
One-Pot Iridium Catalyzed C–H Borylation/Sonogashira Cross-Coupling: Access to Borylated Aryl Alkynes

 , ,
, ,
Abstract
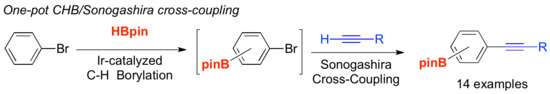
:
1. Introduction
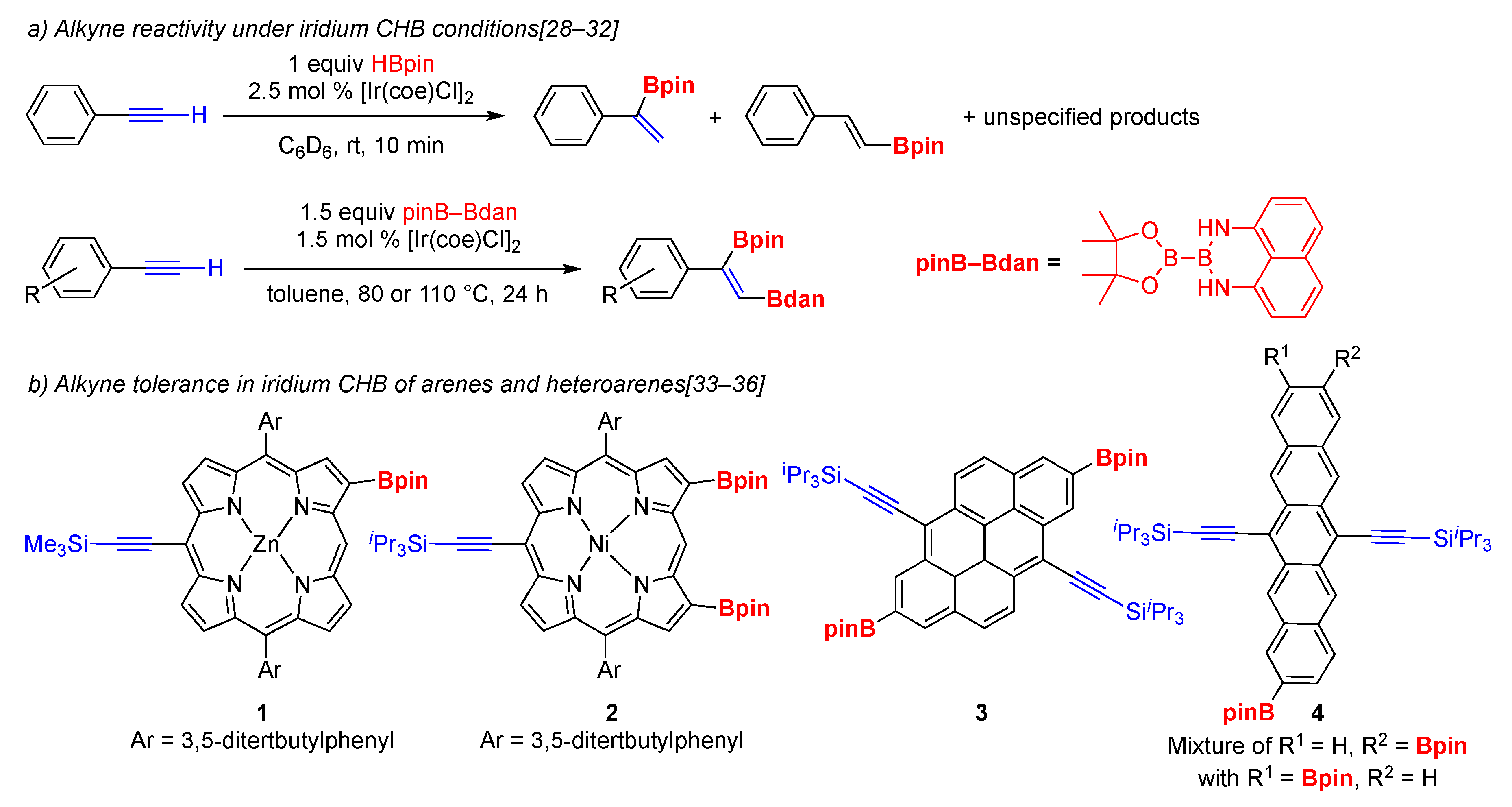
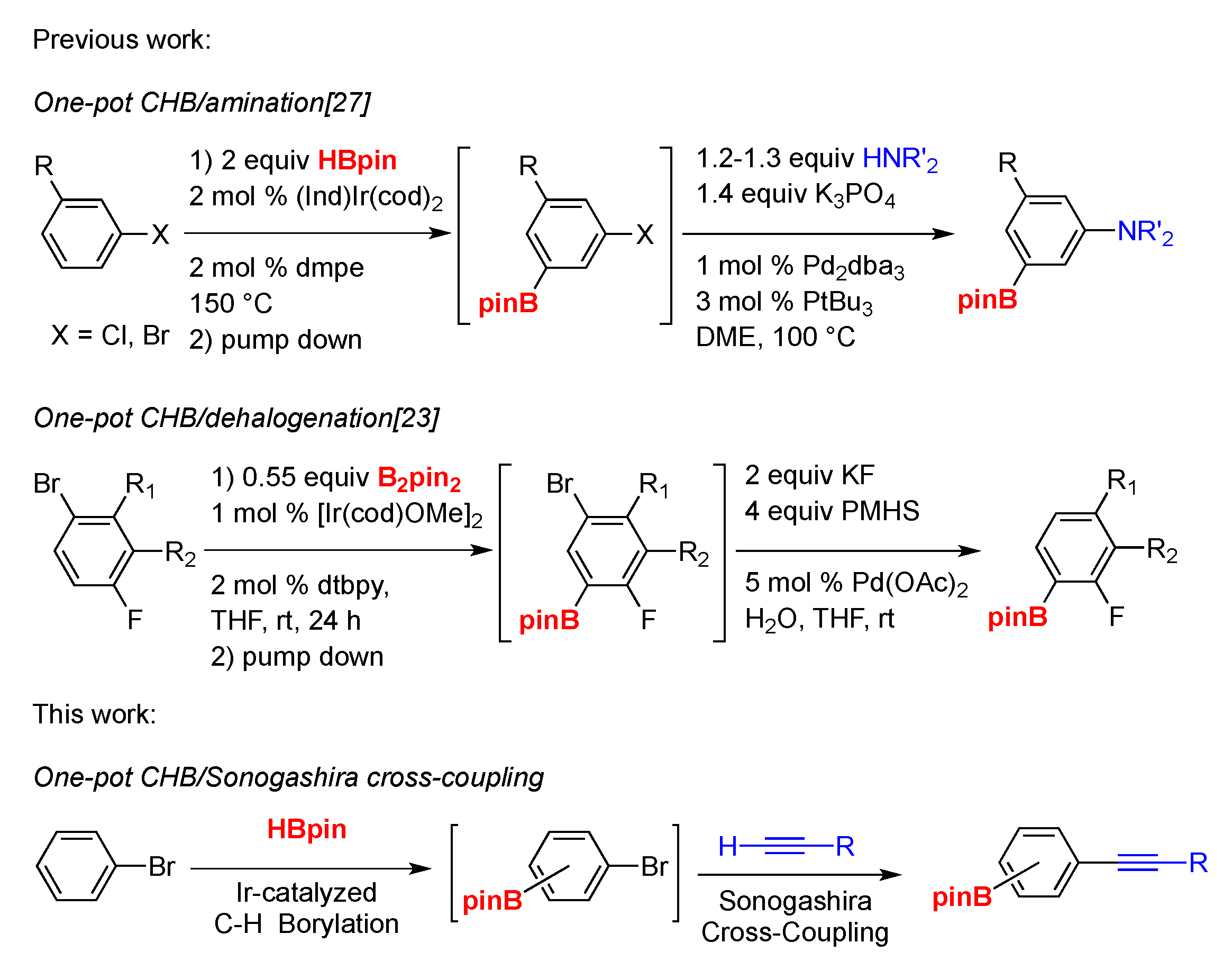
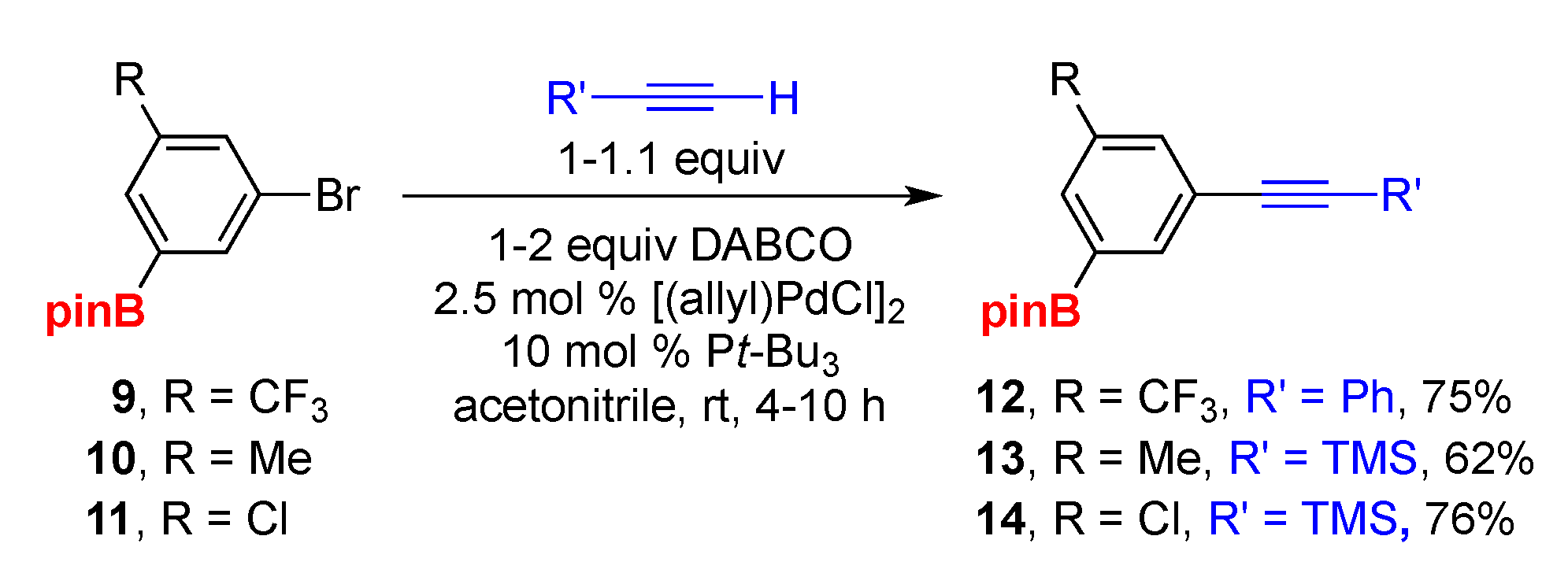
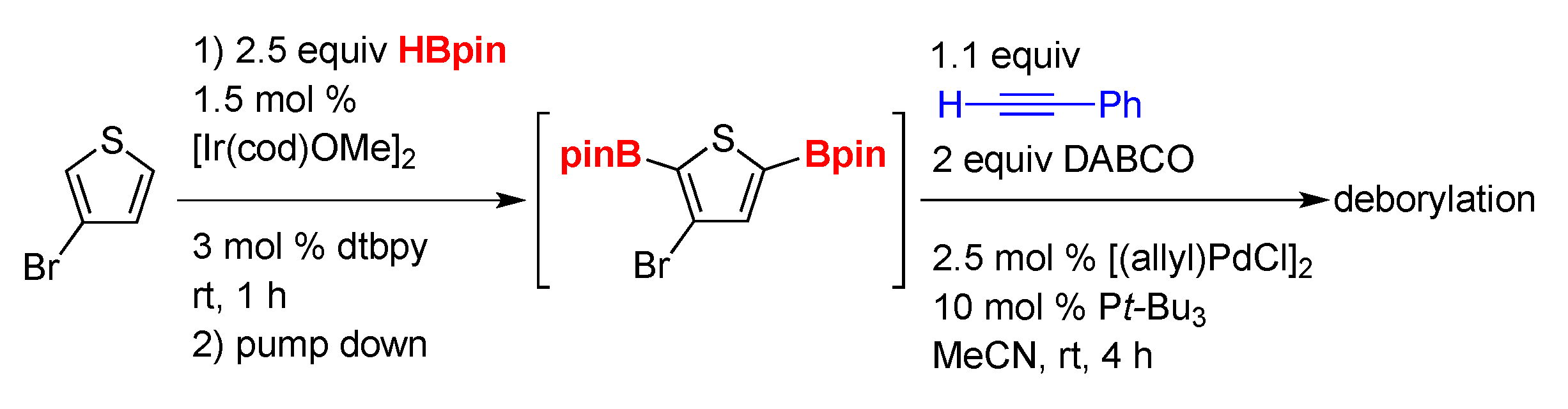
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. General Procedure A: Sonogashira Cross-Coupling of Borylated Aryl Bromides
3.3. General Procedure B: One-Pot CHB/Sonogashira Reaction
3.4. Analytical data of products 12–25
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, C.; Falck, J.R. Transition metal-free ipso-functionalization of arylboronic acids and derivatives. Adv. Synth. Catal. 2014, 356, 2395–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, D.G. Boronic Acids. Preparation and Application in Organic Synthesis, Medicine and Materials, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2011; ISBN 9783527324897. [Google Scholar]
- Cho, J.Y.; Tse, M.K.; Holmes, D.; Maleczka, R.E.; Smith, M.R. Remarkably selective Iridium catalysts for the elaboration of aromatic C-H bonds. Science 2002, 295, 305–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, H.; Yamazaki, H.; Sato, H.; Sakaki, S. Iridium-Catalyzed Borylation of Benzene with Diboron. Theoretical Elucidation of Catalytic Cycle Including Unusual Iridium(V) Intermediate. J. Am. Chem. Soc. 2003, 125, 16114–16126. [Google Scholar] [CrossRef] [PubMed]
- Boller, T.M.; Murphy, J.M.; Hapke, M.; Ishiyama, T.; Miyaura, N.; Hartwig, J.F. Mechanism of the mild functionalization of arenes by diboron reagents catalyzed by iridium complexes. Intermediacy and chemistry of bipyridine-ligated iridium trisboryl complexes. J. Am. Chem. Soc. 2005, 127, 14263–14278. [Google Scholar] [CrossRef] [PubMed]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C− H Activation for the construction of C− B bonds. Chem. Rev. 2009, 110, 890–931. [Google Scholar] [CrossRef]
