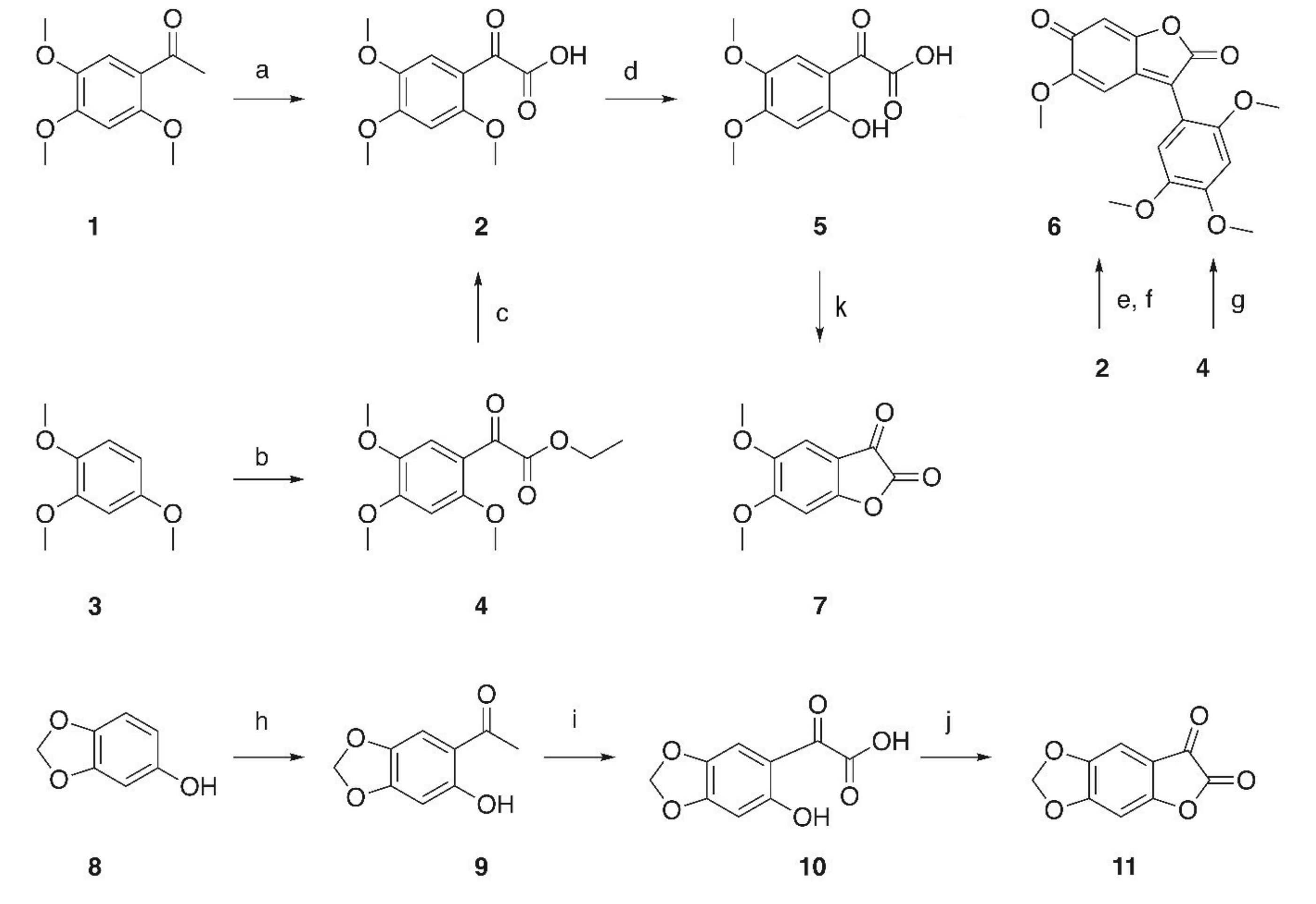
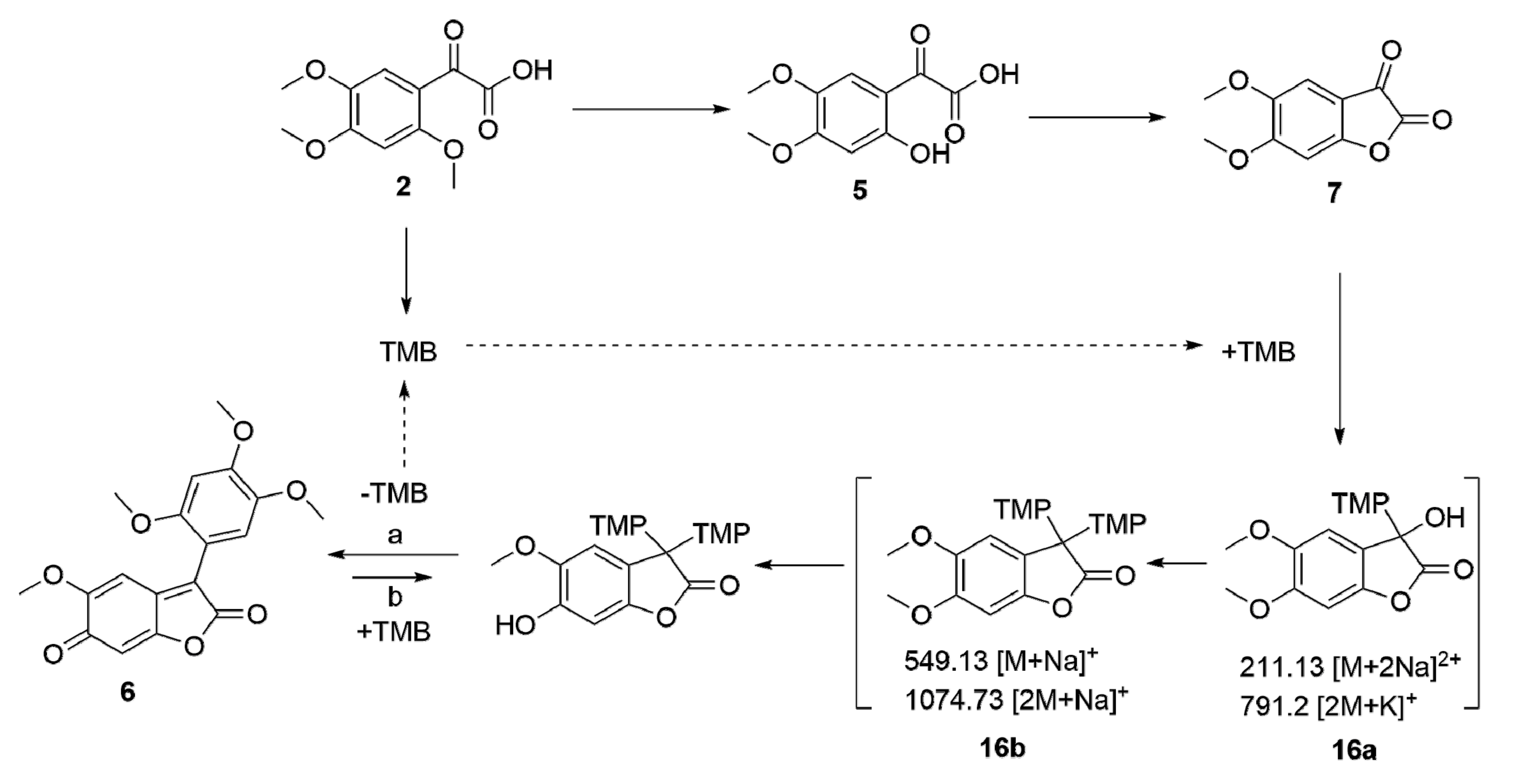
2. Results and Discussion
Oxidation of 2,4,5-trimethoxy-acetophenone (
1,
Scheme 1) with SeO
2 [
31] gave 2,4,5-trimethoxyphenylglyoxylic acid (
2); this compound has previously been isolated from the fungus
Polyporus tumulosus Cooke [
22,
32]. As an alternative, a Friedel–Crafts acylation of 2,4,5-trimethoxybenzene (
3) in the presence of TiCl
4 gave a 94% yield of ester
4 [
33,
34,
35] whose hydrolysis with methanolic KOH for 2 h resulted in an almost quantitative yield of
2. Partial deprotection of the methoxy-groups with AlCl
3 of
2 yielded
5 albeit in low yields, while demethylation with either hydrobromic or hydrochloric acid yielded
6, a red colored solid in almost quantitative yield. A major issue in these reactions is the instability of the α-keto-acids that are readily decarboxylated [
22,
36]. These findings parallel previous reports for trimethoxy-substituted aromatic compounds [
36]. Furthermore, α-keto acids were used for the synthesis of oxadiazolopyrazines—selective antibacterial agents against
Haemophilus influenzae [
37]. Attempts to cyclize
5 under a variety of different conditions invariably led to the formation of
6 in moderate to excellent yields. The formation of
7 was only observed to a rather minor extend by ESI–MS.
