
Complexes Between Adamantane Analogues B4X6 -X = {CH2, NH, O ; SiH2, PH, S} - and Dihydrogen, B4X6:nH2 (n = 1–4)



Abstract
:
1. Introduction
2. Results and Discussion
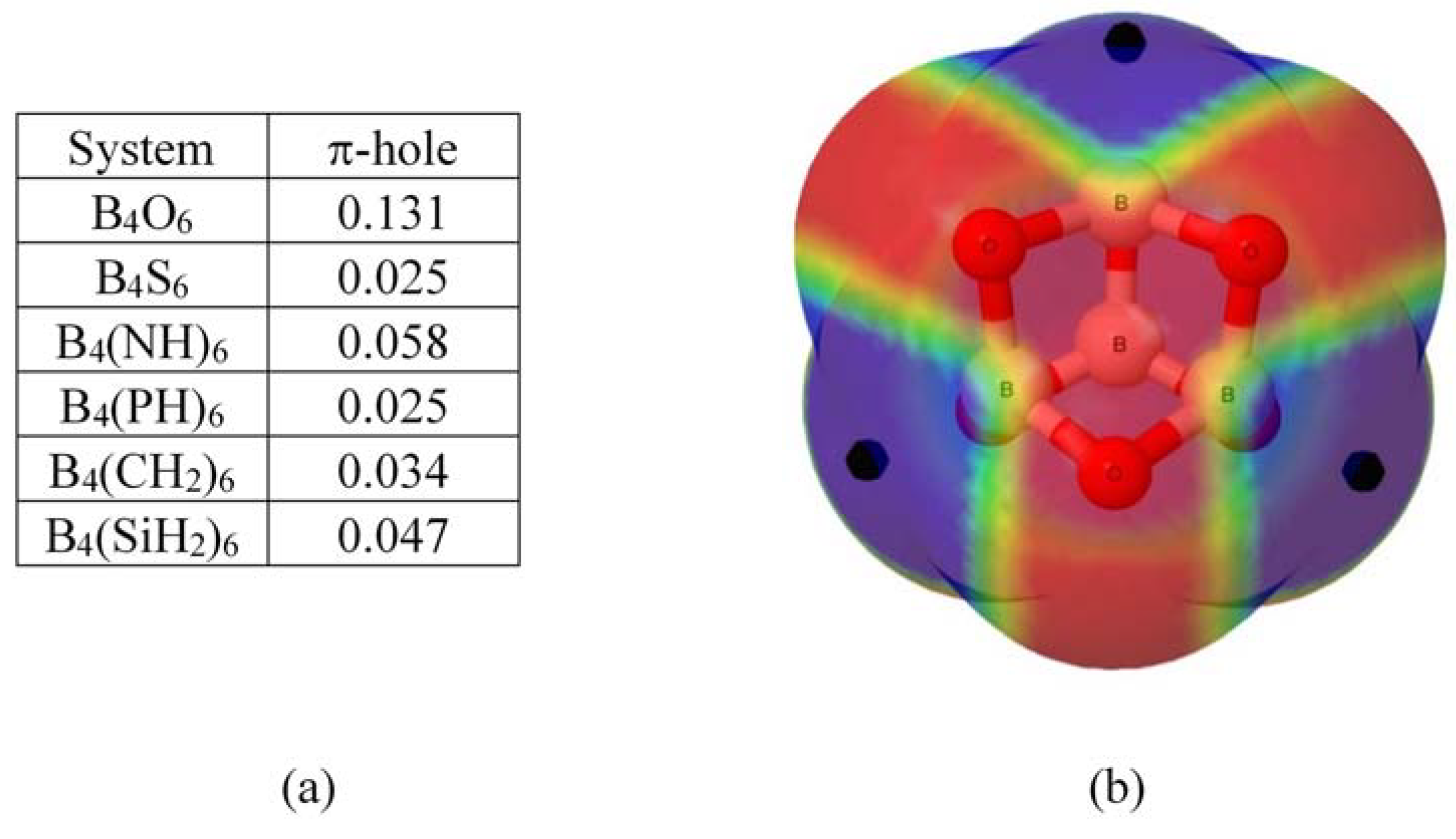
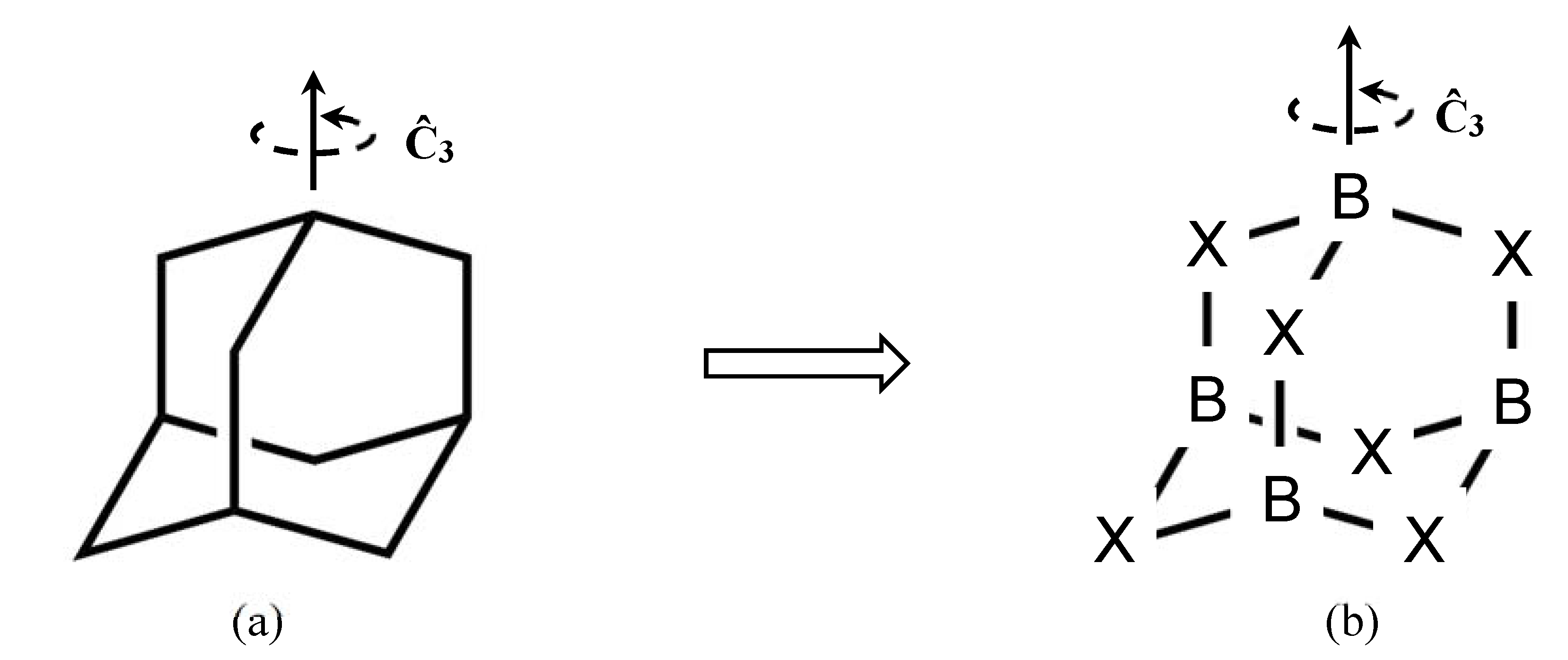
2.1. Molecular Electrostatic Potential (MESP) and π-Holes in B4X6 Systems
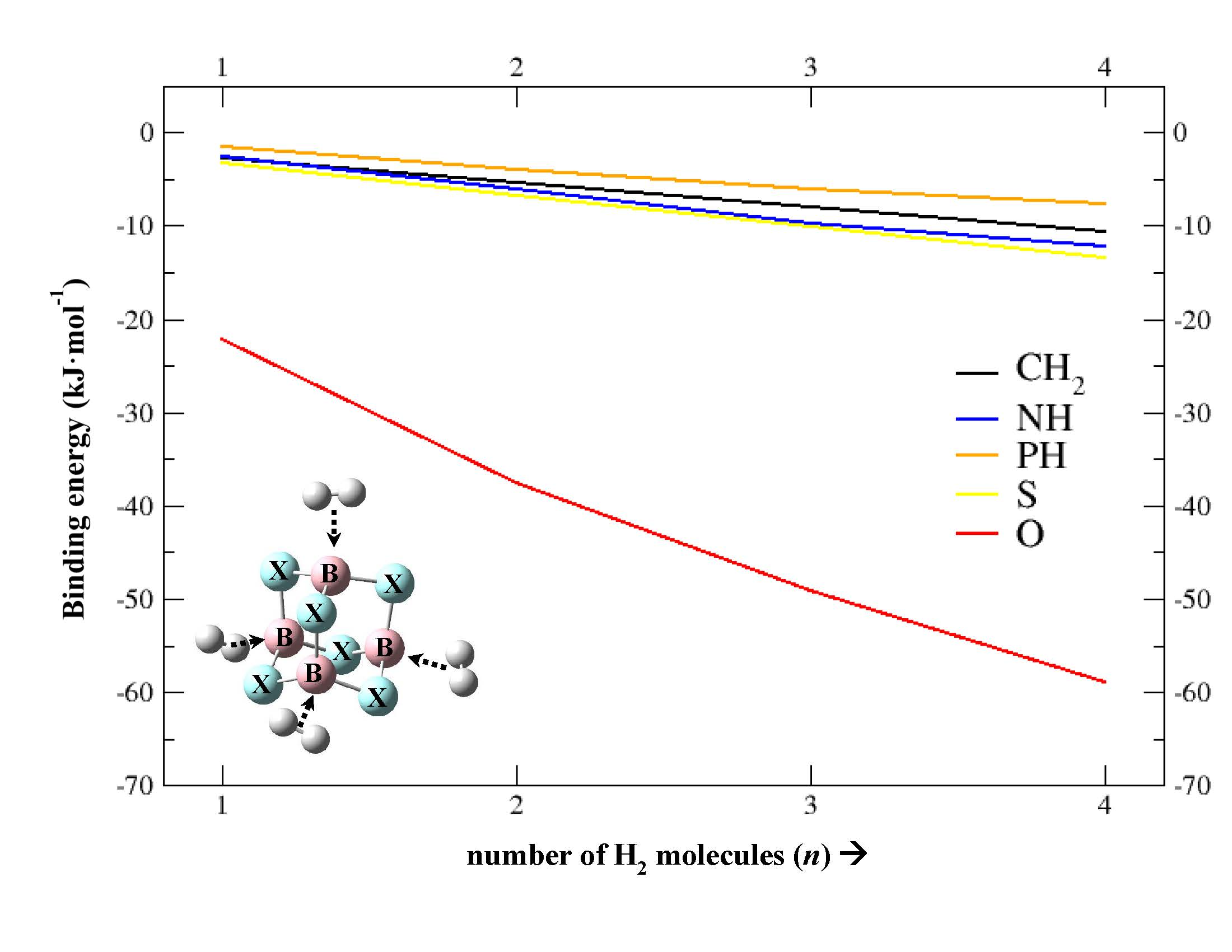
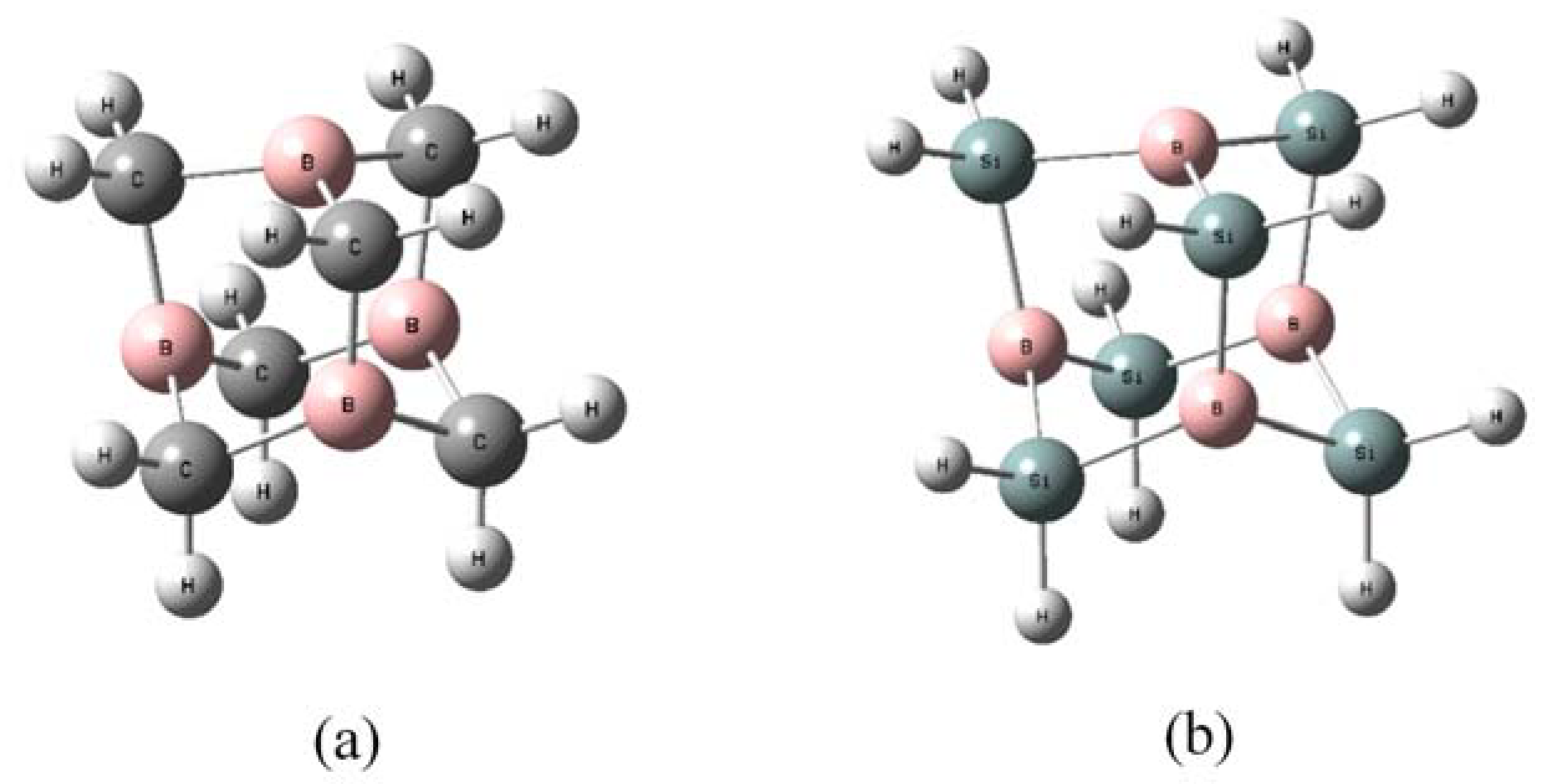
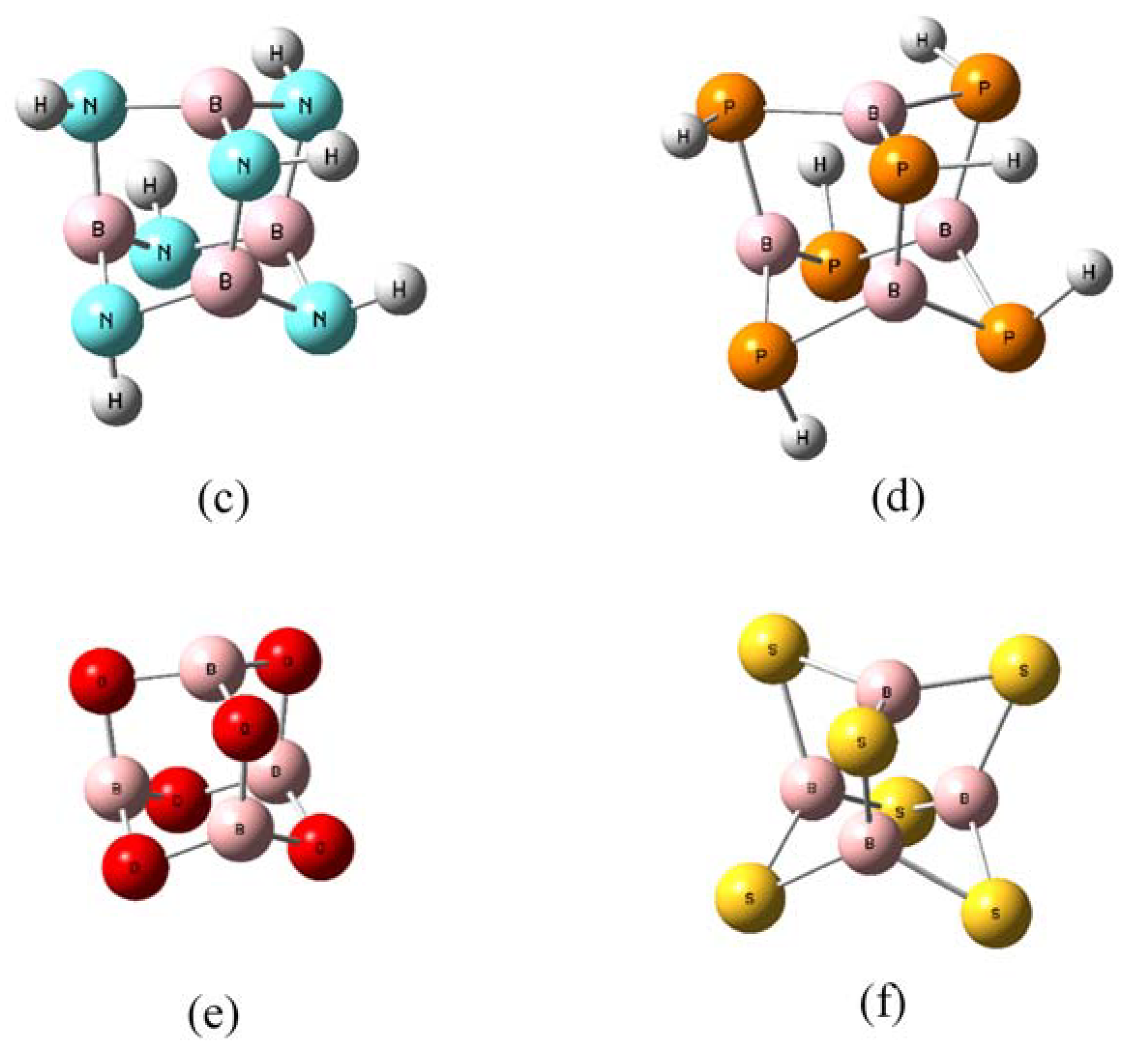
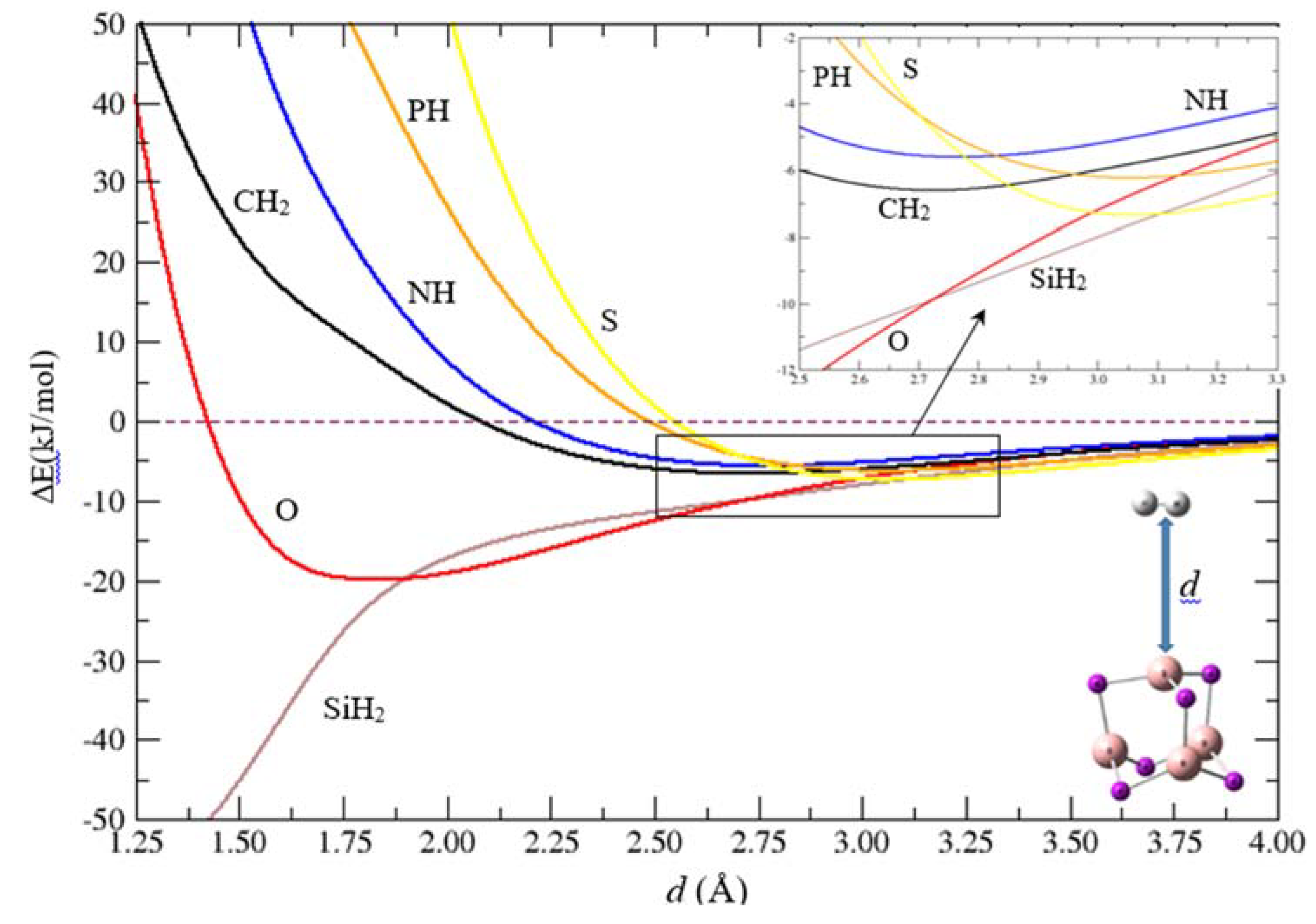
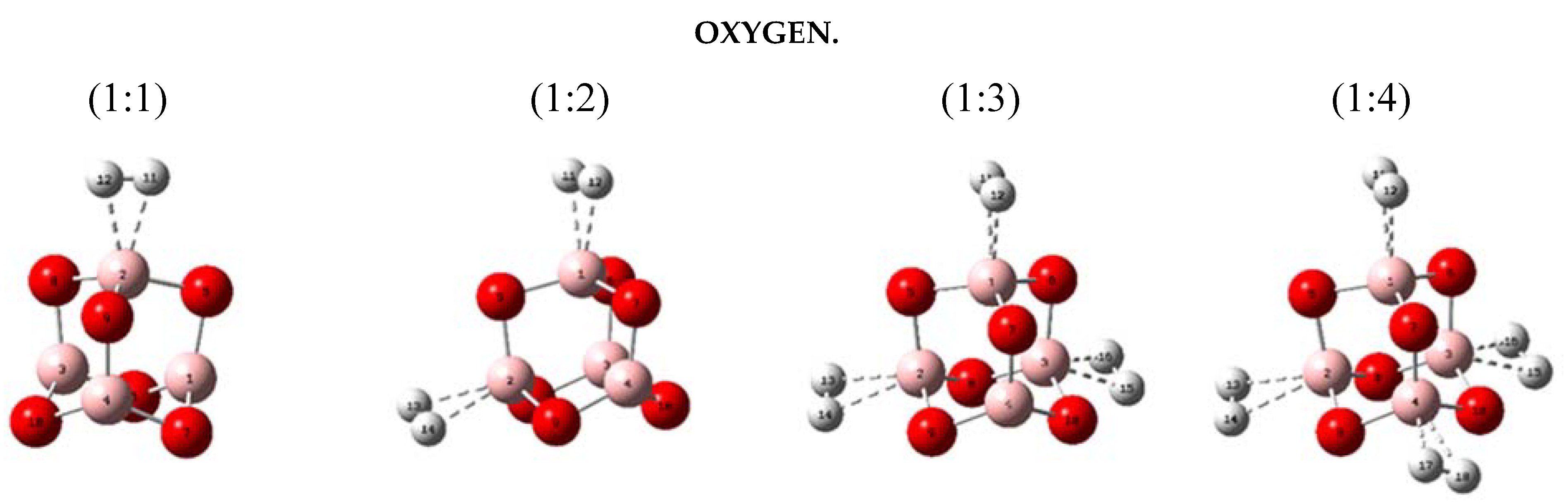
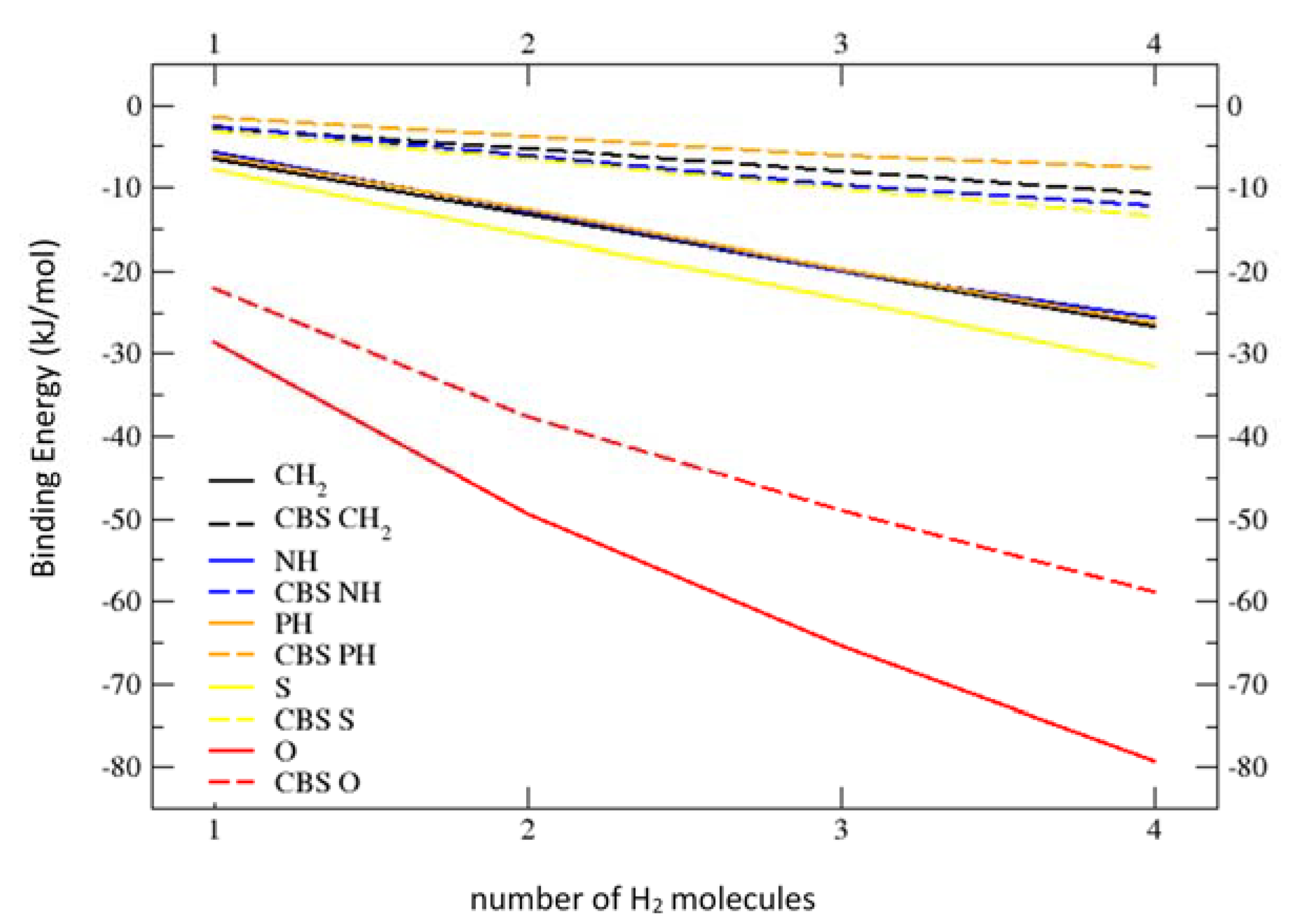
2.2. Geometries and Energies of B4X6:nH2 Complexes (n = 1–4)
3. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zell, T.; Langer, R. Hydrogen storage; Walter de Gruyter: Berlin, Germany, 2019. [Google Scholar]
- Moradi, R.; Groth, K.M. Hydrogen storage and delivery: Review of the state of the art technologies and risk and reliability analysis. Int. J. Hydro. Energy 2019, 44, 12254–12269. [Google Scholar] [CrossRef]