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N.R.; Hartwig, J.F. Mild iridium-catalyzed borylation of arenes. High turnover numbers, room temperature reactions, and isolation of a potential intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. [Google Scholar] [CrossRef]
- Chotana, G.A.; Rak, M.A.; Smith, M.R. Sterically directed functionalization of aromatic C-H bonds: Selective borylation ortho to cyano groups in arenes and heterocycles. J. Am. Chem. Soc. 2005, 127, 10539–10544. [Google Scholar] [CrossRef]
- Iverson, C.N.; Smith, M.R. Stoichiometric and catalytic B-C bond formation from unactivated hydrocarbons and boranes. J. Am. Chem. Soc. 1999, 121, 7696–7697. [Google Scholar] [CrossRef]
- Ros, A.; Fernández, R.; Lassaletta, J.M. Functional group directed C–H borylation. Chem. Soc. Rev. 2014, 43, 3229–3243. [Google Scholar] [CrossRef] [Green Version]
- Haldar, C.; Emdadul Hoque, M.; Bisht, R.; Chattopadhyay, B. Concept of Ir-Catalyzed C–H Bond Activation/Borylation by Noncovalent Interaction. Tetrahedron Lett. 2018, 1–9. [Google Scholar] [CrossRef]
- Mihai, M.T.; Genov, G.R.; Phipps, R.J. Access to the meta position of arenes through transition metal catalysed C-H bond functionalisation: A focus on metals other than palladium. Chem. Soc. Rev. 2018, 47, 149–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.L.; Iwai, T.; Maeda, S.; Sawamura, M. Iridium-Catalyzed Asymmetric Borylation of Unactivated Methylene C(sp 3 )-H Bonds. J. Am. Chem. Soc. 2019, 141, 6817–6821. [Google Scholar] [CrossRef] [PubMed]
- Hyland, S.N.; Meck, E.A.; Tortosa, M.; Clark, T.B. α-Amidoboronate esters by amide-directed alkane C–H borylation. Tetrahedron Lett. 2019, 60, 1096–1098. [Google Scholar] [CrossRef]