Friedel–Crafts acylation of sesamol (
8) with acetic anhydride/BF
3 gave
9 [
38,
39,
40,
41] in 66% isolated yield whose oxidation with SeO
2 in pyridine yielded
10. Treatment of
10 with oxalyl chloride in DCM in the presence of DMF gave a moderate yield of
11 but—interestingly enough - no red-colored by-products (being analogous to
6) were observed during these reactions.
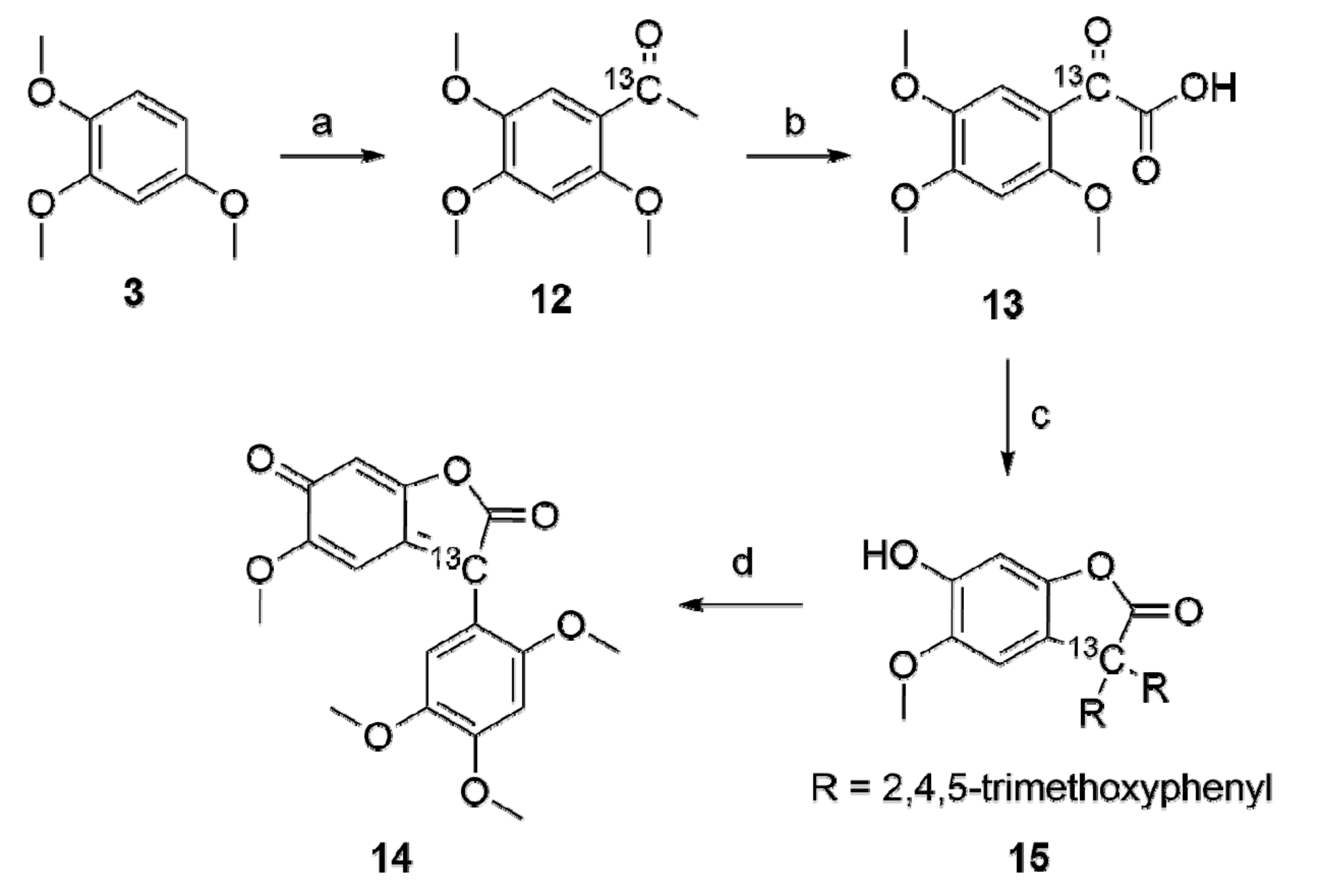
To gain a deeper insight in the structure and formation of
6,
13C-labeling experiments were called for. Thus, trimethoxybenzene (
3) was allowed to react with
13C-labeled acetylchloride (
Scheme 2) in the presence of TiCl
4 and a 97% yield of
13C-labeled
1 (
12) was obtained. From its SeO
2 oxidation compound
13C-labeled
2 (
13) was obtained in 53% yield.
To elucidate the structure of 6, a combination of different analytical techniques had to be applied. An ESI/MS of 6 in MeOH showed a m/z = 345 [M + H]+ corresponding to a molecular composition of C18H16O7 and indicating a “condensation reaction” of 2 having taken place. The 1H-NMR spectra showed the presence of four methoxy groups between δ = 3.81 and 3.97 ppm and four aromatic hydrogens between δ = 6.06 and 7.26 ppm, respectively. The material obtained from the 13C-labeled starting material showed only one labeled carbon in the product as indicated by ESI–MS m/z = 346 for [M + H]+.
