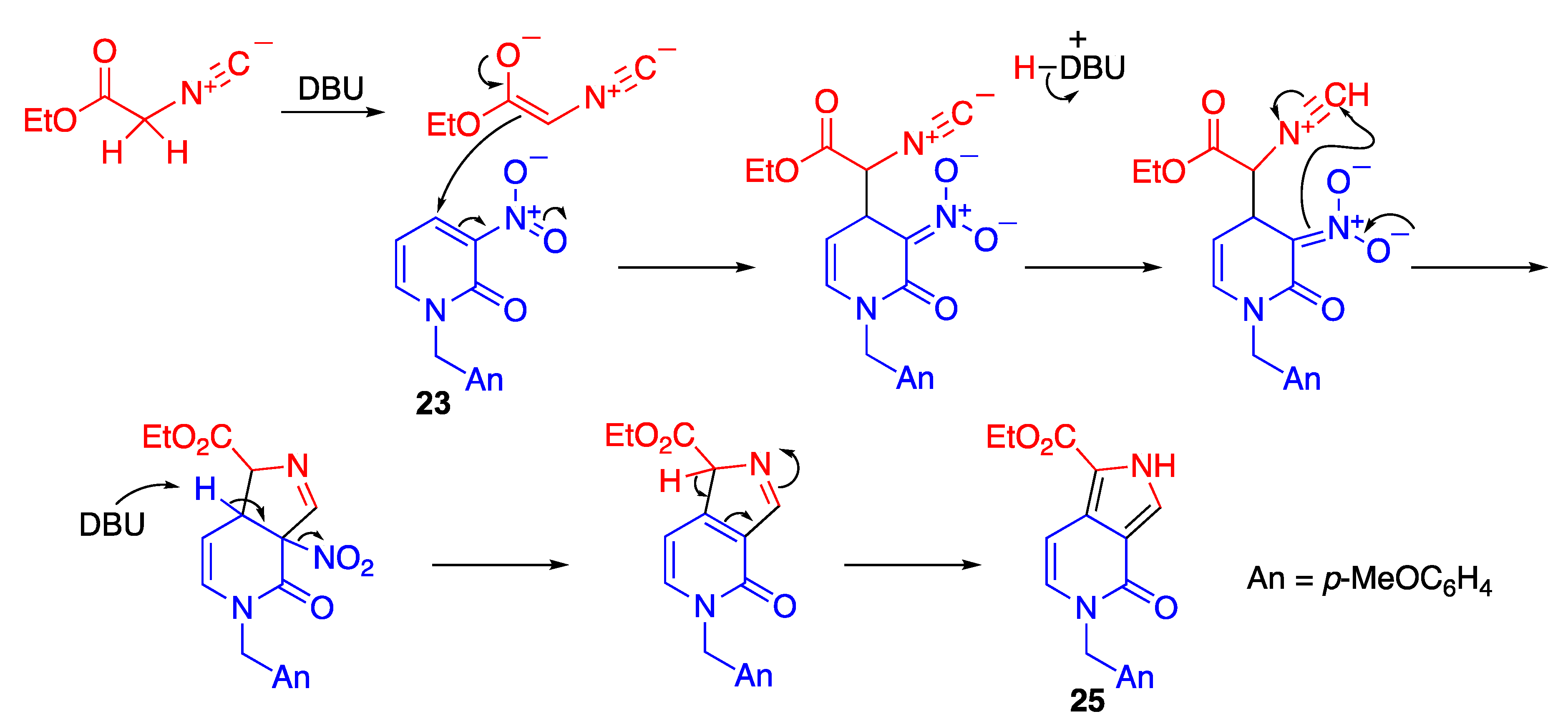
The strongly electron-withdrawing ability of the nitro group activates the scaffold for nucleophilic attack at the vicinal position on the nitroalkene. The nitroalkene moiety of nitropyridones is also susceptible to nucleophilic reaction. Indeed, 1-substituted nitropyridones 23 and 24 react with ethyl isocyanoacetate in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to afford the pyrrolopyridine derivatives 25 and 26, respectively (Scheme 7) [40]. In the latter case, nucleophilic attack of isocyanoacetate occurs regioselectively at the 6-position.
The reaction is initiated by the nucleophilic addition of isocyanoacetate to nitropyridone under basic conditions to produce an anionic intermediate stabilized by the nitro group (Scheme 8). Then, the nucleophilic attack of the nitronate to the protonated isocyano group affords dihydro-2H-pyrrole, from which a pyrrole ring is produced via aromatization by elimination of nitrous acid.
5. Nitro-Promoted Direct Functionalization of Quinolones
5.1. Direct C–C Bond Formation at the 4-Position via Cine-Substitution
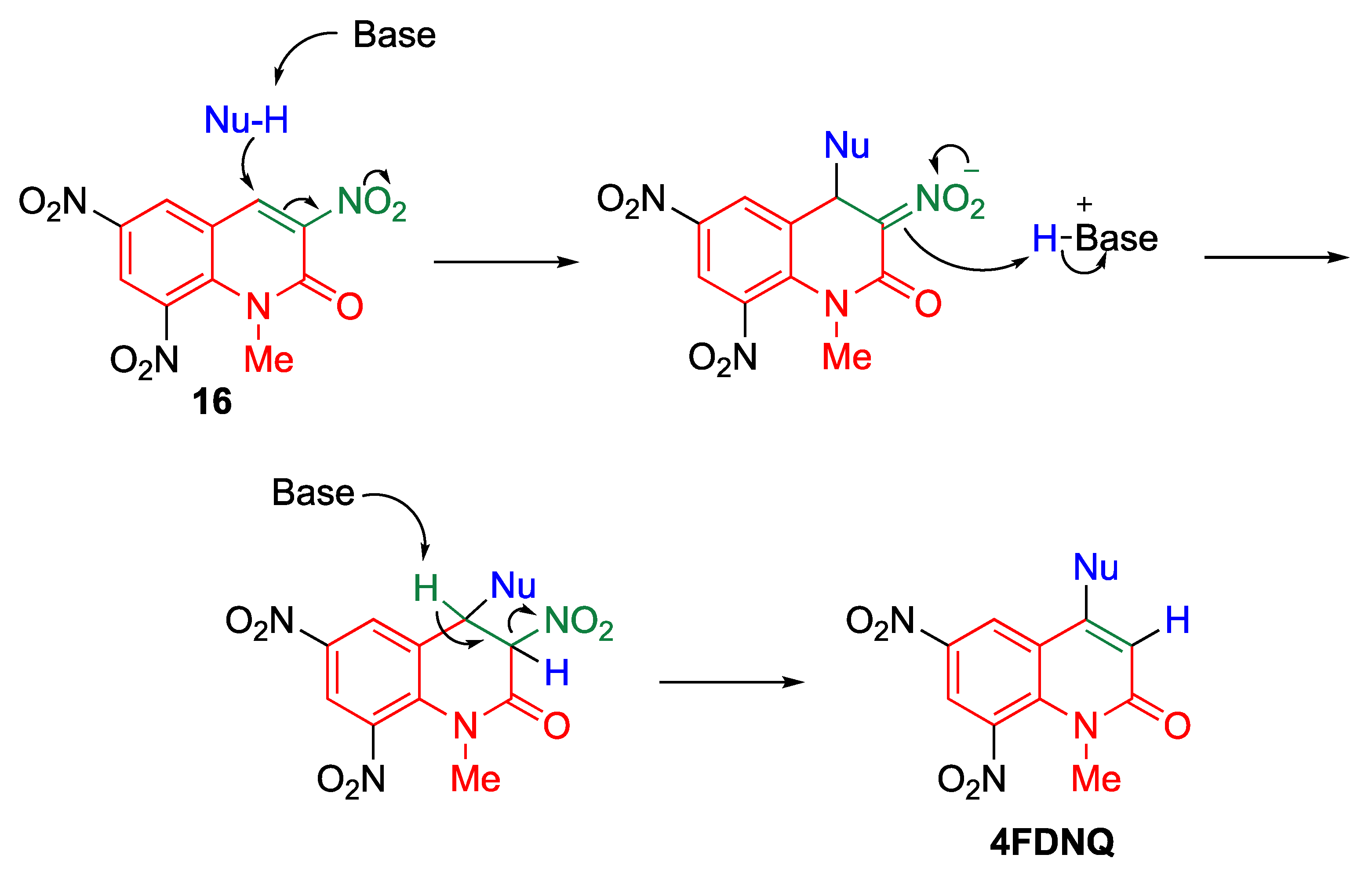
To the best of our knowledge, the currently used methods for direct C–C bond formation in 1-methyl-2-quinolone (MeQone) framework are mainly limited to transition-metal-catalyzed cross-coupling or C–H activation reactions [11,12,13,14,15,16]. As an alternative, the introduction of a nitro group has proved helpful in facilitating direct functionalization of the MeQone framework, affording diversely functionalized MeQones. Indeed, cine-substitution of trinitroquinolone 16 with various nucleophiles can easily proceed to afford 4-functionalized 6,8-dinitro-1-methyl-2-quinolones (4FDNQ) [22]. Initially, the nucleophilic substitution proceeds at the 4-position of 16 to form an adduct intermediate; then, a proton is transferred from the basic group to the 3-position of the adduct intermediate, affording 3,4-dihydroquinolone. The subsequent elimination of nitrous acid, accompanied by aromatization, yields 4FDNQ (Scheme 9). This reaction enables regioselective functionalization at the 4-position of the MeQone framework. Direct C–C bond formation at the 4-position of the MeQone framework is easily achieved upon treatment of 16 with carbon nucleophiles, including 1,3-dicarbonyl compounds, nitroalkanes, aldehydes/ketones, enamines, cyanides, and phenoxides, leading to the formation of versatile skeletons.
5.1.1. cine-Substitution of Trinitroquinolone with 1,3-dicarbonyl Compounds
When trinitroquinolone 16 is reacted with 1,3-dicarbonyl compounds in the presence of triethylamine, 4-position functionalization is efficiently achieved via cine-substitution (Table 6) [41]. Diketones, keto esters, and diesters can be used as nucleophiles in this reaction to afford the corresponding products 27a–e.
When the nitro group at the 8-position is removed, no reaction occurs, even under heating. On the other hand, cine-substitution proceeds smoothly even upon replacement of the electron-withdrawing nitro group of 16 with an electron-donating methyl group (Table 7). These results indicate that the steric repulsion of this substituent with the 1-methyl group activates the MeQone framework, as mentioned in Section 3 [42].
5.1.2. cine-Substitution of Trinitroquinolone with Nitroalkanes
Nitroalkylation of trinitroquinolone 16 is also achieved by using a nitroalkane as a carbon nucleophile in the presence of triethylamine (Table 8) [43]. While primary nitroalkanes undergo cine-substitution efficiently at room temperature, secondary nitroalkanes with steric hindrance are less reactive, requiring longer reaction times and affording relatively low yields.
5.1.3. cine-Substitution of Trinitroquinolone with Aldehyde, Ketones and Enamines
Besides aldehydes, functionalized ketones, such as aliphatic, alicyclic, aromatic, and heteroaromatic ketones work well as carbon nucleophiles in the cine-substitution of trinitroquinolone 16, giving acylmethylated products (Table 9) [44]. Since the acylmethyl group can serve as a scaffold for further chemical transformations, this method can be useful for the construction of a new library of compounds with MeQone framework.
More-reactive enamines can also be used as nucleophiles instead of ketones, which undergo cine-substitution in the presence of water at room temperature. After the addition of enamine to trinitroquinolone 16, hydrolysis of the formed iminium ion forms an acylmethyl group. In this case, the product is obtained as a morpholinium salt 30 (Table 10) [44].
5.1.4. cine-Substitution of Trinitroquinolone with Phenoxides