- Zhong, R.L.; Sakaki, S. Sp3 C-H Borylation Catalyzed by Iridium(III) Triboryl Complex: Comprehensive Theoretical Study of Reactivity, Regioselectivity, and Prediction of Excellent Ligand. J. Am. Chem. Soc. 2019, 141, 9854–9866. [Google Scholar] [CrossRef]
- Kawamorita, S.; Murakami, R.; Iwai, T.; Sawamura, M. Synthesis of primary and secondary alkylboronates through site-selective C(sp3)-H activation with silica-supported monophosphine-Ir catalysts. J. Am. Chem. Soc. 2013, 135, 2947–2950. [Google Scholar] [CrossRef]
- Larsen, M.A.; Cho, S.H.; Hartwig, J. Iridium-Catalyzed, Hydrosilyl-Directed Borylation of Unactivated Alkyl C-H Bonds. J. Am. Chem. Soc. 2016, 138, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Liskey, C.W.; Hartwig, J.F. Iridium-catalyzed C-H borylation of cyclopropanes. J. Am. Chem. Soc. 2013, 135, 3375–3378. [Google Scholar] [CrossRef]
- Liskey, C.W.; Hartwig, J.F. Iridium-catalyzed borylation of secondary C-H bonds in cyclic ethers. J. Am. Chem. Soc. 2012, 134, 12422–12425. [Google Scholar] [CrossRef]
- Lawrence, J.D.; Takahashi, M.; Bae, C.; Hartwig, J.F. Regiospecific functionalization of methyl C-H bonds of alkyl groups in Reagents with heteroatom functionality. J. Am. Chem. Soc. 2004, 126, 15334–15335. [Google Scholar] [CrossRef]
- Mita, T.; Ikeda, Y.; Michigami, K.; Sato, Y. Iridium-catalyzed triple C(sp3)-H borylations: Construction of triborylated sp3-carbon centers. Chem. Commun. 2013, 49, 5601–5603. [Google Scholar] [CrossRef]
