
Direct Analysis of Psilocin and Muscimol in Urine Samples Using Single Drop Microextraction Technique In-Line with Capillary Electrophoresis


Abstract
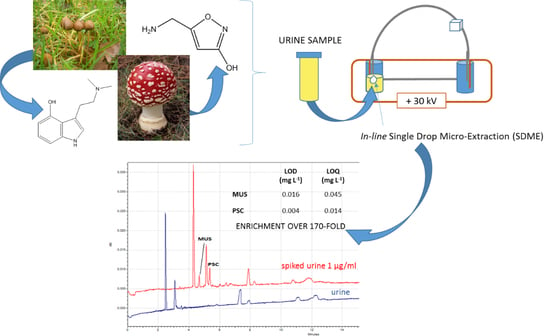
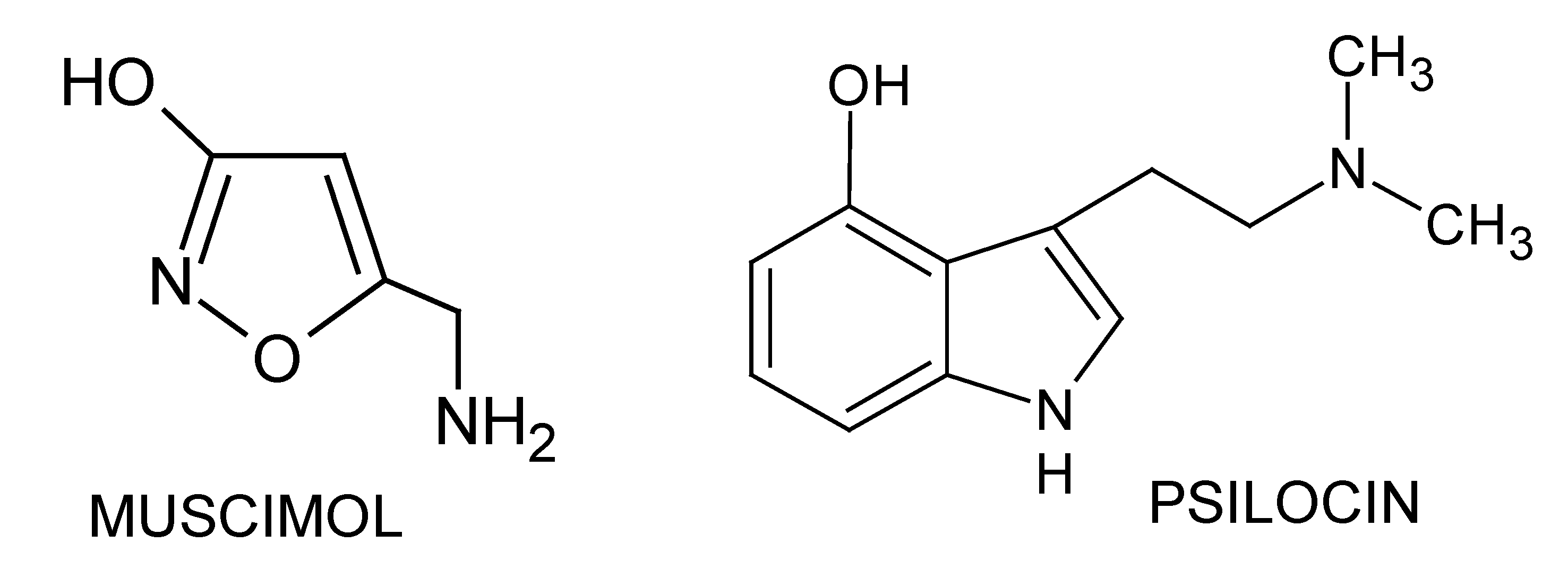
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Optimization of SDME-CE parameters
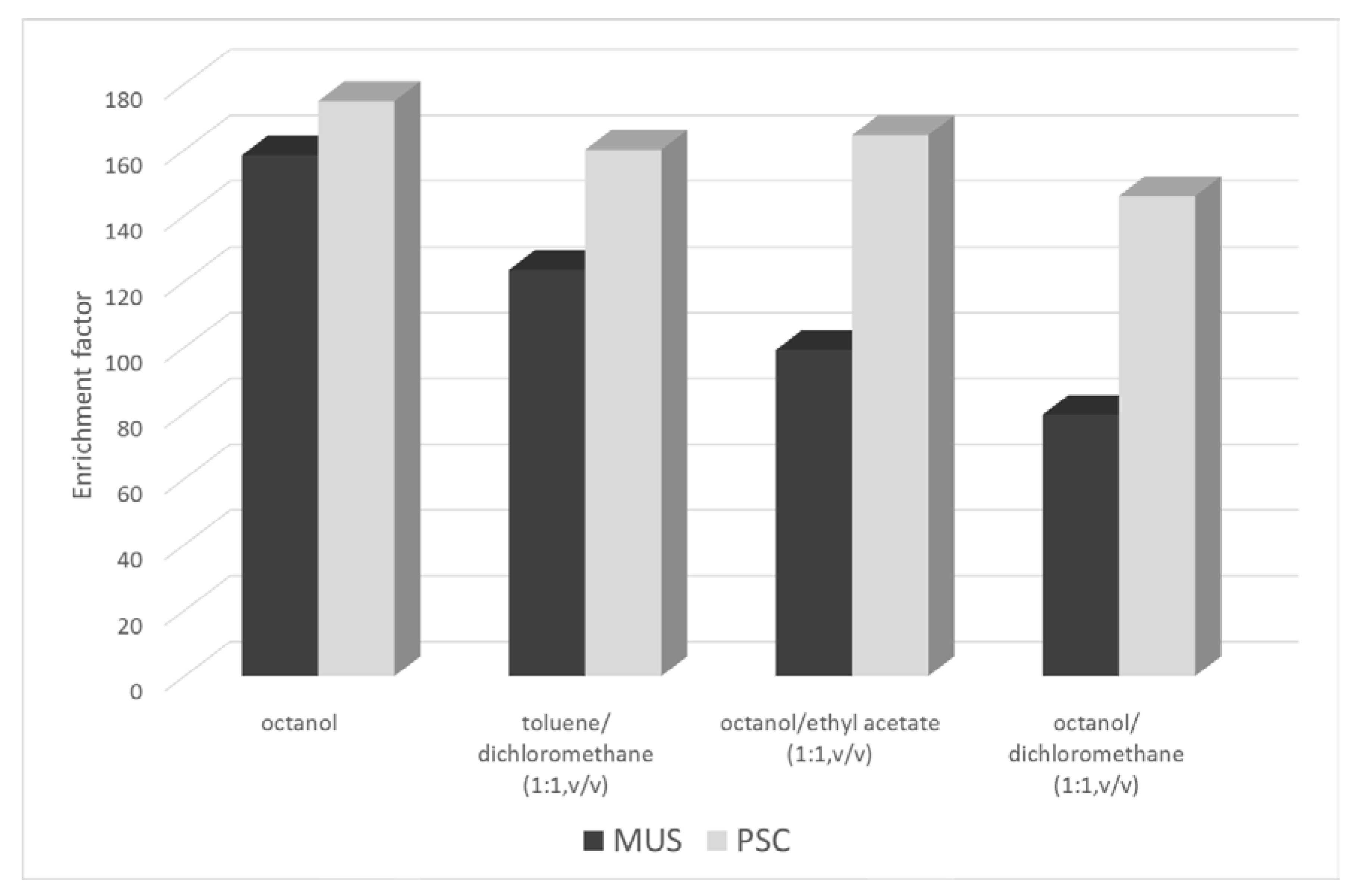
2.1.1. Selection of the Organic Phase and Drop Formation
2.1.2. Adjustment of the Composition of Acceptor and Donor Phase
2.1.3. Influence of the Extraction Time and Injection Parameters
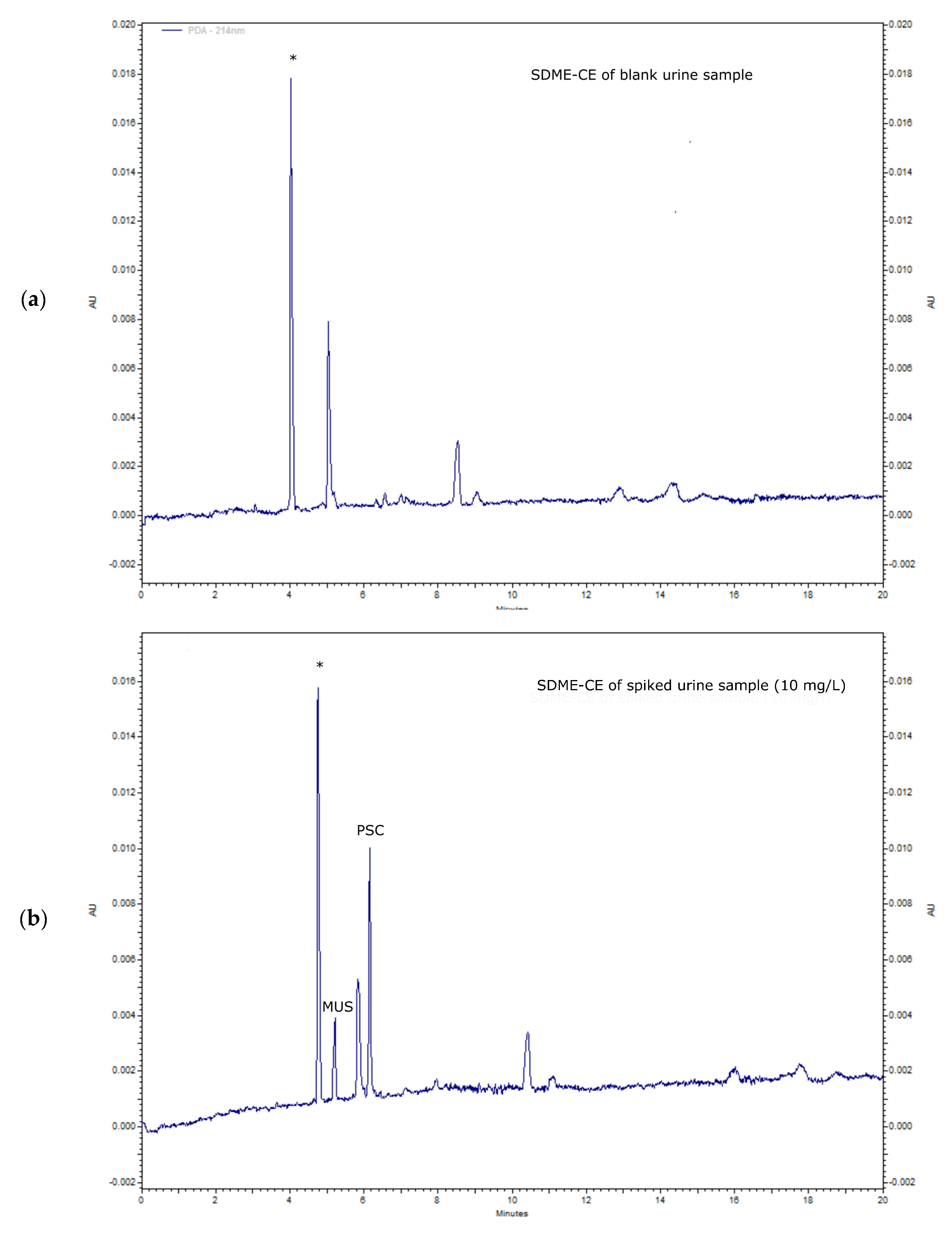
2.2. Real Samples Analysis
3. Materials and Methods
3.1. Chemicals
3.2. SDME–CE Procedures
3.3. Urine Samples Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Michelot, D.; Melendez-Howell, L.M. Amanita muscaria: Chemistry, biology, toxicology, and ethnomycology. Mycol. Res. 2003, 107, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Stebelska, K. Fungal Hallucinogens Psilocin, Ibotenic Acid, and Muscimol: Analytical Methods and Biologic Activities. Ther. Drug Monit. 2013, 35, 420–442. [Google Scholar] [CrossRef] [PubMed]
- Halpern, J.H. Hallucinogens and dissociative agents naturally growing in the United States. Pharmacol. Ther. 2004, 102, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, P.P.; Witkowska, D.; Jasicka-Misiak, I.; Poliwoda, A.; Oterman, M.; Zielińska, K. Chapter 5-Bioactive Alkaloids of Hallucinogenic Mushrooms. In Studies in Natural Products Chemistry; Attaur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 46, pp. 133–168. [Google Scholar]
- Satora, L.; Pach, D.; Ciszowski, K.; Winnik, L. Panther cap Amanita pantherina poisoning case report and review. Toxicon 2006, 47, 605–607. [Google Scholar] [CrossRef]
- Musshoff, F.; Madea, B.; Beike, J. Hallucinogenic mushrooms on the German market-simple instructions for examination and identification. Forensic. Sci. Int. 2000, 113, 389–395. [Google Scholar] [CrossRef]