A combination of electrophilic trinitroquinolone 16 and nucleophilic phenoxide ions results in direct arylation of the MeQone framework (Figure 6) [45]. When 16 is treated with potassium phenoxides possessing electron-donating groups, double cine-substitution proceeds to afford bis(quinolyl)phenols 31 and 32. On the other hand, sterically hindered or electron-deficient phenoxides give monoquinolylphenols 33 and 34 as the only products. Since direct introduction of an aryl group into the MeQone framework is difficult, this method is considered one of the more useful modifications.
From another viewpoint, trinitroquinolone is an aromatic compound. Hence, this reaction can be regarded as an electrophilic arylation, which is not achieved in the usual Friedel–Crafts reaction. This transformation is initiated by the nucleophilic addition of phenoxide at the 4-position of 16 (Scheme 10). The newly introduced benzene ring is aromatized with the assistance of another phenoxide. In addition, proton transfer from the 4-position to an adjacent position of the quinolone ring occurs to afford the dianionic intermediate B. Since B is a highly electron-rich species, it immediately attacks another molecule of 16 to afford bis(quinolyl)phenols 31 (path a). On the other hand, protonation of B followed by elimination of nitrous acid is the preferred route to furnish monoquinolylphenol when electron-deficient or bulky phenoxides are used (path b).
5.1.5. cine-Substitution of Trinitroquinolone with Cyanides
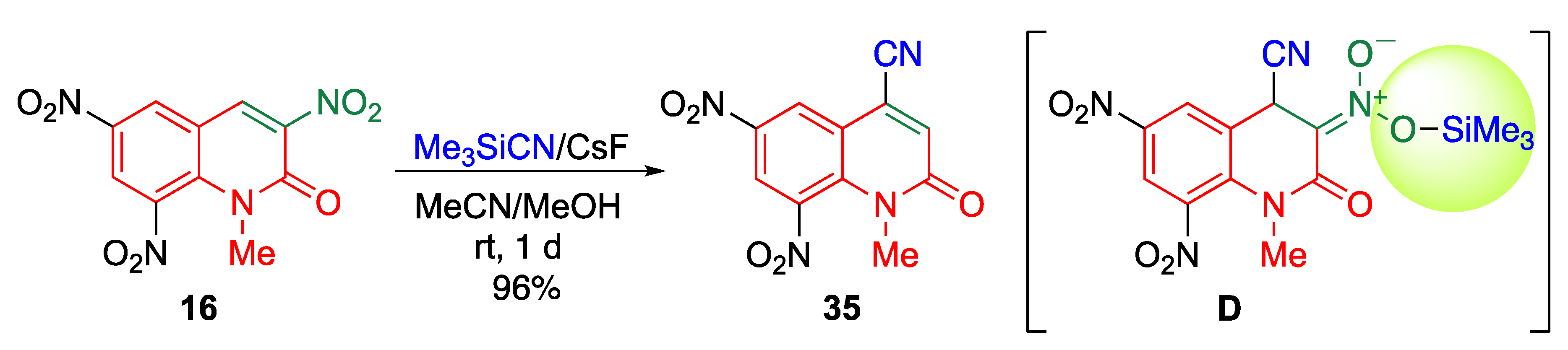
Nitriles represent an important structural motif in medicinal chemistry because of their versatile biological activities [46]. In addition, they have been recognized as extremely useful intermediates for the preparation of other useful building blocks [47,48,49]. Therefore, considerable research effort has been dedicated to the development of methods for introducing cyano groups into organic molecules. Inspired by the above methods for direct C–C bond formation on the MeQone framework, researchers have used potassium cyanide as a carbon nucleophile for reacting with trinitroquinolone 16 to prepare 4-cyano-2-quinolone derivative 35 (Scheme 11) [42]. In this reaction, dimeric product 36 is also obtained. After the addition of a cyanide to 16, the anionic intermediate C is formed, which is a common intermediate for both products 35 and 36. When C is protonated, followed by the elimination of nitrous acid, 35 is obtained (path a). The dimeric product 36 is a result of the addition of C to another molecule of 16 (path b).
The use of trimethylsilyl cyanide/cesium fluoride instead of potassium cyanide is effective in avoiding undesired dimerization due to the steric hindrance of the O-silylated intermediate D, affording cyanoquinolone 35 as the sole product without any detectable dimer 36 (Scheme 12). While conventional strategies for cyanation of the MeQone framework often involve multistep reactions or harsh conditions, the present method makes the cyanation possible under mild reaction conditions with simple experimental manipulations. Thus, this protocol can be used as a powerful tool for constructing a library of versatile MeQone derivatives by further chemical conversion of the cyano and nitro functionalities.
The introduction of a methyl group instead of a nitro group at the 8-position also activates the MeQone framework. Nitrated 1,8-dimethyl-2-quinolones 37 react with potassium cyanide to afford the corresponding 4-cyano MeQones (Table 11).
5.1.6. Reaction of Trinitroquinolone with Tertiary Amines
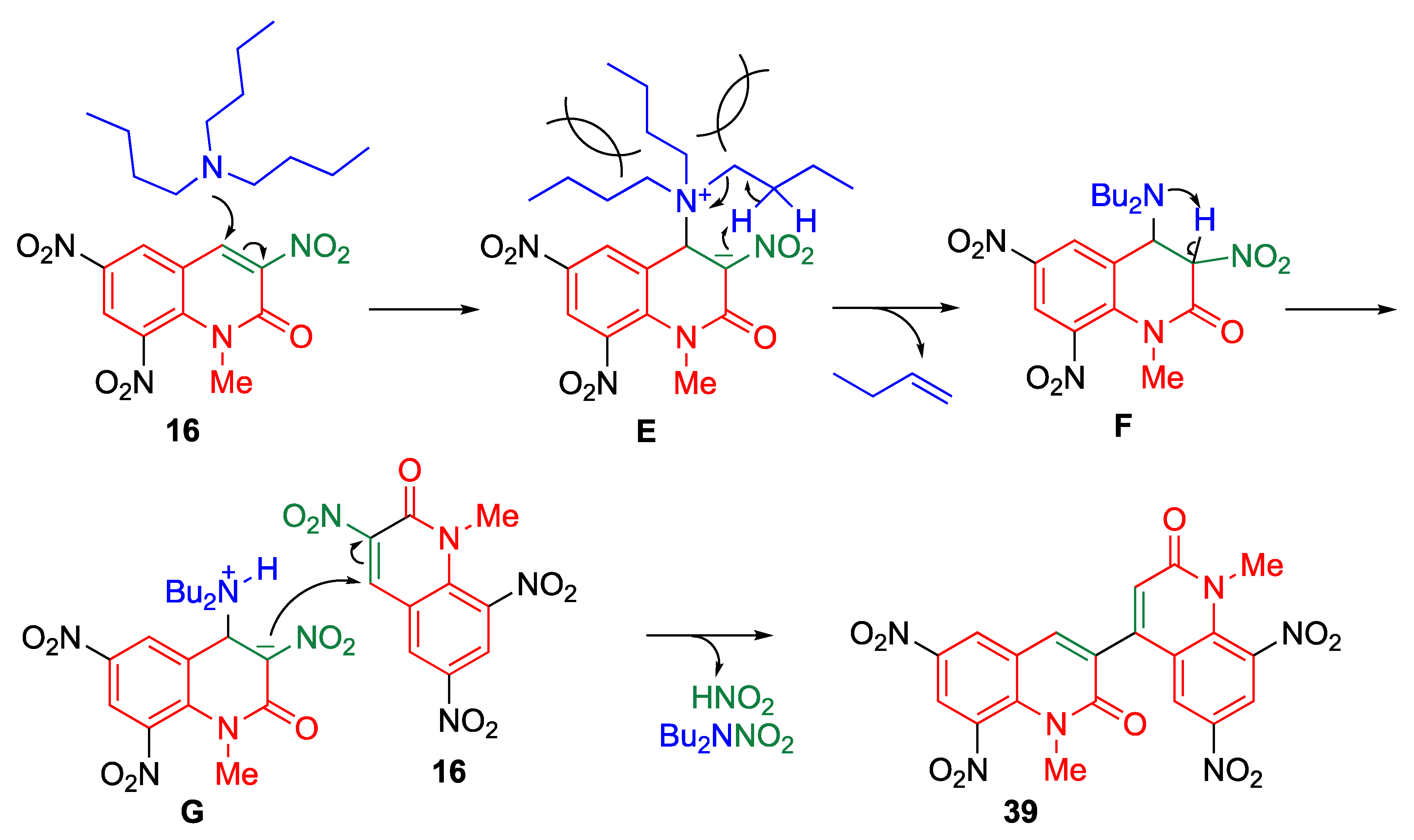
As mentioned in the previous section, the cyanide ion plays two roles: it serves as a nucleophile and it stabilizes anionic intermediate because of its electron-withdrawing nature. Thus, the dimerization of MeQones is also observed. Conversely, introduction of a hetero atom at the 4-position generates a stable anionic intermediate, which undergoes efficient dimerization. The treatment of trinitroquinolone 16 with a tertiary amine causes the dimerization [50]. Interestingly, more than two long alkyl chains possessing β-hydrogens are essential for undergoing this reaction (Table 12).
This reaction is initiated by the nucleophilic addition of tributylamine to trinitroquinolone 16 to produce the zwitterion E. The β-elimination of 1-butene is followed by proton transfer of F to produce the zwitterion G, which reacts with another molecule of 16 to afford dimer 39 (Scheme 13).
5.2. Direct C–N Bond Formation at the 4-Position
5.2.1. cine-Substitution of Trinitroquinolone with Primary Amines