- Robbins, D.W.; Hartwig, J.F. Sterically controlled alkylation of arenes through iridium-catalyzed C-H borylation. Angew. Chemie-Int. Ed. 2013, 52, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Jayasundara, C.R.K.; Unold, J.M.; Oppenheimer, J.; Smith, M.R.; Maleczka, R.E. A catalytic borylation/dehalogenation route to o -fluoro arylboronates. Org. Lett. 2014, 16, 6072–6075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.M.; Tzschucke, C.C.; Hartwig, J.F. One-pot synthesis of arylboronic acids and aryl trifluoroborates by Ir-catalyzed borylation of arenes. Org. Lett. 2007, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Tzschucke, C.C.; Murphy, J.M.; Hartwig, J.F. Arenes to anilines and aryl ethers by sequential iridium-catalyzed borylation and copper-catalyzed coupling. Org. Lett. 2007, 9, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Maleczka, R.E.; Shi, F.; Holmes, D.; Smith, M.R. C-H activation/borylation/oxidation: A one-pot unified route to meta-substituted phenols bearing ortho-/para-directing groups. J. Am. Chem. Soc. 2003, 125, 7792–7793. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.; Chotana, G.A.; Maleczka, R.E.; Smith, M.R. One-pot borylation/amination reactions: Syntheses of arylamine boronate esters from halogenated arenes. Org. Lett. 2006, 8, 1407–1410. [Google Scholar] [CrossRef]
- Olsson, V.J.; Szabó, K.J. Functionalization of unactivated alkenes through iridium-catalyzed borylation of carbon-hydrogen bonds. Mechanism and synthetic applications. J. Org. Chem. 2009, 74, 7715–7723. [Google Scholar] [CrossRef]
- Olsson, V.J.; Szabó, K.J. Selective one-pot carbon-carbon bond formation by catalytic boronation of unactivated cycloalkenes and subsequent coupling. Angew. Chemie-Int. Ed. 2007, 46, 6891–6893. [Google Scholar] [CrossRef]
- Olsson, V.J.; Szabó, K.J. Synthesis of allylsilanes and dienylsilanes by a one-pot catalytic C-H borylation-Suzuki-Miyaura coupling sequence. Org. Lett. 2008, 10, 3129–3131. [Google Scholar] [CrossRef]