Reaction of
2 with AlCl
3 or
13 with hydrochloric acid at low temperatures, however, gave access to an intermediate
15 (from
13); upon warming this reaction mixture,
15 could not be detected any longer but
6 (from
2) or
14 (from
13) was formed. For compound
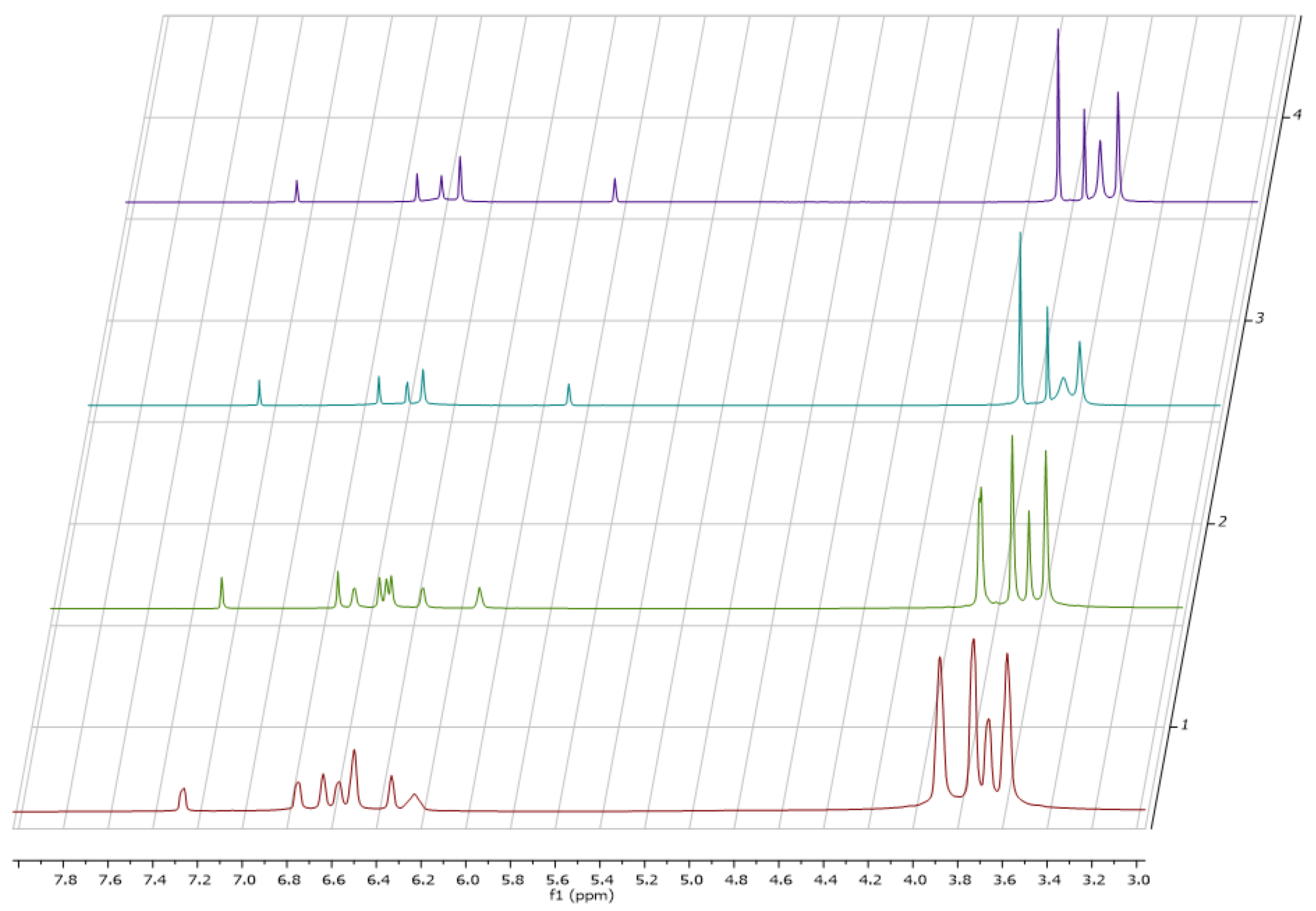
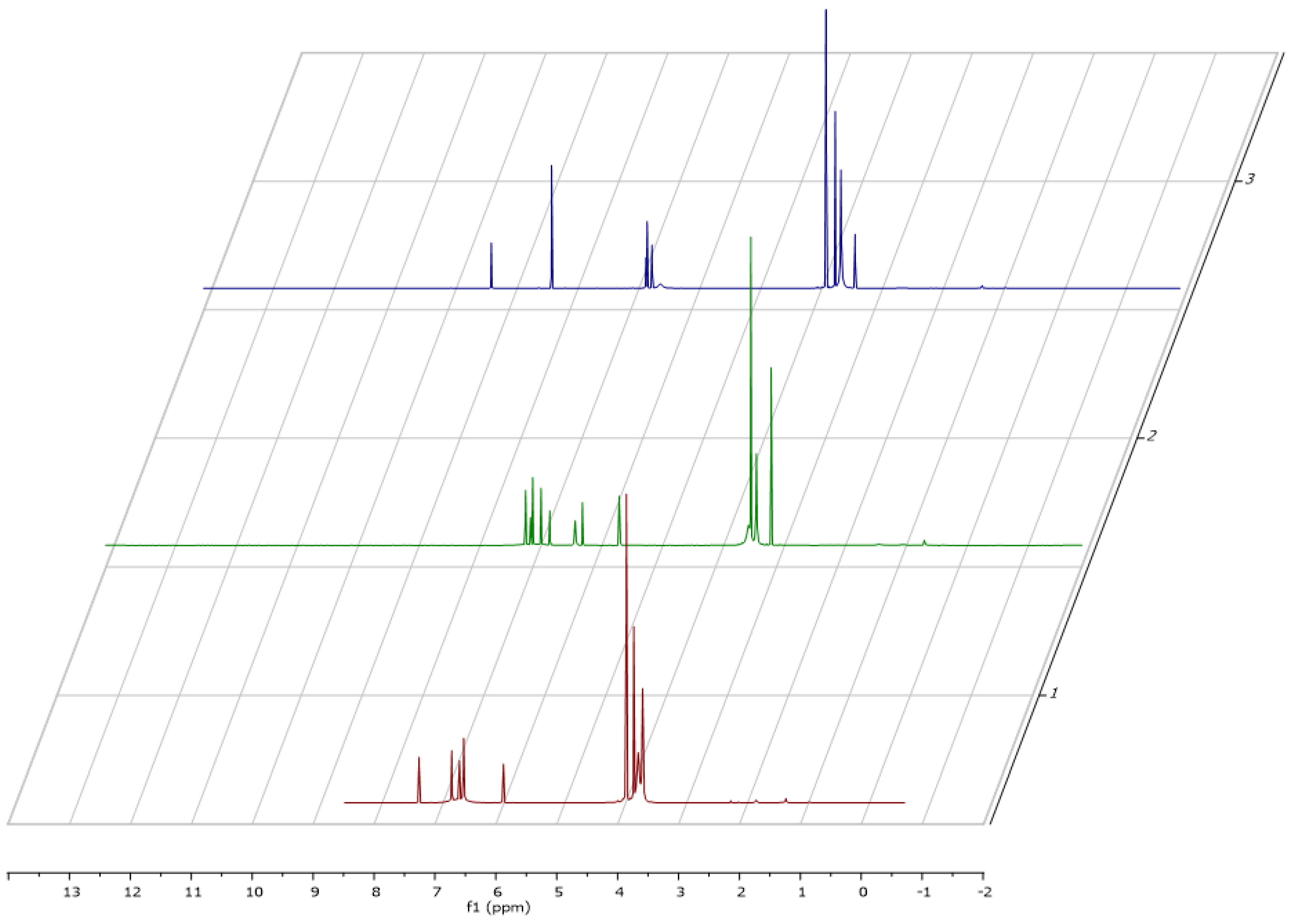
15, different temperatures (−50, −30, 27 and 40 °C) were applied (
Figure 2) and the
1H-NMR spectroscopy revealed a change in the spectra. At room temperature (
Figure 2, cyan) an extensive line broadening of one of the methoxy groups (δ = 3.62 ppm) was observed. In addition, extensive line broadening was seen in the aromatic region. Integration of the signals suggested the presence of seven methoxy groups. Temperature dependent NMR spectroscopy revealed that the line broadening observed at room temperature is due to the presence of a rotational barrier. Furthermore, the NMR spectra of
15 strongly depend on the used solvent (
Figure 3). For example, the aromatic protons in
15 are severely shifted to lower field upon using deuterated toluene as the solvent probably due to an aromatic solvent induced shift behavior. An ESI–MS of
15 showed a quasimolecular ion [M + H]
+ m/z = 513 corresponding well to the proposed structure. NMR spectra of the products are depicted in the
Supplementary File.