A different reactivity is observed when primary amines, instead of tertiary amines, are used as the nucleophiles to react with trinitroquinolone 16. The regioselective C–N bond formation occurs at the 4-position to afford the Meisenheimer complex 40 (Table 13) [51]. When 40 is heated, cine-substituted products 41a and 41b are obtained; however, no cine-substitution is observed for bulky amino substituted derivatives 40c and 40d, accompanied by the recovery of considerable amounts of 16. Upon heating, 40 is converted to dihydroquinolone H, from which nitrous acid is eliminated to afford the cine-substituted products 41 (Scheme 14, path a). However, the elimination of amine proceeds competitively to give the trinitroquinolone 16 (path b), which lowers the yield of 41.
5.2.2. Amino-Halogenation and Imido-Halogenation of Quinolones
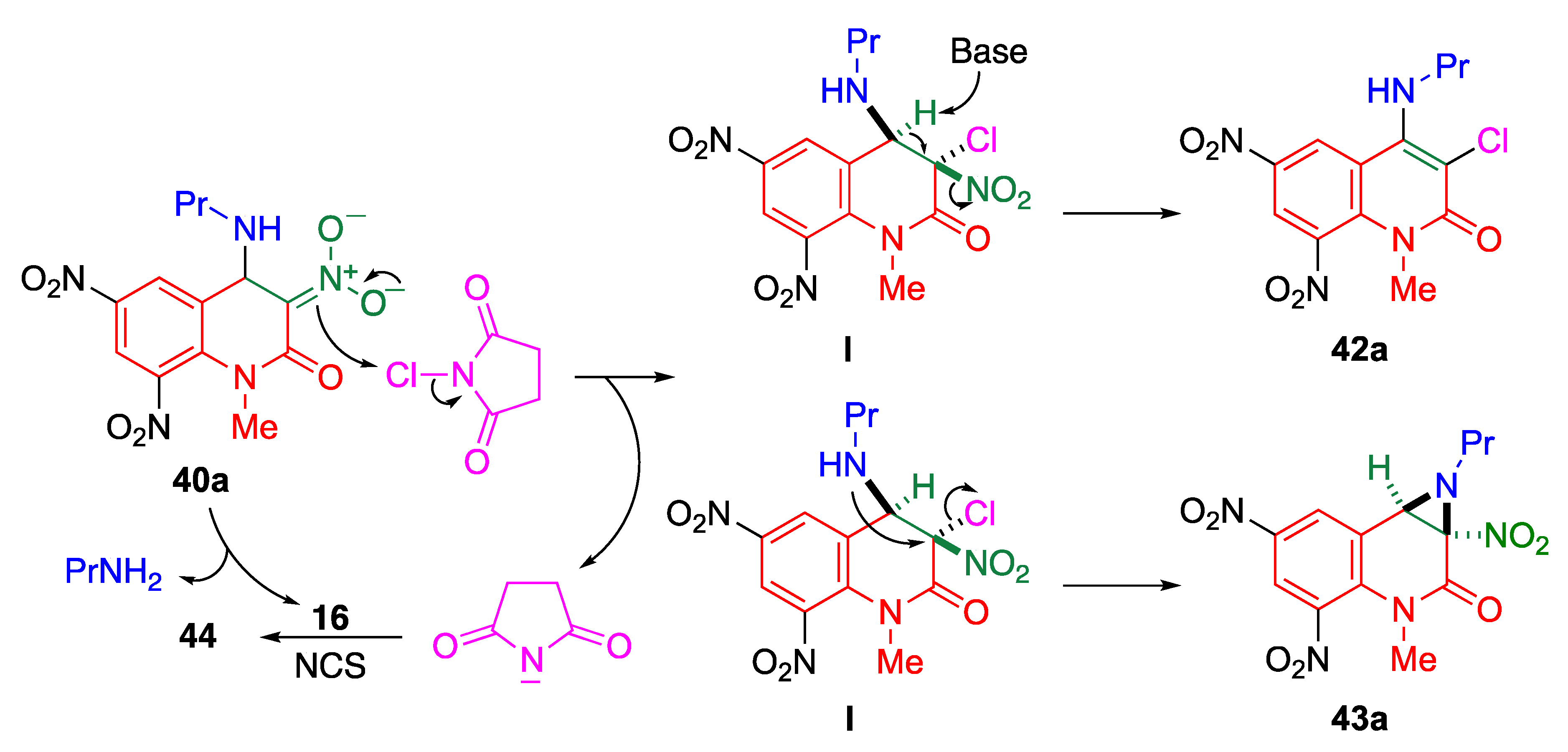
The reaction of trinitroquinolone 16 with excess propylamine in acetonitrile affords the Meisenheimer complex 40a, which can be used for further functionalization of the MeQone framework upon treatment with electrophiles. When the ammonium salt 40a is treated with N-chlorosuccinimide (NCS), three kinds of functionalized quinolone are obtained; the amino-chlorinated product 42, the aziridine-fused quinolone 43, and the imido-chlorinated product 44 (Scheme 15) [52].
A plausible mechanism for these reactions is illustrated in Scheme 16. Chlorination of the Meisenheimer complex 40a affords dihydroquinolone I, which is the common intermediate for 42a and 43a. The amino-chlorinated product 42a is formed by elimination of nitrous acid induced by a base, such as imide anion and amine. When the amino group attacks the vicinal position to substitute chloride, an N-propylaziridine ring is formed to give product 43a. On the other hand, when the eliminated imide anion reacts with trinitroquinolone 16 and NCS, the imido-chlorinated product 44 is formed, which is also formed when 16 is reacted with sodium imide in the presence of NCS (Scheme 17).
The amino-halogenation of trinitroquinolone 16 can be conducted in a one-pot two-step manner, in which the selectivity of 42 is increased by using an excess amount of amine (Table 14). The aliphatic and aromatic primary amines undergo the reaction to afford the corresponding amino-chlorinated products 42a–k in moderate yields. However, less nucleophilic p-nitroaniline shows no change. While the acyclic secondary amine, diethylamine, does not furnish 42m, the cyclic secondary amine, morpholine, yields the corresponding amino-chlorinated product 42n. Ammonia is difficult to handle in this protocol. Instead, the imido group is considered a masked form of an amino group. Indeed, the imido-chlorinated product 44 can be transformed to the amino-chlorinated quinolone 42b by hydrazinolysis (Scheme 17).
When NBS is employed as a halogenating reagent, a small amount of the amino-nitrated product 46 is formed in addition to the amino-brominated product 45, presumably due to the higher leaving ability of bromide than that of chloride (Table 15). Indeed, only amino-nitrated product 46 is obtained without any detectable formation of the iodo-aminated product in the reaction with NIS.
5.2.3. Aziridination of Quinolones
The screening of various 3-nitrated MeQones reveals the tendency of the selectivity between amino-halogenation and aziridination (Table 16). When the electron density of the benzene ring is low, amino-chlorination occurs predominantly to afford 48a–c. On the other hand, for increased electron density, intramolecular substitution exclusively occurs to form an aziridine ring, leading to 49d–g. This tendency is considered to depend on the acidity of the proton at the 4-position in the intermediate J. When the acidity of H4 increases due to the electron-withdrawing group, elimination of a nitrous acid occurs easily via E2 reaction to give the amino-halogenated product 48. In contrast, when the acidity of H4 becomes lower, an intramolecular SN2 reaction proceeds to afford the aziridine 49.
1,8-Dimethyl-3,5-dinitro-2-quinolone 50 exhibits a reactivity different from those of the other nitroquinolones 47. When 50 is subjected to the reaction under the same conditions, cine-substitution takes place, rather than amino-chlorination and aziridination, affording compound 51 quantitatively (Scheme 18). In this reaction, the addition of a propylamine affords the Meisenheimer complex K. However, the steric repulsion with the peri-substituent increases the steric hindrance around the 3-position, thus preventing the attack to NCS. Instead, proton transfer from the 4-position followed by elimination of the nitrite ion affords the cine-substituted product 51.
Aziridine-fused quinolone 49f undergoes a ring-opening reaction followed by rearomatization upon treatment with acid, such as toluenesulfonic acid, hydrochloric acid, and trifluoroborane, to furnish the amino-nitrated MeQone (Scheme 19).