- Zhuk, O.; Jasicka-Misiak, I.; Poliwoda, A.; Kazakova, A.; Godovan, V.V.; Halama, M.; Wieczorek, P.P. Research on acute toxicity and the behavioral effects of methanolic extract from psilocybin mushrooms and psilocin in mice. Toxins 2015, 7, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, N. Hallucinogenic plants of abuse. EMA Emerg. Med. Australas. 2008, 20, 167–174. [Google Scholar] [CrossRef]
- Hasler, F.; Bourquin, D.; Brenneisen, R.; Vollenweider, F.X. Renal excretion profiles of psilocin following oral administration of psilocybin: A controlled study in man. J. Pharmaceut. Biomed. Anal. 2002, 30, 331–339. [Google Scholar] [CrossRef]
- Hasegawa, K.; Gonmori, K.; Fujita, H.; Kamijo, Y.; Nozawa, H.; Yamagishi, I.; Minakata, K.; Watanabe, K.; Suzuki, O. Determination of ibotenic acid and muscimol, the Amanita mushroom toxins, in human serum by liquid chromatography-tandem mass spectrometry. Forensic. Toxicol. 2013, 31, 322–327. [Google Scholar] [CrossRef]
- Støíbrný, J.; Sokol, M.; Merová, B.; Ondra, P. GC/MS determination of ibotenic acid and muscimol in the urine of patients intoxicated with Amanita pantherina. Int. J. Legal Med. 2012, 126, 519–524. [Google Scholar] [CrossRef]
- Ginterová, P.; Sokolová, B.; Ondra, P.; Znaleziona, J.; Petr, J.; Ševčík, J.; Maier, V. Determination of mushroom toxins ibotenic acid, muscimol and muscarine by capillary electrophoresis coupled with electrospray tandem mass spectrometry. Talanta 2014, 125, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Gonmori, K.; Hasegawa, K.; Fujita, H.; Kamijo, Y.; Nozawa, H.; Yamagishi, I.; Minakata, K.; Watanabe, K.; Suzuki, O. Analysis of ibotenic acid and muscimol in Amanita mushrooms by hydrophilic interaction liquid chromatography–tandem mass spectrometry. Forensic. Toxicol. 2012, 30, 168–172. [Google Scholar] [CrossRef]
- Poliwoda, A.; Zielińska, K.; Halama, M.; Wieczorek, P.P. Determination of muscimol and ibotenic acid in mushrooms of Amanitaceae by capillary electrophoresis. Electrophoresis 2014, 35, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, K.; Kuwayama, K.; Miyaguchi, H.; Kanamori, T.; Iwata, Y.; Inoue, H.; Yoshida, T.; Kishi, T. Determination of muscimol and ibotenic acid in Amanita mushrooms by high-performance