- Iwadate, N.; Suginome, M. Differentially Protected Diboron for Regioselective Diboration of Alkynes: Internal-Selective Cross-Coupling of 1-Alkene-1, 2-diboronic Acid Derivatives compounds now provide the most efficient synthetic access to The unsymmetrical diboron was prepared. J. Am. Chem. Soc. 2010, 132, 2548–2549. [Google Scholar] [CrossRef]
- Lee, C.I.; Zhou, J.; Ozerov, O.V. Catalytic dehydrogenative borylation of terminal alkynes by a SiNN pincer complex of iridium. J. Am. Chem. Soc. 2013, 135, 3560–3566. [Google Scholar] [CrossRef] [PubMed]
- Hata, H.; Yamaguchi, S.; Mori, G.; Nakazono, S.; Katoh, T.; Takatsu, K.; Hiroto, S.; Shinokubo, H.; Osuka, A. Regioselective borylation of porphyrins by C-H bond activation under iridium catalysis to afford useful building blocks for porphyrin assemblies. Chem.-An Asian J. 2007, 2, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Akita, M.; Hiroto, S.; Shinokubo, H. Silylethynyl substituents as porphyrin protecting groups for solubilization and selectivity control. Org. Lett. 2014, 16, 1818–1821. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, T.; Kamata, S.; Hitosugi, S.; Isobe, H. Bottom-up synthesis and structures of π-lengthened tubular macrocycles. Chem. Sci. 2013, 4, 3179–3183. [Google Scholar] [CrossRef]
- Koyama, Y.; Hiroto, S.; Shinokubo, H. Synthesis of highly distorted π-extended [2.2]metacyclophanes by intermolecular double oxidative coupling. Angew. Chemie-Int. Ed. 2013, 52, 5740–5743. [Google Scholar] [CrossRef] [PubMed]
- Goldfinger, M.B.; Crawford, K.B.; Swager, T.M. Synthesis of Ethynyl-Substituted Quinquephenyls and Conversion to Extended Fused-Ring Structures. J. Org. Chem. 1998, 63, 1676–1686. [Google Scholar] [CrossRef]
- Maly, K.E.; Maris, T.; Wuest, J.D. Two-dimensional hydrogen-bonded networks in crystals of diboronic acids. CrystEngComm 2006, 8, 33–35. [Google Scholar] [CrossRef]