5.3. Direct C–O Bond Formation at the 4-Position
When trinitroquinolone 16 is treated with a sodium alkoxide at room temperature, nucleophilic addition at the 4-position affords an alkoxylated salt 52 [53], which can be isolated because of stabilization by the adjacent nitro and carbonyl groups. After removal of alcohol, treatment of the adduct 52 with NCS in acetonitrile affords the 4-alkoxy-3-chloro-2-quinolone derivatives 53 in moderate-to-high yields (Table 17). This protocol can be performed in a one-pot manner with simple experimental manipulations.
The reaction proceeds via a similar mechanism, as shown in Scheme 16, for the amino-chlorination (Scheme 20). Chlorination of the alkoxylated salt 52 with NCS affords the dihydroquinolone intermediate L, from which nitrous acid is eliminated to form a bis(functionalized) product 53.
When NBS is used as the halogenating reagent, 4-methoxylated trinitroquinolone 54 is obtained in addition to the methoxy-brominated product 53 (Table 18). In the reaction using NIS, product 54 is furnished without any detectable formation of 53. The different reactivity is due to the higher leaving abilities of bromide and iodide than that of chloride.
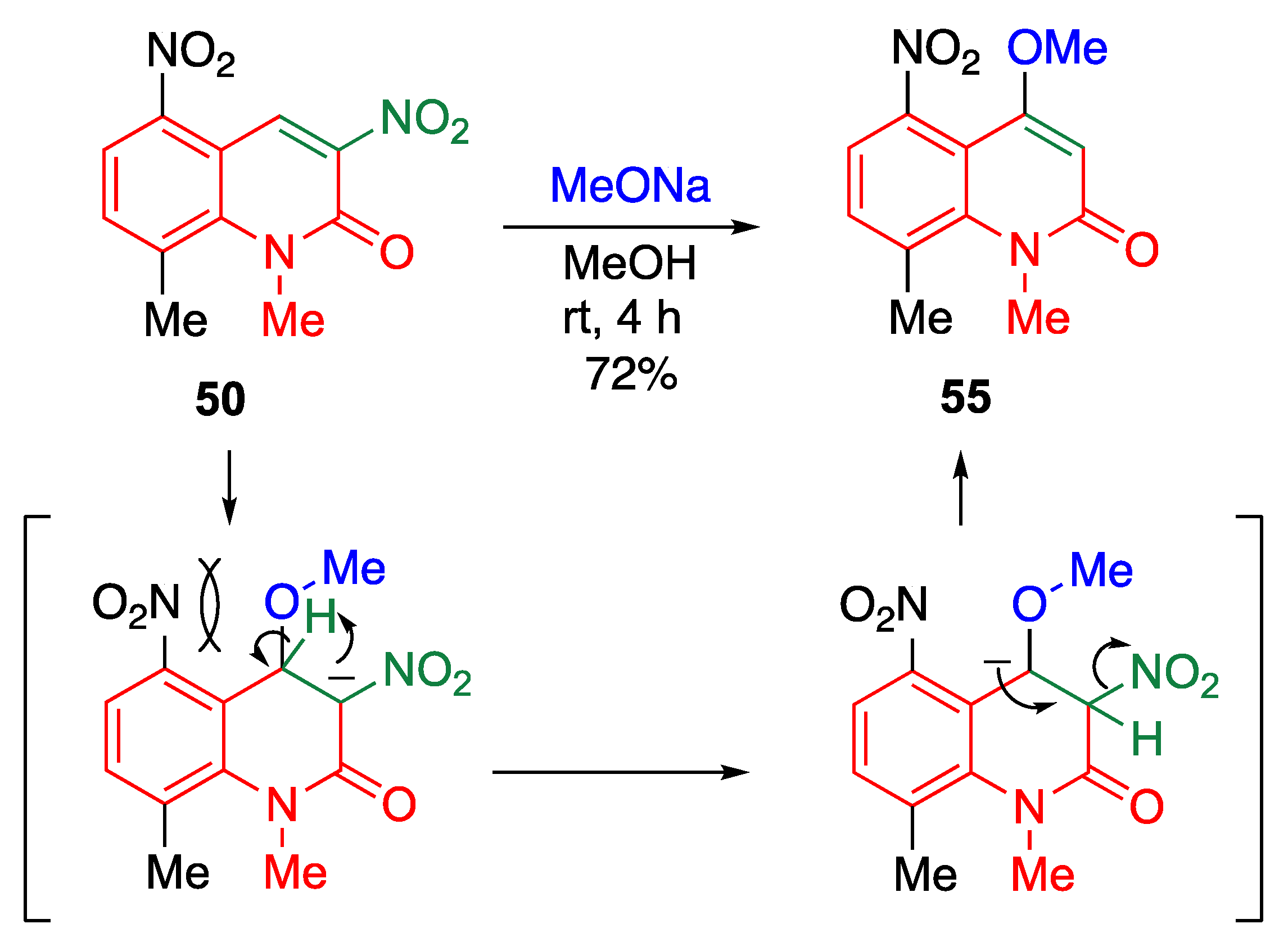
3,5-Dinitro-2-quinolone 50 exhibits a reactivity similar to that observed in amino-chlorination to afford the cine-substituted product 55 (Scheme 21). Although the addition of methoxide to 50 occurs at the 4-position, it cannot react with NCS at all because of steric repulsion between the 4-methoxy and 5-nitro groups. Instead, proton transfer followed by elimination of nitrite anion affords the cine-substituted product 55.
6. Conclusions
In this review, recent progress in the nitro-promoted direct functionalization of pyridones and quinolones was summarized. A variety of functionalities can be easily introduced into pyridone and quinolone frameworks via activation of the nitro group, facilitating the preparation of newly functionalized derivatives. These methods can promote the construction of a library of pyridones and quinolones with potentially interesting and valuable bioactivities. It is expected that more intensive research in this exciting field will establish the nitro-promoted direct functionalization of heterocycles as a powerful and broadly applicable synthetic strategy in organic synthesis.
Author Contributions
Each author contributed to this article equally. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Asif, M. A Mini Review: Biological Significances of Nitrogen Hetero Atom Containing Heterocyclic Compounds. Int. J. Bioorg. Chem. 2017, 2, 146–152. [Google Scholar]
- Joule, J.A. Chapter Four-Natural Products Containing Nitrogen Heterocycles-Some Highlights 1990–2015. Adv. Heterocycl. Chem. 2016, 119, 81–106. [Google Scholar]
- Kvasnica, M.; Urban, M.; Dickinson, N.J.; Sarek, J. Pentacyclic Triterpenoids with Nitrogen- and Sulfur-Containing Heterocycles: Synthesis and Medicinal Significance. Nat. Prod. Rep. 2015, 32, 1303–1330. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.M.; Sperry, J. Natural Products Containing a Nitrogen–Nitrogen Bond. J. Nat. Prod. 2013, 76, 794–812. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, J.; Baraniak, D.; Ostrowski, T. Bioactive Nucleoside Analogues Possessing Selected Five-Membered Azaheterocyclic Bases. Eur. J. Med. Chem. 2015, 97, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Miura, M. A Lesson for Site-Selective C-H Functionalization on 2-Pyridones: Radical, Organometallic, Directing Group and Steric Controls. Chem. Sci. 2018, 9, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Akihisa, T.; Yokokawa, S.; Ogihara, E.; Matsumoto, M.; Zhang, J.; Kikuchi, T.; Koike, K.; Abe, M. Melanogenesis-Inhibitory and Cytotoxic Activities of Limonoids, Alkaloids, and Phenolic Compounds from Phellodendron amurense Bark. Chem. Biodiversity 2017, 14, e1700105. [Google Scholar] [CrossRef]
- Seya, K.; Miki, I.; Murata, K.; Junke, H.; Motomura, S.; Araki, T.; Itoyama, Y.; Oshima, Y. Pharmacological Properties of Pteleprenine, a Quinoline Alkaloid Extracted from Orixa Japonica, on Guinea-Pig Ileum and Canine Left Atrium. J. Pharm. Pharmacol. 1998, 507, 803–807. [Google Scholar] [CrossRef]
- Kamikawa, T.; Hanaoka, Y.; Fujie, S.; Saito, K.; Yamagiwa, Y.; Fukuhara, K.; Kubo, I. SRS-A Antagonist Pyranoquinolone Alkaloids from East African Fagara Plants and Their Synthesis. Bioorg. Med. Chem. 1996, 4, 1317–1320. [Google Scholar] [CrossRef]