- Preuster, P.; Alekseev, A.; Wasserscheid, P. Hydrogen storage technologies for future energy systems. Annu. Rev. Chem. Biomol. 2017, 8, 445–471. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Sharma, V.K.; Kumar, E.A.; Gayathri, V. Hydrogen storage in carbon materials—A review. Energy Stor. 2019, 1, 1–26. [Google Scholar] [CrossRef]
- Khan, N.; Dilshad, S.; Khalid, R.; Kalair, A.R.; Abas, N. Review of energy storage and transportation of energy. Energy Stor. 2019, 1, 1–49. [Google Scholar] [CrossRef]
- Schneemann, A.; White, J.L.; Kang, S.; Jeong, S.; Wan, L.F.; Cho, E.S.; Heo, T.W.; Prendergast, D.; Urban, J.J.; Wood, B.C.; et al. Nanostructured metal hydrides for hydrogen storage. Chem. Rev. 2018, 118, 10775–10839. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.-W. Hydrogen storage in metal-organic frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef]
- Kapelewski, M.T.; Runčevski, T.; Tarver, J.D.; Jiang, H.Z.H.; Hurst, K.E.; Parilla, P.A.; Ayala, A.; Gennett, T.; Fitzgerald, S.A.; Brown, C.M.; et al. Record High Hydrogen Storage Capacity in the Metal–Organic Framework Ni2(m-dobdc) at Near-Ambient Temperatures. Chem. Mater. 2018, 30, 8179–8189. [Google Scholar] [CrossRef]