- Nakamura, H.; Kuroda, H.; Saito, H.; Suzuki, R.; Yamori, T.; Maruyama, K.; Haga, T. Synthesis and biological evaluation of boronic acid containing cis-stilbenes as apoptotic tubulin polymerization inhibitors. ChemMedChem 2006, 1, 729–740. [Google Scholar] [CrossRef]
- Zheng, S.L.; Lin, N.; Reid, S.; Wang, B. Effect of extended conjugation with a phenylethynyl group on the fluorescence properties of water-soluble arylboronic acids. Tetrahedron 2007, 63, 5427–5436. [Google Scholar] [CrossRef] [Green Version]
- Yashima, E.; Nimura, T.; Matsushima, T.; Okamoto, Y. Poly((4-dihydroxyborophenyl)acetylene) as a novel probe for chirality and structural assignments of various kinds of molecules including carbohydrates and steroids by circular dichroism. J. Am. Chem. Soc. 1996, 118, 9800–9801. [Google Scholar] [CrossRef]
- Laus, G.; Müller, A.G.; Schottenberger, H.; Wurst, K.; Buchmeiser, M.R.; Ongania, K.H. Facile synthesis of new areneboronates as terminal ethyne monomers. Monatshefte fur Chemie 2006, 137, 69–75. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Feare, T.E.; Savereide, T.J.; Nazy, J.R. Organoboron Compounds. XIII. Boronic Acids with Neighboring Unsaturated Groups. J. Org. Chem. 1961, 26, 1271–1273. [Google Scholar] [CrossRef]
- Takase, M.; Nakajima, A.; Takeuchi, T. Synthesis of an extended hexagonal molecule as a highly symmetrical ligand. Tetrahedron Lett. 2005, 46, 1739–1742. [Google Scholar] [CrossRef]
- Perttu, E.K.; Arnold, M.; Iovine, P.M. The synthesis and characterization of phenylacetylene tripodal compounds containing boroxine cores. Tetrahedron Lett. 2005, 46, 8753–8756. [Google Scholar] [CrossRef]
- Zheng, S.L.; Reid, S.; Lin, N.; Wang, B. Microwave-assisted synthesis of ethynylarylboronates for the construction of boronic acid-based fluorescent sensors for carbohydrates. Tetrahedron Lett. 2006, 47, 2331–2335. [Google Scholar] [CrossRef]