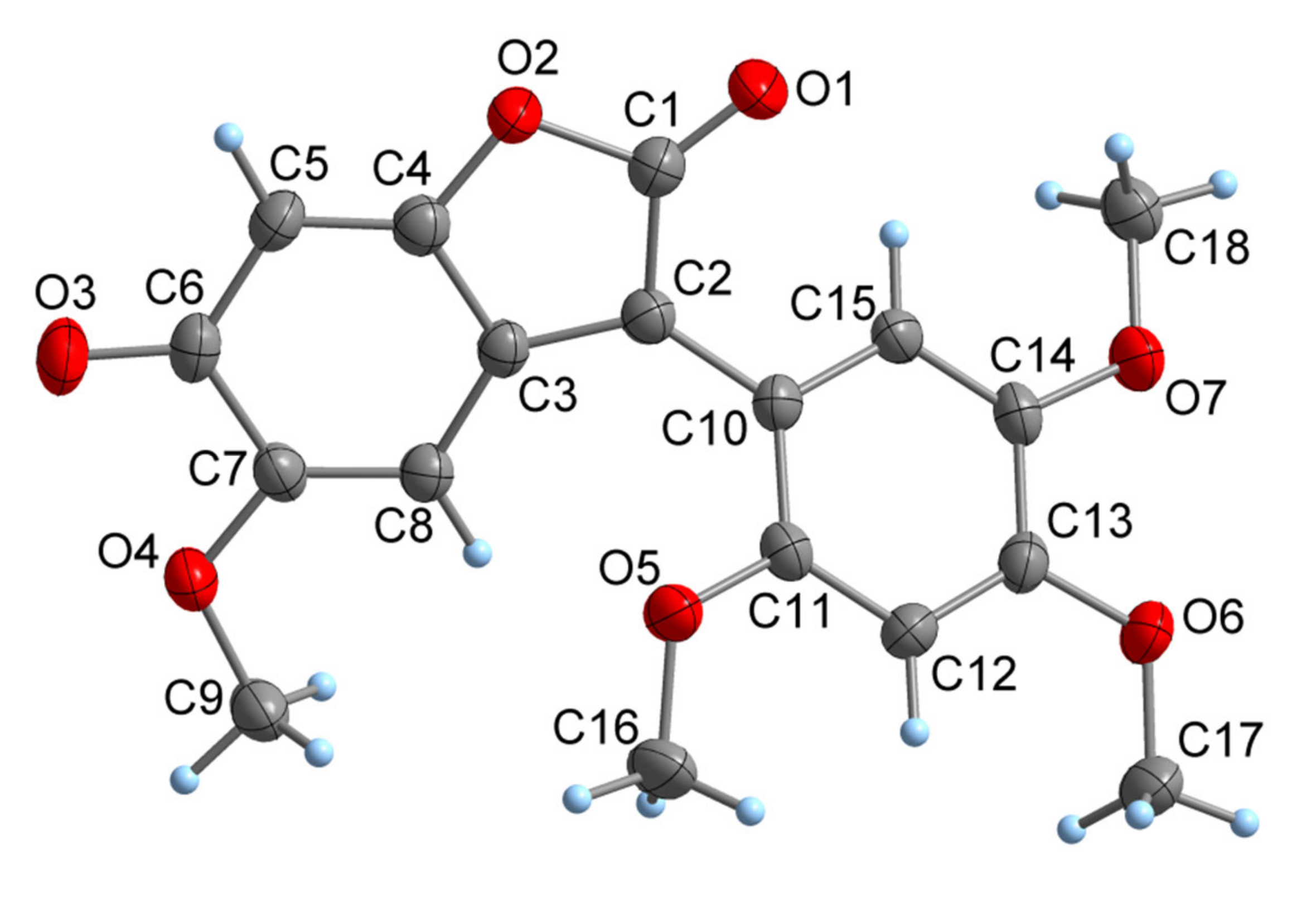
As
6 crystallizes readily from ethyl acetate, crystals suitable for a single crystal X ray analysis could be obtained. The crystal structure of the black orthorhombic prisms (space group P2
12
12
1) is consistent with the NMR data comprising C
18H
16O
7 molecules. The molecular structure is depicted in
Figure 4.
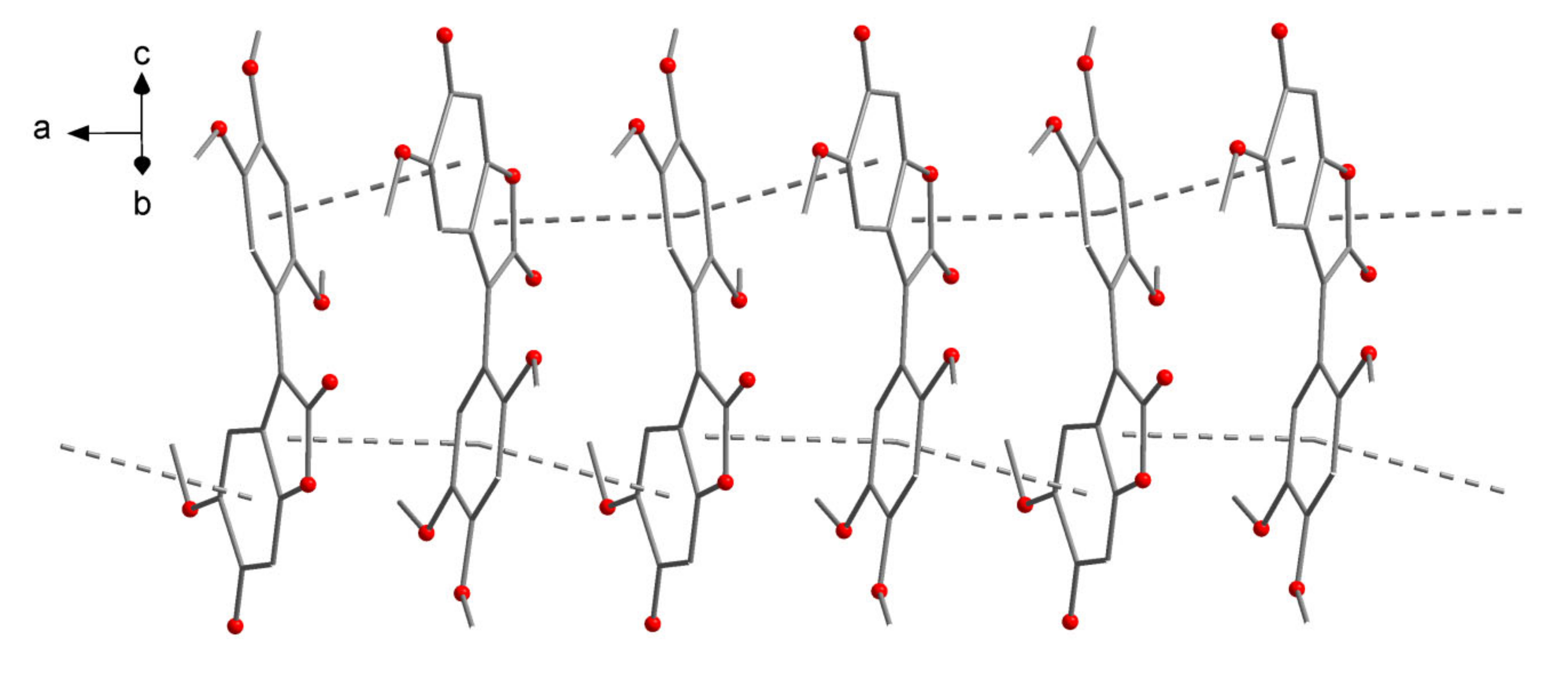
The bond lengths within the benzofurane-derived bicyclic system (C1–C8, O2) cover a wide range of 133.2(3)–150.1(4) pm. C1–C2 (147.4(4) pm), C3–C4 (144.0(4) pm), C3–C8 (142.8(4) pm), C5–C6 (145.8(4) pm), C6-C7 (150.1(4) pm), C1–O2 (140.6(3) pm) and C4–O2 (138.2(3) pm) can be described as single bonds, while C2–C3 (136.0(3) pm), C4–C5 (133.2(3) pm) and C7–C8 (134.2(3) pm) have double bond character. The interconnection between the two ring systems, C2–C10, is a single bond at 147.0(4) pm. The C–C separations within the benzene ring (C10–C15) are in a typical narrow range of 137.3(4)–141.1(3) pm. The molecule is not strictly planar as the two ring systems are twisted around the C2–C10 vector about 41.0(1)°. In the crystal, the molecules are stacked together to a one-dimensional supramolecular array by π interactions (
Figure 5), with the closest intermolecular contacts being C13∙∙∙O2 (342.2(4) pm) and C8∙∙∙C15 (344.7(4) pm). The distances between the ring centroids are 356.81(3) pm ((C1–C4, O2)∙∙∙(C10–C15)) and 369.58(3) pm ((C3–C8)∙∙∙(C10–C15)), respectively.