- Zhu, B.; Zou, R.; Xu, Q. Metal–organic framework based catalysts for hydrogen evolution. Adv. Energy Mater. 2018, 8, 1801193. [Google Scholar] [CrossRef]
- Yan, Y.; da Silva, I.; Blake, A.J.; Dailly, A.; Manuel, P.; Yang, S.; Schröder, M. High volumetric hydrogen adsorption in a porous anthracene-decorated metal–organic framework. Inorg Chem. 2018, 57, 12050–12055. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, K.; Nobusada, K.; Boero, M. Hydrogen storage mechanism and diffusion in metal–organic frameworks. Phys. Chem. Chem. Phys. 2019, 21, 7756–7764. [Google Scholar] [CrossRef]
- Hobza, P.; Müller-Dethlefs, K. Non-Covalent Interactions. Theory and Experiment; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Chakraborty, A.; Giri, S.; Chattaraj, P.K. Analyzing the efficiency of Mn–(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) complexes as effective hydrogen storage materials. Struct. Chem. 2011, 22, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.-J.; Jia, J.; Wu, H.-S. Computational investigation of hydrogen storage on scandium–acetylene system. Int. J. Hydro. Energy 2015, 40, 420–428. [Google Scholar] [CrossRef]
- Konda, R.; Titus, E.; Chaudhari, A. Adsorption of molecular hydrogen on inorganometallic complexes B2H4M (M.=Li, Be, Sc, Ti, V). Struct. Chem. 2018, 29, 1593–1599. [Google Scholar] [CrossRef]
- Konda, R.; Kalamse, V.; Deshmukh, A.; Chaudhari, A. Closoborate-transition metal complexes for hydrogen storage. RSC Adv. 2015, 5, 99207–99216. [Google Scholar] [CrossRef]
- Naumkin, F.Y.; Wales, D.J. Hydrogen trapped in Ben cluster cages: The atomic encapsulation option. Chem. Phys. Lett. 2012, 545, 44–49. [Google Scholar] [CrossRef]
- Meng, Y.; Han, Y.; Zhu, H.; Yang, Z.; Shen, K.; Suo, B.; Lei, Y.; Zhai, G.; Wen, Z. Two dimetallocenes with vanadium and chromium: Electronic structures and their promising application in hydrogen storage. Int. J. Hydro. Energy 2015, 40, 12047–12056. [Google Scholar] [CrossRef]
- Grabowski, S.; Ruiperez, F. Dihydrogen bond interactions as a result of H2 cleavage at Cu, Ag and Au centres. Phys. Chem. Chem. Phys. 2016, 18, 12810–12818. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Solimannejad, M.; Grabowski, S.J. Dihydrogen bonding vs metal−σ interaction in complexes between H2 and metal hydride. J. Phys. Chem. A 2011, 115, 201–210. [Google Scholar] [CrossRef]
- Alkorta, I.; Montero-Campillo, M.M.; Elguero, J.; Yáñez, M.; Mó, O. Complexes between H2 and neutral oxyacid beryllium derivatives. The role of angular strain. Mol. Phys. 2019, 117, 1142–1150. [Google Scholar] [CrossRef]
- Alkorta, I.; Montero-Campillo, M.M.; Elguero, J.; Yáñez, M.; Mó, O. Complexes between neutral oxyacid beryllium salts and dihydrogen: A possible way for hydrogen storage? Dalton Trans. 2018, 47, 12516–12520. [Google Scholar] [CrossRef]
- Kayal, S.; Manna, U.; Das, G. Steric influence of adamantane substitution in tris-urea receptor: Encapsulation of sulphate and fluoride-water cluster. J. Chem. Sci. 2018, 130, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hooley, R.J.; Shenoy, S.R.; Rebek, J., Jr. Electronic and steric effects in binding of deep cavitands. Org. Lett. 2008, 10, 5397–5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fort, R.C. Adamantane: The chemistry of diamond molecules; Marcel Dekker: New York, NY, USA, 1976. [Google Scholar]