- Joseph, B.; Darro, F.; Béhard, A.; Lesur, B.; Collignon, F.; Decaestecker, C.; Frydman, A.; Guillaumet, G.; Kiss, R. 3-Aryl-2-Quinolone Derivatives: Synthesis and Characterization of In Vitro and In Vivo Antitumor Effects with Emphasis on a New Therapeutical Target Connected with Cell Migration. J. Med. Chem. 2002, 45, 2543–2555. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, F.; Jia, A.; Li, X. Palladium-Catalyzed Selective Oxidative Olefination and Arylation of 2-Pyridones. Chem. Sci. 2012, 3, 3231–3236. [Google Scholar] [CrossRef]
- Nakatani, A.; Hirano, K.; Satoh, T.; Miura, M. Nickel-Catalyzed Direct Alkylation of Heterocycles with α-Bromo Carbonyl Compounds: C3-Selective Functionalization of 2-Pyridones. Chem. Eur. J. 2013, 19, 7691–7695. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Min, Q.; Zhao, H.; Gu, J.; Zhang, X. A General Synthesis of Fluoroalkylated Alkenes by Palladium-Catalyzed Heck-Type Reaction of Fluoroalkyl Bromides. Angew. Chem. Int. Ed. 2014, 53, 1–6. [Google Scholar]
- Nakatani, A.; Hirano, K.; Satoh, T.; Miura, M. Manganese-Mediated C3-Selective Direct Alkylation and Arylation of 2-Pyridones with Diethyl Malonates and Arylboronic Acids. J. Org. Chem. 2014, 79, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Elenicha, O.V.; Lytvyn, R.Z.; Skripskaya, O.V.; Lyavinets, O.S.; Pitkovych, K.E.; Yagodinets, P.I.; Obushak, M.D. Synthesis of Nitrogen-Containing Heterocycles on the Basis of 3-(4-Acetylphenyl)-1-methylquinolin-2(1H)-one. Russ. J. Org. Chem. 2016, 52, 373–378. [Google Scholar] [CrossRef]
- Li, J.; Hu, D.; Liang, X.; Wang, Y.; Wang, H.; Pan, Y. Praseodymium(III)-Catalyzed Regioselective Synthesis of C3-N-Substituted Coumarins with Coumarins and Azides. J. Org. Chem. 2017, 82, 9006–9011. [Google Scholar] [CrossRef]
- Prendergast, A.M.; McGlacken, G.P. Transition Metal Mediated C–H Activation of 2-Pyrones, 2-Pyridones, 2-Coumarins and 2-Quinolones. Eur. J. Org. Chem. 2018, 6068–6082. [Google Scholar] [CrossRef]
- Maity, S.; Das, D.; Sarkar, S.; Samanta, R. Direct Pd(II)-Catalyzed Site-Selective C5-Arylation of 2-Pyridone Using Aryl Iodides. Org. Lett. 2018, 20, 5167–5171. [Google Scholar] [CrossRef]
- Diesel, J.; Finogenova, A.M.; Cramer, N. Nickel-Catalyzed Enantioselective Pyridone C–H Functionalizations Enabled by a Bulky N-Heterocyclic Carbene Ligand. J. Am. Chem. Soc. 2018, 140, 4489–4493. [Google Scholar] [CrossRef]
- Hazra, S.; Hirano, K.; Miura, M. Solvent-Controlled Rhodium-Catalyzed C6-Selective C–H Alkenylation and Alkynylation of 2-Pyridones with Acrylates. Asian J. Org. Chem. 2019, 8, 1097–1101. [Google Scholar] [CrossRef]
- Zhao, H.; Xu, X.; Luo, Z.; Cao, L.; Li, B.; Li, H.; Xu, L.; Fan, Q.; Walsh, P.J. Rhodium(I)-Catalyzed C6-Selective C–H Alkenylation and Polyenylation of 2-Pyridones with Alkenyl and Conjugated Polyenyl Carboxylic Acids. Chem. Sci. 2019, 10, 10089–10096. [Google Scholar] [CrossRef]
- Nishiwaki, N. Chemistry of Nitroquinolones and Synthetic Application to Unnatural 1-Methyl-2-quinolone Derivatives. Molecules 2010, 15, 5174–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderari, G.; Seebach, D. Asymmetric Michael-Additions. Stereoselective Alkylation of Chiral, Non-racemic Enolates by Nitroolefins. Preparation of Enantiomerically Pure γ-Aminobutyric and Succinic Acid Derivatives. Helv. Chim. Acta 1985, 68, 1592–1604. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G.; Fiorini, D.; Petrini, M. Unprecedented, Selective Nef Reaction of Secondary Nitroalkanes Promoted by DBU under Basic Homogeneous Conditions. Tetrahedron Lett. 2002, 43, 5233–5235. [Google Scholar] [CrossRef]
- Wehrli, P.A.; Schaer, B. Direct Transformation of Primary Nitro Compounds into Nitriles. New Syntheses of α,β-Unsaturated Nitriles and Cyanohydrin Acetates. J. Org. Chem. 1977, 42, 3956–3958. [Google Scholar] [CrossRef]
- Orlandi, M.; Brenna, D.; Harms, R.; Jost, S.; Benaglia, M. Recent Developments in the Reduction of Aromatic and Aliphatic Nitro Compounds to Amines. Org. Process Res. Dev. 2018, 22, 430–445. [Google Scholar] [CrossRef]
- Yadav, M.R.; Nagaoka, M.; Kashihara, M.; Zhong, R.-L.; Miyazaki, T.; Sakai, S.; Nakao, Y. The Suzuki–Miyaura Coupling of Nitroarenes. J. Am. Chem. Soc. 2017, 139, 9423–9426. [Google Scholar] [CrossRef]
- Inoue, F.; Kashihara, M.; Yadav, M.R.; Nakao, Y. Buchwald–Hartwig Amination of Nitroarenes. Angew. Chem. Int. Ed. 2017, 56, 13307–13309. [Google Scholar] [CrossRef]
- Halimehjani, A.Z.; Namboothiri, I.N.N.; Hooshmand, S.E. Nitroalkenes in the Synthesis of Carbocyclic Compounds. RSC Adv. 2014, 4, 31261–31299. [Google Scholar] [CrossRef]
- Hao, F.; Nishiwaki, N. Chemistry of Nitroaziridines. Heterocycles 2019, 99, 54–72. [Google Scholar]