- Schwier, T.; Rubin, M.; Gevorgyan, V. B(C6F5)3-catalyzed allylation of propargyl acetates with allylsilanes. Org. Lett. 2004, 6, 1999–2001. [Google Scholar] [CrossRef]
- Hundertmark, T.; Littke, A.F.; Buchwald, S.L.; Fu, G.C. Pd(PhCN)2Cl2/P(t-Bu)3: A versatile catalyst for Sonogashira reactions of aryl bromides at room temperature. Org. Lett. 2000, 2, 1729–1731. [Google Scholar] [CrossRef]
- Gelman, D.; Buchwald, S.L. Efficient Palladium-Catalyzed Coupling of Aryl Chlorides and Tosylates with Terminal Alkynes: Use of a Copper Cocatalyst Inhibits the Reaction. Angew. Chemie-Int. Ed. 2003, 42, 5993–5996. [Google Scholar] [CrossRef]
- Soheili, A.; Albaneze-Walker, J.; Murry, J.A.; Dormer, P.G.; Hughes, D.L. Efficient and General Protocol for the Copper-Free Sonogashira Coupling of Aryl Bromides at Room Temperature. Org. Lett. 2003, 5, 4191–41941. [Google Scholar] [CrossRef]
- Marigo, M.; Marsich, N.; Farnetti, E. Polymerization of phenylacetylene catalyzed by organoiridium compounds. J. Mol. Catal. A Chem. 2002, 187, 169–177. [Google Scholar] [CrossRef]
- Kallepalli, V.A.; Gore, K.A.; Shi, F.; Sanchez, L.; Chotana, G.A.; Miller, S.L.; Maleczka, R.E.; Smith, M.R. Harnessing C-H Borylation/Deborylation for Selective Deuteration, Synthesis of Boronate Esters, and Late Stage Functionalization. J. Org. Chem. 2015, 80, 8341–8353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, F.; Tyagarajan, S.; Perera, D.; Krska, S.W.; Maligres, P.E.; Smith, M.R.; Maleczka, R.E. Bismuth Acetate as a Catalyst for the Sequential Protodeboronation of Di- and Triborylated Indoles. Org. Lett. 2016, 18, 1554–1557. [Google Scholar] [CrossRef] [Green Version]
- Uson, R.; Orto, L.A.; Cabeza, J.A. Dinuclear methoxy, cyclooctadiene, and barrelene complexes of rhodium and iridium. Inorg. Synth. 1985, 23, 126–130. [Google Scholar]