- Spilovska, K.; Zemek, F.; Korabecny, J.; Nepovimova, E.; Soukup, O.; Windisch, M.; Kuca, K. Adamantane –A lead structure for drugs in clinical practice. Curr. Med. Chem. 2016, 23, 3245–3266. [Google Scholar] [CrossRef]
- Schwertfeger, H.; Fokin, A.A.; Schreiner, P.R. Diamonds are a chemist’s best friend: Diamondoid chemistry beyond adamantane. Angew. Chem. Int. Ed. 2008, 47, 1022–1036. [Google Scholar] [CrossRef]
- Bagrii, E.I.; Nekhaev, A.I.; Maksimov, A.L. Oxidative functionalization of adamantanes. Petrol. Chem. 2017, 57, 183–197. [Google Scholar] [CrossRef]
- Štimac, A.; Šekutor, M.; Mlinarić-Majerski, K.; Frkanec, L.; Frkanec, R. Adamantane in drug delivery systems and surface recognition. Molecules 2017, 22, 297. [Google Scholar] [CrossRef] [Green Version]
- Speckamp, W.N.; Dijkink, J.; Huisman, H.O. 1-Aza-adamantanes. J. Chem. Soc. D: Chem. Comm. 1970, 4, 197–198. [Google Scholar] [CrossRef]