- Hao, F.; Yokoyama, S.; Nishiwaki, N. Direct Dihalo-Alkoxylation of Nitroalkenes Leading to β,β-Dihalo-β-nitroethyl Alkyl Ethers. Org. Biomol. Chem. 2018, 16, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Asahara, H.; Sofue, A.; Kuroda, Y.; Nishiwaki, N. Alkynylation and Cyanation of Alkenes Using Diverse Properties of a Nitro Group. J. Org. Chem. 2018, 83, 13691–13699. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, E.J.; Bakke, J.M.; Sletvold, I.; Svensen, H. Nucleophilic Alkylations of 3-Nitropyridines. Org. Biomol. Chem. 2004, 2, 2671–2676. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Watanabe, K.; Nishiuchi, Y.; Honda, R.; Matsuzaki, H.; Hongo, H. Diels-Alder Reactions of Nitro-2(1H)-pyridones with 2,3-Dimethyl-1,3-butadiene. Chem. Pharm. Bull. 2001, 49, 601–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, R.; Yoshisuji, T.; Wakayanagi, S.; Wakamatsu, H.; Matsuzaki, H. Synthesis of 5(6H)-Phenanthridones Using Diels-Alder Reaction of 3-Nitro-2(1H)-quinolones Acting Dienophiles. Chem. Pharm. Bull. 2006, 54, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, R.; Watanabe, K.; Yoshisuji, T.; Kabuto, C.; Matsuzaki, H.; Hongo, H. Diels-Alder Reaction of 2(1 H)-quinolones Having an Electron-Withdrawing Group at the 3-Position Acting as Dienophiles with Dienes. Chem. Pharm. Bull. 2001, 49, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Asahara, M.; Nagamatsu, M.; Tohda, Y.; Nishiwaki, N.; Ariga, M. Diels-Alder Reaction of 1-Methyl-3,6,8-trinitro-2-quinolone. J. Heterocycl. Chem. 2004, 41, 803–805. [Google Scholar] [CrossRef]
- Asahara, M.; Shibano, C.; Koyama, K.; Tamura, M.; Tohda, Y.; Ariga, M. The Nitroalkene Showing Dual Behaviors in the Same Reaction System. Tetrahedron Lett. 2005, 46, 7519–7521. [Google Scholar] [CrossRef]
- Nishiwaki, N.; Tanaka, C.; Asahara, M.; Asaka, N.; Tohda, Y.; Ariga, M. A Nitro Group Distorting 2-Quinolone Skeleton. Heterocycles 1999, 51, 567–574. [Google Scholar]
- Murashima, T.; Nishi, K.; Nakamoto, K.; Kato, A.; Tamai, R.; Uno, H.; Ono, N. Preparation of Novel Heteroisoindoles from Nitropyridines and Nitropyridones. Heterocycles 2002, 58, 301–310. [Google Scholar] [CrossRef]
- Nishiwaki, N.; Tanaka, A.; Uchida, M.; Tohda, Y.; Ariga, M. cine-Substitution of 1-Methyl-3,6,8-trinitro-2-quinolone. Bull. Chem. Soc. Jpn. 1996, 69, 1337–1381. [Google Scholar] [CrossRef]
- Chen, X.; Kobiro, K.; Asahara, H.; Kakiuchi, K.; Sugimoto, R.; Saigo, K.; Nishiwaki, N. Reactive 2-Quinolones Dearomatized by Steric Repulsion between 1-Methyl and 8-Substituted Groups. Tetrahedron 2013, 69, 4624–4630. [Google Scholar] [CrossRef] [Green Version]
- Asahara, M.; Ohtsutsumi, M.; Ariga, M.; Nishiwaki, N. Regioselective Nitroalkylation of the 1-Methyl-2-quinolone Framework. Heterocycles 2009, 78, 2851–2854. [Google Scholar]
- Asahara, M.; Katayama, T.; Tohda, Y.; Nishiwaki, N.; Ariga, M. Synthesis of Unnatural 1-Methyl-2-quinolone Derivatives. Chem. Pharm. Bull. 2004, 52, 1134–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasahara, M.; Ohtsutsumi, M.; Tamura, M.; Nishiwaki, N.; Ariga, M. Electrophilic Arylation of Phenols: Construction of a New Family of 1-Methyl-2-quinolones. Bull. Chem. Soc. Jpn. 2005, 78, 2235–2237. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Li, Y.; Xiang, S.; Fan, W.; Jin, J.; Huang, D. Utilization of Nitriles as the Nitrogen Source: Practical and Economical Construction of 4-Aminopyrimidine and β-Enaminonitrile Skeletons. Org. Chem. Front. 2019, 6, 3071–3077. [Google Scholar] [CrossRef]
- Ghosh, T.; Si, A.; Misra, A.K. Facile Transformation of Nitriles into Thioamides: Application to C-Glycosyl Nitrile Derivatives. ChemistrySelect 2017, 2, 1366–1369. [Google Scholar] [CrossRef]
- Xi, F.; Kamal, F.; Schenerman, M.A. A Novel and Convenient Transformation of Nitriles to Aldehydes. Tetrahedron Lett. 2002, 43, 1395–1396. [Google Scholar] [CrossRef]
- Nishiwaki, N.; Sakashita, M.; Azuma, M.; Tanaka, C.; Tamura, M.; Asaka, N.; Hori, K.; Tohda, Y.; Ariga, M. Novel Functionalization of 1-Methyl-2-quinolone; Dimerization and Denitration of Trinitroquinolone. Tetrahedron 2002, 58, 473–478. [Google Scholar] [CrossRef]
- Asahara, M.; Nagamatsu, M.; Tohda, Y.; Nishiwaki, N.; Ariga, M. Effective C-N Bond Formation on the 1-Methyl-2-quinolone Skeleton. ARKIVOC 2005, 1, 1–6. [Google Scholar]
- Hao, F.; Asahara, H.; Nishiwaki, N. Direct Amino-halogenation and Aziridination of the 2-Quinolone Framework by Sequential Treatment of 3-Nitro-2-quinolone with Amine and N-Halosuccinimide. Tetrahedron 2017, 73, 1255–1264. [Google Scholar] [CrossRef]
- Hao, F.; Asahara, H.; Nishiwaki, N. A Direct and Vicinal Functionalization of the 1-Methyl-2-quinolone Framework: 4-Alkoxylation and 3-Chlorination. Org. Biomol. Chem. 2016, 14, 5128–5135. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Biological activities of pyridones and quinolones.