- Merola, J.S.; Kacmarcik, R.T. Synthesis and Reaction Chemistry of (η5-Indenyl)(cycloactadiene)iridium: Migration of Indenyl from Iridium to Cycloodadiene. Organometallics 1989, 8, 778–784. [Google Scholar] [CrossRef]
- Juliette, J.J.J.; Rutherford, D.; Horváth, I.T.; Gladysz, J.A. Transition metal catalysis in fluorous media: Practical application of a new immobilization principle to rhodium-catalyzed hydroborations of alkenes and alkynes. J. Am. Chem. Soc. 1999, 121, 2696–2704. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
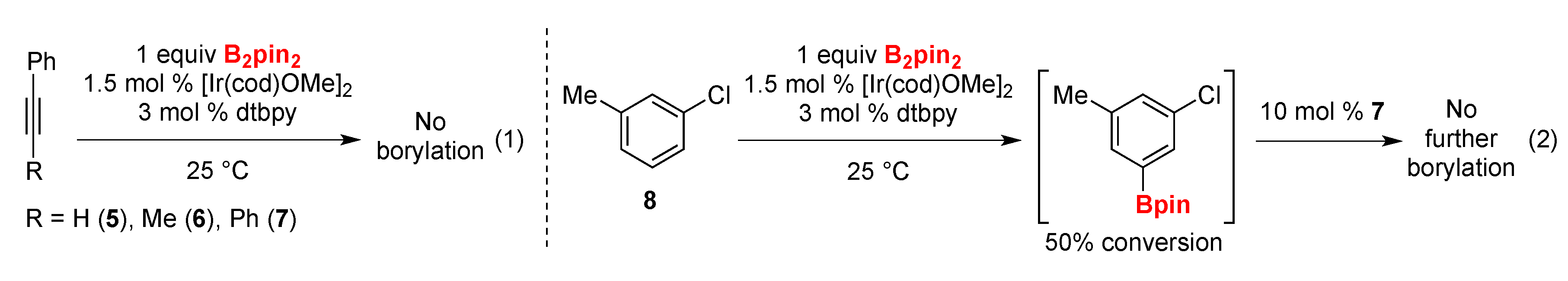{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Reagent | Product | Entry | Reagent | Product |
| 1 |  |  | 8b |  |  |
| 2 |  |  | 9b,c |  |  |
| 3 |  |  | 10 |  |  |
| 4 |  |  | 11 |  |  |
| 5 |  |  | 12 |  |  |
| 6 |  |  | 13 |  |  |
| 7 |  |  | 14 |  |  |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chotana, G.A.; Montero Bastidas, J.R.; Miller, S.L.; Smith, M.R., III; Maleczka, R.E., Jr. One-Pot Iridium Catalyzed C–H Borylation/Sonogashira Cross-Coupling: Access to Borylated Aryl Alkynes. Molecules 2020, 25, 1754. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071754
Chotana GA, Montero Bastidas JR, Miller SL, Smith MR III, Maleczka RE Jr. One-Pot Iridium Catalyzed C–H Borylation/Sonogashira Cross-Coupling: Access to Borylated Aryl Alkynes. Molecules. 2020; 25(7):1754. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071754
Chicago/Turabian StyleChotana, Ghayoor A., Jose R. Montero Bastidas, Susanne L. Miller, Milton R. Smith, III, and Robert E. Maleczka, Jr. 2020. "One-Pot Iridium Catalyzed C–H Borylation/Sonogashira Cross-Coupling: Access to Borylated Aryl Alkynes" Molecules 25, no. 7: 1754. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071754