While the reaction of
2 with trifluoroacetic acid (TFA) in dry DCM did not lead to the formation of
6 and with conc. sulfuric or phosphoric acid only low yields (<10%) were observed, the reaction of
2 with HBr (48%) gave 42% of
6 and with conc. HCl (37%) an 56% yield was observed. Reaction of
2 with 37% HCl in the presence of 5 equivalents 1,2,4-trimethoxybenzene finally led to a 96% yield of
6. From these observations as well as from the
13C-labeling experiments (vide supra), a tentative mechanism for this reaction was deduced (as depicted in
Scheme 3). In the course of the reaction of
2 with TFA in dry DCM an intermediate
16a was observed. ESI-MS experiments showed the presence of a quasimolecular ions
m/z = 211.1 ([M + 2Na]
2+) and
m/z = 791.2 ([2M + K]
+) corresponding to M = 376. Subsequently from
16a intermediate
16b is formed; the latter compound was detected in ESI–MS spectra showing
m/z = 549.13 ([M + Na]
+) and 1074.73 ([2M + Na]
+). Friedel–Crafts reactions are known to be reversible [
42,
43,
44,
45,
46,
47,
48] and compound
6 is very insoluble in the reaction mixture thus explaining the very high yield of
6 in these reactions. Previously, a retro Friedel–Crafts alkylation was used to access a fungal pigment from
Peniophora sanguinea Bres [
13].
To verify these assumptions, to a solution of 6 in dry 2M HCl (in ethyl acetate) 3 was added in 10-fold excess and within several hours of stirring at room temperature, the reaction mixture turned from black to slightly red. ESI–MS investigations of this reaction mixture showed the formation of 15 (m/z = 513, [M + H]+).
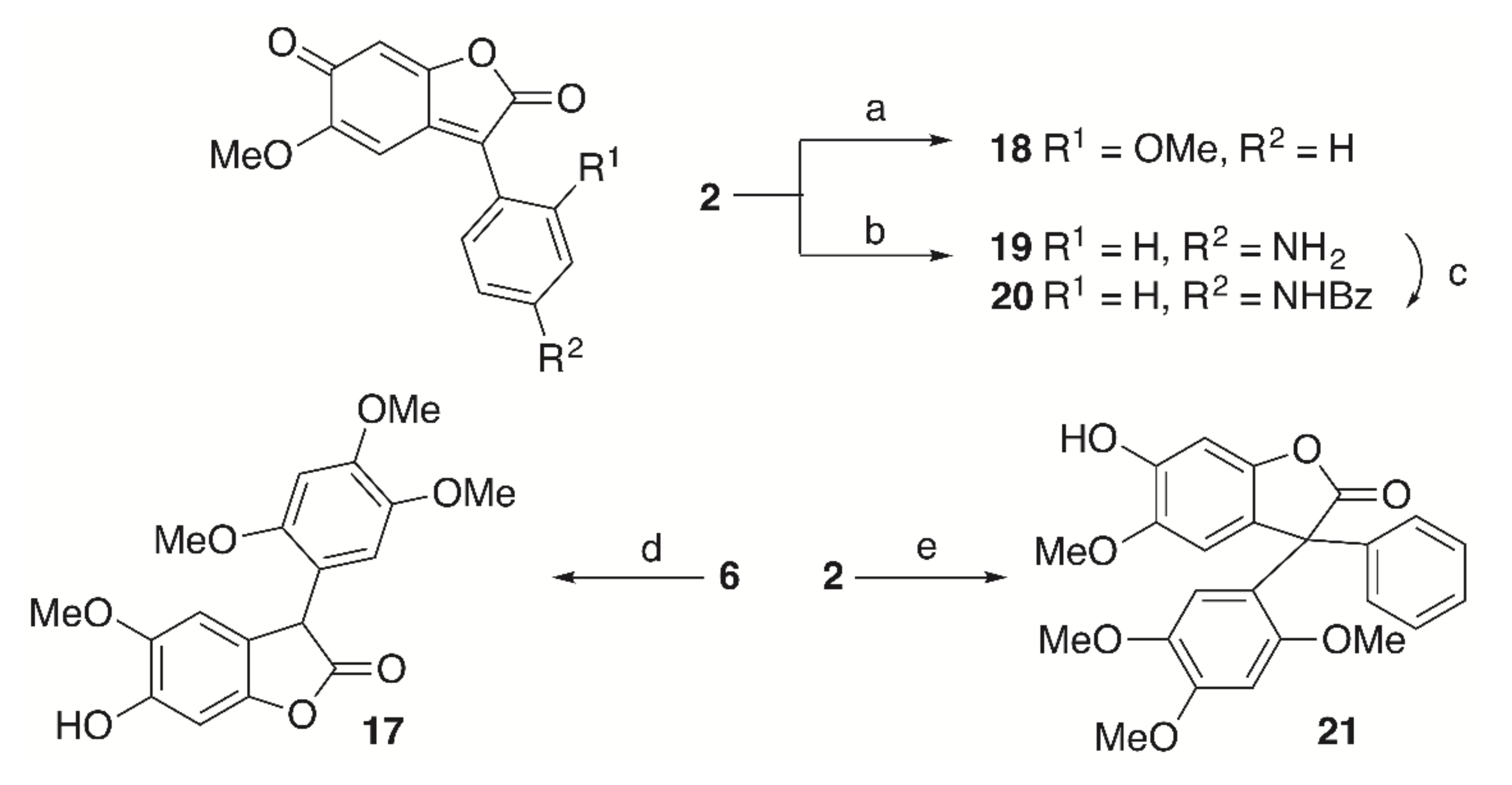
Treatment of
6 with Zn/HCl led to the formation of
17. To have a first insight into the scope of this reaction (
Scheme 4),
2 was allowed to react with anisole or BOC-aniline in the presence of hydrochloric acid and products
18 and
19 were obtained, respectively. Benzoylation of
19 gave
20. Interestingly enough, reaction of
2 with benzene in the presence of hydrochloric acid furnished a 18% yield of
21.