Figure 2.
Conventional strategies for the functionalization of aza-heterocycles.

Figure 3.
Resonance structure of pyridone framework.

Scheme 1.
Properties of a nitro group.

Scheme 2.
Two plausible pathways for cycloadducts 5 and 6 including denitration.

Scheme 3.
Diels-Alder cycloaddition of trinitroquinolone 16 with cyclopentadiene.

Scheme 4.
Cycloaddition of 16 with α,β-unsaturated oxime.

Figure 4.
ORTEP (30% probability ellipsoids) view of trinitroquinolone 16.

Figure 5.
Dual behaviors of a nitroalkene in the cycloaddition reaction.

Scheme 5.
Cycloaddition of 16 with ethoxyethane.

Scheme 6.
A plausible mechanism for the formation of product 22.

Scheme 7.
Cyclization of nitropyridones 23 and 24.

Scheme 8.
A plausible mechanism for cyclization of nitropyridone 23 with isocyanoacetate.

Scheme 9.
cine-Substitution of trinitroquinolone 16.

Figure 6.
cine-Substituted products from 16 and potassium phenoxides.

Scheme 10.
A plausible mechanism for the reaction of 16 with phenoxide.

Scheme 11.
cine-Substitution of 16 with potassium cyanide.

Scheme 12.
cine-Substitution of 16 with trimethylsilyl cyanide.

Scheme 13.
A plausible mechanism for dimerization of 16.

Scheme 14.
Two reaction paths leading to 41 and 16.

Scheme 15.
Reaction of Meisenheimer complex 40a with NCS.

Scheme 16.
A plausible mechanism for the formation of 42a and 43a.

Scheme 17.
Synthesis of imido-chlorinated product 44 and the hydrazinolysis.

Scheme 18.
Different reactivity of 3,5-dinitro-2-quinolone 50 for the amino-chlorination.

Scheme 19.
Aziridine ring opening leading to vicinally functionalized quinolone.

Scheme 20.
A plausible mechanism for alkoxy-chlorination of MeQones.