- Shibuya, M.; Tomizawa, M.; Suzuki, I.; Iwabuchi, Y. 2-Azaadamantane N-Oxyl (AZADO) and 1-Me-AZADO: highly efficient organocatalysts for oxidation of alcohols. J. Am. Chem. Soc. 2006, 128, 8412–8413. [Google Scholar] [CrossRef]
- Kirillo, A.M. Hexamethylenetetramine: An old new building block for design of coordination polymers. Coordin. Chem. Rev. 2011, 255, 1603–1622. [Google Scholar] [CrossRef]
- Dreyfors, J.M.; Jones, S.B.; Sayed, Y. Hexamethylenetetramine: A review. Am. Ind. Hyg. Assoc. J. 1989, 50, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, I.; Guella, G.; Frostin, M.; Hnawia, E.; Laurent, D.; Debitus, C.; Pietra, F. On the first polyarsenic organic compound from Nature: Arsenicin A from the new caledonian marine sponge echinochalina bargibanti. Chem. Eur. J. 2006, 12, 8989–8994. [Google Scholar] [CrossRef]
- Whitlow, K.S.; Belson, M.; Barrueto, F.; Nelson, L.; Henderson, A.K. Tetramethylenedisulfotetramine: Old agent and new terror. Ann. Emerg. Med. 2005, 45, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://en.wikipedia.org/wiki/Phosphorus_pentasulfide (accessed on 12 December 2019).