liquid chromatography and liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2007, 852, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, K.; Mohri, H.; Kuwayama, K.; Miyaguchi, H.; Iwata, Y.; Gohda, A.; Fukushima, S.; Inoue, H.; Kishi, T. Analysis of hallucinogenic constituents in Amanita mushrooms circulated in Japan. Forensic. Sci. Int. 2006, 164, 172–178. [Google Scholar] [CrossRef]
- Kamata, T.; Nishikawa, M.; Katagi, M.; Tsuchihashi, H. Direct detection of serum psilocin glucuronide by LC/MS and LC/MS/MS: Time-courses of total and free (unconjugated) psilocin concentrations in serum specimens of a “magic mushroom” user. Forensic. Toxicol. 2006, 24, 36–40. [Google Scholar] [CrossRef]
- Bogusz, M.J. Liquid chromatography-mass spectrometry as a routine method in forensic sciences: A proof of maturity. J. Chromatogry. B 2000, 748, 3–19. [Google Scholar] [CrossRef]
- Elian, A.A.; Hackett, J.; Telepchak, M.J. Analysis of psilocybin and psilocin in urine using SPE and LC-tandem mass spectrometry. LC GC N. Am. 2011, 29, 854–859. [Google Scholar]
- Fernandez, M.D.M.R.; Laloup, M.; Wood, M.; De Boeck, G.; Lopez-Rivadulla, M.; Wallemacq, P.; Samyn, N. Liquid chromatography-tandem mass spectrometry method for the simultaneous analysis of multiple hallucinogens, chlorpheniramine, ketamine, ritalinic acid, and metabolites, in urine. J. Anal. Toxicol. 2007, 31, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Björnstad, K.; Beck, O.; Helander, A. A multi-component LC–MS/MS method for detection of ten plant-derived psychoactive substances in urine. J. Chromatogr. B 2009, 877, 1162–1168. [Google Scholar] [CrossRef]
- Martin, R.; Schürenkamp, J.; Pfeiffer, H.; Köhler, H. A validated method for quantitation of psilocin in plasma by LC–MS/MS and study of stability. Int. J. Legal Med. 2012, 126, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Schürenkamp, J.; Gasse, A.; Pfeiffer, H.; Köhler, H. Determination of psilocin, bufotenine, LSD and its metabolites in serum, plasma and urine by SPE-LC-MS/MS. Int. J. Legal Med. 2013, 127, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, M.; Yan, X.; Wu, E.; Zhu, H.; Lee, K.J.; Chu, V.M.; Zhan, L.; Lee, W.; Kang, J.S. Determining the pharmacokinetics of psilocin in rat plasma using ultra-performance liquid chromatography coupled with a photodiode array detector after orally administering an extract of Gymnopilus spectabilis. J. Chromatogr. B 2011, 879, 2669–2672. [Google Scholar] [CrossRef] [PubMed]
- Lindenblatt, H.; Krämer, E.; Holzmann-Erens, P.; Gouzoulis-Mayfrank, E.; Kovar, K.A. Quantitation of psilocin in human plasma by high-performance liquid chromatography and electrochemical detection: Comparison of liquid-liquid extraction with automated on-line solid-phase extraction. J. Chromatogry. B 1998, 709, 255–263. [Google Scholar] [CrossRef]
- Albers, C.; Kohler, H.; Lehr, M.; Brinkmann, B.; Beike, J. Development of a psilocin immunoassay for serum and blood samples. Int. J. Legal Med. 2004, 118, 326–331. [Google Scholar] [CrossRef]
- Sticht, G.; Kaferstein, H. Detection of psilocin in body fluids. Forensic. Sci. Int. 2000, 113, 403–407. [Google Scholar] [CrossRef]
- Alnajjar, A.; Idris, A.M.; Multzenberg, M.; McCord, B. Development of a capillary electrophoresis method for the screening of human urine for multiple drugs of abuse. J. Chromatogr. B 2007, 856, 62–67. [Google Scholar] [CrossRef]
- Cheng, K.; Choi, K.; Kim, J.; Sung, I.H.; Chung, D.S. Sensitive arsenic analysis by carrier-mediated counter-transport single drop microextraction coupled with capillary electrophoresis. Microchem. J. 2013, 106, 220–225. [Google Scholar] [CrossRef]
- Choi, J.; Choi, K.; Kim, J.; Ahmed, A.Y.B.H.; Al-Othman, Z.A.; Chung, D.S. Sensitive analysis of amino acids with carrier-mediated single drop microextraction in-line coupled with capillary electrophoresis. J. Chromatogr. A 2011, 1218, 7227–7233. [Google Scholar] [CrossRef]
- Choi, K.; Kim, J.; Jang, Y.O.; Chung, D.S. Direct chiral analysis of primary amine drugs in human urine by single drop microextraction in-line coupled to CE. Electrophoresis 2009, 30, 2905–2911. [Google Scholar] [CrossRef]