4. Experimental
4.1. 2,4,5-Trimethoxyphenylglyoxylic Acid (2)
From1. A suspension of 2,4,5-trimethoxybenzene (1, 840 mg, 4.00 mmol) and selenium dioxide (800 mg, 7.21 mmol) in pyridine (5 mL) was stirred for 4 h at 80 °C. The mixture was poured into NaOH (100 mL, 0.05 m) and extracted with EtOAc (3 × 50 mL). The aqueous phase was acidified with 2 m hydrochloric acid and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with brine, dried (MgSO4), the solvent was evaporated and the residue re-crystallized from EtOAc to yield 2 (550 mg, 57%) as a yellow solid.
From4. To a solution of 4 (1.0 g, 3.73 mmol) in methanol (50 mL), powdered KOH (1.5 g, 26.7 mmol) was added and the mixture was stirred for 2 h. Water (100 mL) was added and the aqueous phase was extracted with chloroform (3 × 70 mL). The combined organic phases were dried (MgSO
4) and the solvent was removed to afford
2 (886 mg, 99%) as a yellow solid; an analytical sample showed m.p. 182–185 °C (lit.:[
49] 186 °C); R
F = 0.35 (SiO
2, CHCl
3/MeOH; 2:1); IR (KBr): ν = 3432br, 1736s, 1610s, 1517s, 1480m, 1423w, 1384w, 1288s, 1288s, 1230s, 1190m, 1142s, 1018s cm
−1; UV-Vis (MeOH): λ
max (log ε) = 237 (4.22), 279 (4.09), 341 (4.01) nm;
1H-NMR (400 MHz, DMSO-
d6): δ = 7.22 (s, 1H, 8-H), 6.82 (s, 1H, 5-H), 3.92 (s, 3H, 9-H), 3.84 (s, 3H, 10-H), 3.76 (s, 3H, 11-H) ppm;
13C-NMR (100 MHz, DMSO-d
6): δ = 186.0 (C-2), 167.6 (C-1), 157.2 (C-6), 156.4 (C-4), 143.7 (C-7), 113.1 (C-3), 110.2 (C-8), 98.1 (C-5), 57.1 (C-10), 56.2 (C-9), 55.8 (C-11) ppm; MS (ESI, MeOH):
m/z (%) = 238.9 ([M − H]
−, 100), 478.8 ([2M − H]
−, 8), 501.0 ([2M − 2H + Na]
−, 3); analysis calcd for C
11H
12O
6 (240.21): C 55.00, H 5.04; found: C 54.79, H 5.18.
4.2. 2,4,5-Trimethoxyphenyl-glyoxylic Acid Ethyl-ester (4)
To a solution of 1,2,4-trimethoxybenzene (3, 5.0 g, 29.7 mmol) in DCM (200 mL) at −20 °C, TiCl
4 (3.6 mL, 32.8 mmol) was added followed by slowly adding ethyl chlorooxoacetate (3.5 mL, 31.3 mmol) and the mixture was stirred for 2 h. Aqueous work-up (2M HCl) followed by extraction with chloroform (3 × 100 mL) and column chromatography (SiO
2, hexanes/CHCl
3, 1:1) gave
4 (7.48 g, 94%) as pale yellow crystalline solid; m.p. 89–91 °C (lit.:[
50] 89.5 °C); R
F = 0.78 (SiO
2, hexanes/CHCl
3); IR (ATR): ν = 3004w, 2991w, 2943w, 2908w, 2840w, 1732m, 1633m, 1599s, 1581m, 1513s, 1482m, 1461m, 1450m, 1440m, 1418m, 1390w, 1371m, 1298s, 1268s, 1259s, 1228s, 1218s, 1191s, 1179s, 1141vs, 1112m, 1038m, 1021s, 1007s, 954m, 862m, 816m, 805m, 770m, 765m, 743m, 710m, 655m cm
−1; UV-Vis (methanol): λ
max (logε) 237 (3.52), 281 (3.40), 346 (3.34) nm;
1H-NMR (400 MHz, CDCl
3) δ = 7.38 (s, 1H, 2′-H), 6.46 (s, 1H, 5′-H), 4.36 (q,
J = 7.1 Hz, 2H, Et
CH2), 3.95 (s, 3H, OMe(6′)), 3.87 (s, 3H, OMe(3′)), 3.84 (s, 3H, OMe(4′)), 1.38 (t,
J = 7.1 Hz, 3H, Et
CH3) ppm;
13C-NMR (101 MHz, CDCl
3) δ = 184.8 (C-2), 166.1 (C-1), 157.3 (C-4‘), 156.5 (C-6‘), 144.2 (C-3‘), 114.2 (C-1‘), 111.0 (C-2‘), 96.5 (C-5‘), 61.6 (C-Et
CH2), 56.9 (OMe(C4‘)), 56.3 (OMe(C3‘)), 56.3 (OMe(C2‘)), 14.1 (CEt
CH3) ppm; MS (ESI, MeOH):
m/z (%) = 269.1 ([M + H]
+, 100); analysis calcd for C
13H
16O
6 (268.26): C 58.20, H 6.01; found: C 57.95, H 6.22.
4.3. 2-Hydroxy-4,5-dimethoxyphenyl-glyoxylic Acid (5)
A solution of AlCl
3 (166 mg, 1.25 mmol) and 2 (150 mg, 0.62 mmol) in DCM (5 mL) was heated for 3 h under microwave irradiation at 50 °C. Usual aqueous work-up (aq. HCl, 80 mL, 0.125
m) followed by extraction (ethyl acetate, 2 × 50 mL) and re-crystallization from chloroform gave 5 (17 mg, 13%) as yellow solid; m.p. 163–165 °C (lit:[
22] 142–144 °C); R
F = 0.25 (SiO
2, CHCl
3/MeOH 2:1); IR (KBr): ν = 3150br, 1713s, 1573s, 1526s, 1482m, 1400m, 1352w, 1308m, 1267s, 1240m, 1205s, 1181m, 1165s, 1035m cm
−1; UV-Vis (MeOH): λ
max (log ε = 238 (4.07), 283 (3.99), 344 (3.93) nm;
1H-NMR (400 MHz, DMSO-d
6): δ = 7.24 (s, 1H, 8-H), 6.61 (s, 1H, 5-H), 3.78 (s, 3H, 10-H), 3.74 (s, 3H, 9-H) ppm;
13C-NMR (101 MHz, DMSO-d
6): δ = 185.6 (C-2), 167.8 (C-1), 157.1 (C-6), 155.4 (C-4), 142.8 (C-7), 112.3 (C-3), 111.2 (C-8), 100.7 (C-5), 56.6 (C-10), 56.0 (C-9) ppm; MS (ESI, MeOH):
m/z (%) = 224.9 ([M − H]
−, 100), 472.9 ([2M − 2H + Na]
−, 16); analysis calcd for C
10H
10O
6 (226.18): C 53.10, H 4.46; found: C 52.96, H 4.60.
4.4. 5-Methoxy-3-(2,4,5-trimethoxyphenyl)benzofuran-2,6-dione (6)
Method A. Compound 2 (250 mg, 1.04 mmol) was suspended in conc. hydrochloric acid (25 mL) and the mixture was heated to 40 °C for 3 h, diluted with water (25 mL) and extracted with chloroform (3 × 25 mL). The organic extracts were dried (MgSO4), the solvent was removed and the remaining residue was subjected to column chromatography (SiO2, CHCl3/MeOH 9:1) to afford 6 (199 mg, 56%) as a dark red solid.