Scheme 21.
cine-Substitution of 3,5-dinitro-2-quinolone 50 by sodium methoxide.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
D-A cycloaddition of 5-nitropyridones 1 with diene 2.

| R1 | R3 | Yield/% | |
|---|---|---|---|
| H | H | a | 26 |
| Me | H | b | 30 |
| Me | COOMe | c | 22 |
Table 2.
D-A reaction of 5-nitropyridones 4 accompanying elimination of the nitro group.

| R3 | Yield/% | ||
|---|---|---|---|
| 5 | 6 | ||
| H | a | 27 | 10 |
| NO2 | b | 33 | 15 |
Table 3.
Cycloaddition of 3-nitropyridones 7 with diene 2.

| R1 | R4 | Yield/% | ||
|---|---|---|---|---|
| 8 | 9 | |||
| H | H | a | 0 | 15 |
| Me | H | b | 20 | 22 |
| Me | COOMe | c | 36 | 0 |
Table 4.
Cycloaddition of 5-substituted 3-nitropyridones 10 with diene 2.

| R1 | R5 | Yield/% | |||
|---|---|---|---|---|---|
| 11 | 12 | 13 | |||
| H | NO2 | a | 13 | 15 | 0 |
| Me | NO2 | b1 | 36 | 33 | 0 |
| Me | COOMe | c | 31 | 0 | 14 |
| H | COOMe | d | 13 | 0 | 5 |
1 8% of 8b is obtained.
Table 5.
Cycloaddition of 3-nitrated quinolones 14 with dienes 2.

| R | R1 | R2 | R3 | Yield/% | |
|---|---|---|---|---|---|
| H | OMe | H | H | a | 83 |
| NO2 | OMe | H | H | b | 68 |
| H | H | Me | Me | c | 95 |
| NO2 | H | Me | Me | d | 64 |
| H | H | OMe | OMe | e | 45 |
| NO2 | H | OMe | OMe | f | 13 |
Table 6.
cine-Substitution of 16 with 1,3-dicarbonyl compounds.

| R1 | R2 | Yield/% | |
|---|---|---|---|
| Me | Me | a | 88 |
| -(CH2)3- | b | 68 | |
| Me | OEt | c | 93 |
| CH2COOEt | OEt | d | 26 |
| OEt | OEt | e | 93 |
Table 7.
Effect of the substituent at the 8-position for the cine-substitution.

| R | Yield/% |
|---|---|
| NO2 | 88 |
| H | 0 1 |
| Me | 92 |
1 At 80 °C.
Table 8.
cine-Substitution of 16 with nitroalkanes.

| R1 | R2 | Yield/% |
|---|---|---|
| Me | H | 80 |
| Et | H | 98 |
| Me | Me | 77 1 |
1 For 1 d.
Table 9.
cine-Substitution of 16 with ketones.

| R1 | R2 | R3 | Yield/% |
|---|---|---|---|
| H | Me | Me | 41 |
| Me | H | H | 83 |
| Ph | H | H | 83 |
| Et | Me | H | 18 |
| -(CH2)4- | H | 82 | |
| Ph | Me | H | 77 |
| Ph | Ph | H | 69 |
| 2-furyl | H | H | 45 |
| 2-pyridyl | H | H | 74 |
Table 10.
cine-Substitution of 16 with enamines.

| R1 | R2 | R3 | Yield/% |
|---|---|---|---|
| H | Me | Me | 98 |
| -(CH2)4- | H | 40 | |
| Ph | Me | H | 43 |
| Ph | Ph | H | 98 |
Table 11.
cine-Substitution of nitrated 1,8-dimethyl-2-quinolones with trimethylsilyl cyanide.

| R5 | R6 | R7 | Yield/% | |
|---|---|---|---|---|
| NO2 | H | NO2 | a | 83 |
| NO2 | H | H | b | 47 |
| H | NO2 | H | c | quant. |
Table 12.
Reactions of 16 with tertiary amines.

| R1 | R2 | R3 | Yield/% |
|---|---|---|---|
| Me | Me | Me | 0 |
| Et | Et | Et | 34 |
| Pr | Pr | Pr | 76 |
| Bu | Bu | Bu | 93 |
| Bu | Bu | Me | 79 |
| Bu | Me | Me | 18 |
| PhCH2 | PhCH2 | PhCH2 | 0 |
Table 13.
cine-Substitution of 16 with primary amines.

| R | Yield/% | ||
|---|---|---|---|
| 40 | 41 | ||
| Pr | a | 71 | 36 |
| i-Bu | b | 74 | 29 |
| s-Bu | c | 56 | 0 |
| t-Bu | d | 74 | 0 |
Table 14.
One-pot amino-chlorination of trinitroquinolone 16.

| R1 | R2 | Yield/% | |
|---|---|---|---|
| Pr | H | a | 62 |
| i-Bu | H | c | 70 |
| s-Bu | H | d | 49 |
| PhCH2 | H | e | 54 |
| HOCH2CH2 | H | f | 56 |
| CH2=CHCH2 | H | g | 35 |
| Ph | H | h | 54 |
| 4-MeOC6H4 | H | i | 37 |
| 4-BuC6H4 | H | j | 41 |
| 4-IC6H4 | H | k | 62 |
| 4-NO2C6H4 | H | l | trace |
| Et | Et | m | 0 |
| -(CH2)2-O-(CH2)2- | n | 62 | |
Table 15.
Scanning of halogenating agents.

| X | Yield/% | |
|---|---|---|
| 45 | 46 | |
| Cl | 62 | 0 |
| Br | 63 | 16 |
| I | 0 | 62 |
Table 16.
Amino-chlorination and aziridination of various 3-nitrated MeQones.

| R1 | R6 | R7 | R8 | Yield/% | ||
|---|---|---|---|---|---|---|
| 48 | 49 | |||||
| Me | NO2 | H | NO2 | a | 62 | 0 |
| Me | NO2 | H | Me | b | 13 | 21 |
| Me | NO2 | H | H | c | 13 | 49 |
| Me | Br | H | H | d | trace | 68 |
| Me | H | H | H | e | 0 | 65 |
| Me | H | H | H | f | 0 | 71 |
| H | H | H | H | g | 0 | 61 |
Table 17.
Alkoxy-chlorination of 16 leading to 53.

| R | R6 | R8 | Yield/% | ||
|---|---|---|---|---|---|
| 52 | 53 | ||||
| Me | NO2 | NO2 | a | 81 | 85 |
| Et | NO2 | NO2 | b | quant. | 73 |
| i-Bu | NO2 | NO2 | c | - | 46 |
| i-Pr | NO2 | NO2 | d | - | 45 |
| PhCH2CH2 | NO2 | NO2 | e | - | 55 |
| CH2=CHCH2 | NO2 | NO2 | f | - | 51 |
| HC≡CCH2 | NO2 | NO2 | g | - | 29 |
| Me | NO2 | Me | h | - | 78 |
| Me | NO2 | H | i | - | 73 |
| Me | H | H | j | - | 65 |
Table 18.
Scanning of halogenating agents.

| X | Yield/% | |
|---|---|---|
| 53 | 54 | |
| Cl | 85 | 0 |
| Br | 62 | 27 |
| I | 0 | 29 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hao, F.; Nishiwaki, N. Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones. Molecules 2020, 25, 673. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25030673
AMA Style
Hao F, Nishiwaki N. Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones. Molecules. 2020; 25(3):673. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25030673
Chicago/Turabian StyleHao, Feiyue, and Nagatoshi Nishiwaki. 2020. "Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones" Molecules 25, no. 3: 673. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25030673