- Gao, W.; Chen, G.; Chen, Y.; Zhang, X.; Yin, Y.; Hu, Z. Application of single drop liquid–liquid–liquid microextraction for the determination of fluoroquinolones in human urine by capillary electrophoresis. J. Chromatogr. B 2011, 879, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Choi, K.; Ahmed, A.Y.B.H.; Alothman, Z.A.; Chung, D.S. Highly sensitive chiral analysis of amino acids by in-line single drop microextraction and capillary electrophoresis with laser-induced fluorescence detection. Anal. Chim. Acta 2010, 677, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Choi, K.; Chung, D.S. Double sample preconcentration by in-line coupled large volume single drop microextraction and sweeping in capillary electrophoresis. Electrophoresis 2009, 30, 1953–1957. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-K.; Choi, K.; Ahmed, A.Y.B.H.; Alothman, Z.A.; Chung, D.S. Selective preconcentration of amino acids and peptides using single drop microextraction in-line coupled with capillary electrophoresis. J. Chromatogr. A 2010, 1217, 3357–3361. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qiu, H.; Li, J.; Han, H.; Liu, X.; Jiang, S. Novel approach to improve the detection of colchicine via online coupling of ionic liquid-based single-drop microextraction with capillary electrophoresis. J. Sep. Sci. 2011, 34, 594–600. [Google Scholar] [CrossRef]
- Wang, Q.; Qiu, H.; Li, J.; Liu, X.; Jiang, S. On-line coupling of ionic liquid-based single-drop microextraction with capillary electrophoresis for sensitive detection of phenols. J. Chromatogr. A 2010, 1217, 5434–5439. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhou, X.; Yan, N.; Zhou, L.; Chen, X. On-line combination of single-drop liquid–liquid–liquid microextraction with capillary electrophoresis for sample cleanup and preconcentration: A simple and efficient approach to determining trace analyte in real matrices. J. Chromatogr. A 2010, 1217, 1856–1861. [Google Scholar] [CrossRef]
- Alothman, Z.A.; Dawod, M.; Kim, J.; Chung, D.S. Single-drop microextraction as a powerful pretreatment tool for capillary electrophoresis: A review. Anal. Chim. Acta 2012, 739, 14–24. [Google Scholar] [CrossRef]