Method B. Compound 2 (200 mg, 0.83 mmol) and 1,2,4-trimethoxybenzene (0.62 mL, 4.16 mmol) were suspended in conc. hydrochloric acid (25 mL) and the mixture was heated to 40 °C for 4 h, diluted with water (25 mL) and extracted with chloroform (3 × 25 mL). The organic extracts were dried (MgSO4), the solvent was removed and the remaining residue was subjected to column chromatography (SiO2, CHCl3/MeOH 9:1) to afford 6 (274 mg, 96%) as a dark red solid.
Method C. Compound 4 (1.0 g, 3.73 mmol) was suspended in hydrochloric acid (50 mL) and heated to 40 °C for 4 h; workup as described above gave 6 (636 mg, 49%) as dark red solid; m.p. 238–239 °C; RF = 0.68 (SiO2, CHCl3/MeOH 9:1); IR (ATR): ν = 2958w, 2941w, 2841w, 1772s, 1735w, 1643s, 1610m, 1588vs, 1559s, 1527m, 1517m, 1506m, 1461s, 1454s, 1437m, 1416m, 1388m, 1350s, 1316w, 1274s, 1234vs, 1223s, 1209vs, 1192s, 1180vs, 1167vs, 1134s, 1052w, 1040m, 1023s, 1001s, 985s, 934s, 864s, 854s, 844s, 822m, 814m, 778m, 774m, 766m 701m, 696m, 669m cm−1, UV–Vis (MeOH): λmax (log ε = 311 (3.31), 357 (3.08), 498 (3.00) nm; 1H-NMR (400 MHz, CDCl3): δ = 7.09 (s, 1H, 6′-H), 6.61 (s, 1H, 3′-H), 6.36 (s, 1H, 4-H), 6.06 (s, 1H, 7-H), 3.97 (s, 3H, OMe(4′)), 3.88 (s, 3H, OMe(5′), 3.87 (s, 3H, OMe(2′)), 3.82 (s, 3H, OMe(5)) ppm; 13C NMR (101 MHz, CDCl3): δ = 181.3 (C-6), 167.4 (C-2), 160.5 (C-7a), 153.82 (C-2′), 152.78 (C-4′), 152.74 (C-5), 143.74 (C-5′), 138.80 (C-3a), 125.59 (C-3), 114.3 (C-6′), 109.3 (C-1′), 104.2 (C-7), 100.7 (C-4), 97.6 (C-3′), 56.8 + 56.6 (OMe(2′) + OMe (5′)), 56.3 (OMe(4′)), 56.3 (OMe(5)) ppm; MS (ESI, MeOH): m/z (%) = 345.2 ([M + H]+, 100); analysis calcd for C18H16O7 (344.32): C 62.79, H 4.68; found: C 62.55, H 4.83.
4.5. 1-(6-Hydroxybenzo[d][1,3]dioxol-5-yl)ethan-1-one (9)
To a suspension of sesamol (8, 5.52 g, 40.00 mmol) in acetic anhydride (20 mL, 0.21 mol) at 0 °C BF
3∙OEt
2 (10 mL, 81.03 mmol) was added and the reaction mixture was heated at 85 °C for 1 h. An aqueous solution of sodium acetate sol. (satd., 40 mL) was added and the mixture was extracted with diethylether (3 × 50 mL). The combined organic layers were washed with sat. NaHCO
3 sol. (4 × 100 mL), water (3 × 100 mL), brine (1 × 100 mL) and dried (MgSO
4). The solvent was removed and recrystallization from ethanol gave
9 (4.74 g, 66%) as an off-white solid; m.p. 111–113 °C (lit.:[
51] 114 °C); R
F = 0.81 (SiO
2, CHCl
3/MeOH, 9:1); C; IR (KBr): ν = 3446br, 2919m, 1633s, 1485s, 1425s, 1366m, 1322s, 1257s, 1182s, 1118m, 1037s, 958m, 924s, 856m, 844m, 787m, 773m, 540m cm
−1; UV–Vis (CHCl
3): λ
max (log ε) = 241 (4.16), 278 (3.86), 349 (3.92) nm;
1H-NMR (400 MHz, CDCl
3): δ = 7.04 (s, 1H, 6-H), 6.43 (s, 1H, 3-H), 5.97 (s, 2H, 7-H), 2.51 (s, 3H, 9-H) ppm;
13C NMR (100 MHz, CDCl
3): δ = 202.0 (C-8), 162.2 (C-2), 154.5 (C-4), 140.6 (C-5), 112.4 (C-1), 107.3 (C-6), 102.0 (C-7), 98.8 (C-3), 26.6 (C-9) ppm; MS (ESI, MeOH):
m/z (%) = 181.1 ([M + H]
+, 100); analysis calcd for C
9H
8O
4 (344.32): C 62.79, H 4.68; found: C 62.57, H 4.90.
4.6. 2-(6-Hydroxybenzo[d][1,3]dioxol-5-yl)-2-oxoacetic Acid (10)
Compound 9 (2.5 g, 13.88 mmol) and selenium dioxide (3.08 g, 27.75 mmol) were suspended in dry pyridine (45 mL) and stirred at 80 °C for 20 h. Aqueous work-up (400 mL, 1 m), followed by extraction with EtOAc (8 × 100 mL) and evaporation of the solvent gave 10 as a brownish solid (1.96 g, 67%). An analytical sample was obtained by re-dissolving 10 in EtOAc (100 mL), washing with NaOH (100 mL, 0.2 m), separation of the phases, acidification of the aqueous phase with HCl (2 m, 2.5 mL) followed by extraction with EtOAc (2 × 50 mL). The combined organic extracts were washed with brine, dried (MgSO4) and the solvent was removed at room temperature to yield analytically pure 10 (1.27 g, 44%) as an orange solid; m.p. 138–141 °C; RF = 0.22 (SiO2, CHCl3/MeOH, 4:1); IR (KBr): ν = 3422br, 3284m, 2925w, 1765m, 1734w, 1616m, 1593m, 1481m, 1416m, 1376w, 1289m, 1237s, 1176m, 1102w, 1038m cm−1; UV–vis (MeOH): λmax (log ε) = 246 (4.04), 286 (3.80), 358 (8.87) nm; 1H-NMR (400 MHz, DMSO-d6): δ = 7.08 (s,1H, 8-H), 6.53 (s, 1H, 5-H), 6.08 (s, 2H, 9-H) ppm; 13C-NMR (101 MHz, DMSO-d6): δ = 187.9 (C-2), 167,4 (C-1), 159.8 (C-4), 155.3 (C-6), 141.7 (C-7), 111.7 (C-3), 106.6 (C-8), 102.8 (C-9), 98.3 (C-5) ppm; MS (ESI, MeOH): m/z (%) = 209.0 ([M − H]−, 100), 441.0 ([2M-H + Na]−, 3); analysis calcd for C9H6O6 (210.14): C 51.44, H 2.88; found: C 51.32, H 3.03.