- Shi, Y.X.; Xu, K.; Clegg, J.K.; Ganguly, R.; Hirao, H.; Friščić, T.; García, F. The first synthesis of the sterically encumbered adamantoid phosphazane P4(NtBu)6: Enabled by mechanochemistry. Angew. Chem. Int. Ed. 2016, 55, 12736–12740. [Google Scholar] [CrossRef] [PubMed]
- Kutsumura, N.; Ohshita, R.; Horiuchi, J.; Tateno, K.; Yamamoto, N.; Saitoh, T.; Nagumo, Y.; Kawai, H.; Nagase, H. Synthesis of heterocyclic compounds with adamantane-like cage structures consisting of phosphorus, sulfur, and carbon. Tetrahedron 2017, 73, 5214–5219. [Google Scholar] [CrossRef]
- Mikhailov, B.M. Reactions of allylboranes with unsaturated compounds. Pure Appl. Chem. 1980, 52, 691–704. [Google Scholar] [CrossRef]
- El-Hamdi, M.; Solà, M.; Poater, J.; Timoshkin, A.Y. Complexes of adamantane-based group 13 Lewis acids and superacids: Bonding analysis and thermodynamics of hydrogen splitting. J. Comput. Chem. 2016, 37, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- García, J.C.; Justo, J.F.; Machado, W.V.M.; Assali, L.V.C. Functionalized adamantane: Building blocks for nanostructure self-assembly. Phys. Rev. B 2009, 80, 125421. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Mohajeri, A.; Pakiari, A.H.; Bagheri, N. Theoretical studies on the nature of bonding in σ-hole complexes. Chem. Phys. Lett. 2009, 467, 393–397. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.; Concha, M. σ-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Solimannejad, M.; Alkorta, I.; Elguero, J. Orthogonal interactions between nitryl derivatives and electron donors: Pnictogen bonds. Phys. Chem. Chem. Phys. 2013, 15, 14310–14318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solimannejad, M.; Ramezani, V.; Trujillo, C.; Alkorta, I.; Sánchez-Sanz, G.; Elguero, J. Competition and interplay between σ-hole and π-hole interactions: A computational study of 1:1 and 1:2 complexes of nitryl halides (O2NX) with ammonia. J. Phys. Chem. A 2012, 116, 5199–5206. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Lane, P.; Clark, T.; Riley, K.; Politzer, P. σ-holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Saida, A.B.; Chardon, A.; Osi, A.; Tumanov, N.; Wouters, J.; Adjieufack, A.I.; Champagne, B.; Berionni, G. Pushing the Lewis acidity boundaries of boron compounds with non-planar triarylboranes derived from triptycenes. Angew. Chem. Int. Ed. 2019, 58, 1–6. [Google Scholar]
- Feyereisen, M.W.; Feller, D.; Dixon, D.A. Hydrogen bond energy of the water dimer. J. Phys. Chem. 1996, 100, 2993–2997. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Gaussian 09, Revision, E.01; Gaussian Inc.: Wallingford, CT, USA, 2019.
- Kumar, A.; Yeole, S.D.; Gadre, S.R.; López, R.; Rico, J.F.; Ramirez, G.; Ema, I.; Zorrilla, D. DAMQT 2.1.0: A new version of the DAMQT package enabled with the topographical analysis of electron density and electrostatic potential in molecules. J. Comput. Chem. 2015, 36, 2350–2359. [Google Scholar] [CrossRef]
- López, R.; Fernández Rico, J.; Ramírez, G.; Ema, I.; Zorrilla, D. DAMQT: A package for the analysis of electron density in molecules. Comput. Phys. Commun. 2009, 180, 1654–1660. [Google Scholar] [CrossRef]
- Feller, D. The use of systematic sequences of wave functions for estimating the complete basis set, full configuration interaction limit in water. J. Chem. Phys. 1993, 98, 7059. [Google Scholar] [CrossRef]
- Williams, T.G.; DeYonker, N.J.; Wilson, A.K. Hartree-Fock complete basis set limit properties for transition metal diatomics. J. Chem. Phys. 2008, 128, 044101. [Google Scholar] [CrossRef] [PubMed]
- Halkier, A.; Helgaker, T.; Jorgensen, P.; Klopper, W.; Olsen, J. Basis-set convergence of the energy in molecular Hartree–Fock calculations. Chem. Phys. Lett. 1999, 302, 437–446. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B···H2 | H-H | |||||||
|---|---|---|---|---|---|---|---|---|
| X | 1:1 | 1:2 | 1:3 | 1:4 | 1:1 | 1:2 | 1:3 | 1:4 |
| CH2 | 2.737 | 2.740 | 2.745 | 2.750 | 0.755 | 0.755 | 0.754 | 0.754 |
| NH | 2.777 | 2.727 | 2.705 | 2.727 | 0.755 | 0.755 | 0.755 | 0.755 |
| PH | 3.080 | 3.045 | 3.046 | 3.049 | 0.755 | 0.755 | 0.755 | 0.755 |
| O | 1.624 | 1.687 | 1.766 | 1.850 | 0.774 | 0.770 | 0.766 | 0.763 |
| S | 3.071 | 3.070 | 3.068 | 3.068 | 0.755 | 0.755 | 0.755 | 0.755 |
| X | ΔE(1:1) | ΔE(1:2) | ΔE(1:3) | ΔE(1:4) |
|---|---|---|---|---|
| MP2 CBS | MP2 CBS | MP2 CBS | MP2 CBS | |
| CH2 | −6.6 −2.8 | −13.3 −5.4 | −20.1 −8.0 | −26.8 −10.7 |
| NH | −5.8 −2.5 | −12.9 −6.1 | −20.0 −9.7 | −25.8 −12.2 |
| PH | −6.2 −1.6 | −13.3 −3.9 | −19.9 −6.1 | −26.4 −7.7 |
| O | −28.6 −22.1 | −49.5 −37.6 | −65.5 −49.1 | −79.2 −58.9 |
| S | −7.9 −3.3 | −15.8 −6.7 | −23.4 −10.1 | −31.6 −13.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva-Enrich, J.M.; Alkorta, I.; Elguero, J. Complexes Between Adamantane Analogues B4X6 -X = {CH2, NH, O ; SiH2, PH, S} - and Dihydrogen, B4X6:nH2 (n = 1–4). Molecules 2020, 25, 1042. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051042
Oliva-Enrich JM, Alkorta I, Elguero J. Complexes Between Adamantane Analogues B4X6 -X = {CH2, NH, O ; SiH2, PH, S} - and Dihydrogen, B4X6:nH2 (n = 1–4). Molecules. 2020; 25(5):1042. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051042
Chicago/Turabian StyleOliva-Enrich, Josep M., Ibon Alkorta, and José Elguero. 2020. "Complexes Between Adamantane Analogues B4X6 -X = {CH2, NH, O ; SiH2, PH, S} - and Dihydrogen, B4X6:nH2 (n = 1–4)" Molecules 25, no. 5: 1042. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051042