- Shibue, M.; Mant, C.T.; Hodges, R.S. Effect of anionic ion-pairing reagent hydrophobicity on selectivity of peptide separations by reversed-phase liquid chromatography. J. Chromatogr. A 2005, 1080, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, J.Å.; Mathiasson, L. Liquid membrane extraction in analytical sample preparation: I. Principles. TRAC Trend Anal. Chem. 1999, 18, 318–325. [Google Scholar] [CrossRef]
- Dżygiel, P.; Wieczorek, P.P. Chapter 3-Supported Liquid Membranes and Their Modifications: Definition, Classification, Theory, Stability, Application and Perspectives A2-Kislik, Vladimir S. In Liquid Membranes; Elsevier: Amsterdam, The Netherlands, 2010; pp. 73–140. [Google Scholar]
- Kiani, F.; Abbaszadeh, M.; Pousti, M.; Koohyar, F. Theoretically nanoscale study on ionization of muscimol nano drug in aqueous solution. Braz. J. Pharm. Sci. 2015, 51, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Chen, G.; Chen, T.; Zhang, X.; Chen, Y.; Hu, Z. Directly suspended droplet microextraction combined with single drop back-extraction as a new approach for sample preparation compatible with capillary electrophoresis. Talanta 2011, 83, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poliwoda, A.; Zielińska, K.; Wieczorek, P.P. Direct Analysis of Psilocin and Muscimol in Urine Samples Using Single Drop Microextraction Technique In-Line with Capillary Electrophoresis. Molecules 2020, 25, 1566. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071566
Poliwoda A, Zielińska K, Wieczorek PP. Direct Analysis of Psilocin and Muscimol in Urine Samples Using Single Drop Microextraction Technique In-Line with Capillary Electrophoresis. Molecules. 2020; 25(7):1566. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071566
Chicago/Turabian StylePoliwoda, Anna, Katarzyna Zielińska, and Piotr P. Wieczorek. 2020. "Direct Analysis of Psilocin and Muscimol in Urine Samples Using Single Drop Microextraction Technique In-Line with Capillary Electrophoresis" Molecules 25, no. 7: 1566. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071566