4.7. [1,3]Dioxolo[4,5-f]benzofuran-6,7-dione (11)
A solution of 10 (4.25 g, 22.12 mmol) was suspended in dry DCM (200 mL) and at 0 °C oxalyl chloride (1.9 mL, 22.00 mmol) and dry DMF (200 µL) were added. The reaction mixture was stirred at r.t. for 14 h. The volatiles were removed under reduced pressure, the residue is re-dissolved in dry THF (50 mL) and evaporated to dryness. Column chromatography (SiO2, DCM) gave 11 (1.55 g, 36%) as slightly orange solid; m.p. 203–205 °C; RF = 0.69 (SiO2, DCM); IR (KBr): v = 3446br, 1818w, 1765m, 1716m, 1616s, 1480s, 1415m, 1299s, 1233s, 1176m, 1095m, 1037m cm–1; UV–vis (CHCl3): λmax (log ε = 260 (3.82), 310 (3.56), 414 (3.33) nm; 1H-NMR (500 MHz, DMSO-d6): δ = 7.24 (s, 1H, 4-H), 7.14 (s, 1H, 7-H), 6.23 (s, 2H, 9-H) ppm; 13C-NMR (126 MHz, DMSO-d6): δ = 174.9 (C-2), 161.8 (C-8), 157.3 (C-1), 156.9 (C-6), 145.2 (C-5), 112.5 (C-3), 103.4 (C-9), 102.7 (C-4), 95.6 (C-7) ppm; MS (ESI, MeOH): m/z (%) = 225.0 ([M + H + MeOH]+, 68), 247.1 ([M + Na + MeOH]+, 100), 356.0 ([3(M + MeOH) + K + H]2+, 18), 467.7 ([4(M + MeOH) + K + H]2+, 30); analysis calcd for C9H4O5 (192.13): C 56.26, H 2.10; found: C 55.97, H 2.27.
4.8. 1-(2,4,5-Trimethoxyphenyl)ethan-1-one-1-13C (12)
To a solution of 1,2,4-trimethoxybenzene (3, 2.0 g, 11.9 mmol) in DCM (50 mL) at −20 °C TiCl4 (1.44 mL, 13.1 mmol) was added followed by adding dropwise 13C-acetylchloride (0.93 mL, 13 mmol). The resulting mixture was stirred for 2 h, poured into 2M hydrochloric acid and extracted with chloroform (3 × 50 mL). The combined organic extracts were washed with brine and dried (MgSO4). Evaporation followed by column chromatography gave 12 (2.4 g, 95%) as a pale yellow crystalline solid.; m.p. 189–192 °C; RF = 0.25 (SiO2, hexanes/ethyl acetate, 1:1); 1H-NMR (400 MHz, CDCl3): δ = 7.42 (d, J = 4.2 Hz, 1H, 6-H), 6.50 (d, J = 1.6 Hz, 1H, 3-H), 3.94 (s, 3H, OMe), 3.91 (s, 3H, OMe), 3.87 (s, 3H, OMe), 2.59 (d, J = 6.2 Hz, 3H, 2′-H) ppm; 13C-NMR (101 MHz, CDCl3): δ = 197.2 (C-1′), 155.6 (d, J = 2.2 Hz, C-4), 153.9 (C-2), 143.0 (d, J = 4.0 Hz, C-5), 119.2 (d, J = 54.5 Hz, C-1), 112.54 (d, J = 1.5 Hz, C-6), 96.42 (d, J = 3.4 Hz, C-3), 56.3 (OMe), 56.1 (OMe), 56.1 (OMe), 32.03 (d, J = 42.7 Hz, C-2′); MS (ESI): m/z (%) = 212.37 ([M + H]+, 100).
4.9. 2-Oxo-2-(2,4,5-trimethoxyphenyl)acetic-2-13C Acid (13)
A suspension of 12 (2.0 g, 9.47 mmol) and selenium dioxide (2.0 g, 18 mmol) in pyridine (20 mL) was stirred for 4 h at 80 °C. Work-up as described above followed by re-crystallization from EtOAc gave 13 (1.20 g, 53%) as yellow solid; m.p. 163–165 °C; RF = 0.35 (SiO2, CHCl3/MeOH, 2:1); 1H-NMR (400 MHz, DMSO-d6): δ = 7.22 (d, J = 4.2 Hz, 1H, 6-H), 6.82 (d, J = 1.8 Hz, 1H, 3-H), 3.92 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.76 (s, 3H, OMe) ppm; 13C-NMR (101 MHz, DMSO-d6): δ = 186.5 (C-2′), 168.1 (d, J = 74.3 Hz, C-1′), 157.7 (d, J = 2.0 Hz, C-2), 156.9 (d, J = 0.8 Hz, C-4), 144.2 (d, J = 4.3 Hz, C-5), 113.6 (d, J = 59.5 Hz, C-1), 110.6 (d, J = 1.7 Hz, C-6), 98.6 (d, J = 3.0 Hz, C-3), 57.6 (OMe), 56.7 (OMe), 56.3 (OMe) ppm; MS (ESI, MeOH): m/z (%) = 239.8 ([M − H]−, 100).
4.10. 6-Hydroxy-5-methoxy-3,3-bis(2,4,5-trimethoxyphenyl)benzofuran-2(3H)-one-3-13C (15)
A suspension of 13 (200 mg, 0.83 mmol) in conc. hydrochloric acid (25 mL) was stirred 0 °C for 2 h. The reaction mixture was neutralized with an aqueous sat. soln. of NaHCO3 and extracted with chloroform (3 × 25 mL). The organic extracts were dried (MgSO4), the solvent was removed under diminished pressure and the remaining residue was subjected to column chromatography (SiO2, CHCl3/MeOH, 9:1) to afford 15 as a slightly yellow solid (13 mg, 3%); m.p. 103–105 °C; RF = 0.66 (SiO2, CHCl3/MeOH, 9:1); 1H-NMR (500 MHz, toluene-d8): δ = 7.17–6.83 (m, 2H, 2 × 6′-H), 7.01 (s, 1H, 7-H), 6.70 (d, J = 2.6 Hz, 1H, 4-H 6.28 (s, 2H, 2 × 2′-H), 3.49 – 3.38 (m, 6H, 2 × OMe(2′)), 3.40 (s, 6H, 2 × OMe(5′)), 3.30 (s, 6H, 2 × OMe(4′)), 3.06 (s, 3H, OMe(5)) ppm; 13C-NMR (126 MHz, toluene-d8): δ = 175.91 (d, J = 52.5 Hz, C-2), 148.13 (C-2′), 146.40 (C-4′), 142.8 (C-5), 142.8(C-5′), 137.10 (C-7a), 124.4 (d, J = 57.3 Hz, C-1′), 122.72 (d, J = 45.6 Hz, C-3b), 108.80 (d, J = 1.7 Hz, C-4), 99.8 (C-3′), 97.80 (C-7), 57.41 (C-3), 56.58 (OMe(2′)), 55.65 (OMe(5′)), 55.55 (OMe(4′)), 55.34 (OMe(5)) ppm; MS (ESI, MeOH): m/z (%) = 512.1 ([M − H]−, 100), 536.1 ([M + Na]+, 100), 782.0 ([3M + Na + H]2+, 33), 791.1 ([3M + K + H]2+, 26).
4.11. 6-Hydroxy-5-methoxy-3-(2,4,5-trimethoxyphenyl)benzofuran-2(3H)-one (17)
Compound 6 (250 mg, 0.73 mmol) was suspended in hydrochloric acid (25 mL) and zinc (250 mg, 3.82 mmol) was added at 0 °C. The reaction was stirred for 4 h at room temperature, diluted with water (20 mL) and extracted with EtOAc (3 × 50mL). The combined organic extracts were washed with brine and dried (MgSO4). Purification by column chromatography (SiO2, hexanes/CHCl3, 1:1) gave 17 (112 mg, 43%) as slightly yellow solid; m.p. 143–147 °C; RF = 0.32 (SiO2, hexanes/CHCl3, 1:1); 1H-NMR (500 MHz, CDCl3): δ = 6.78 (s, 1H, 7-H), 6.68 (s, 1H, 6′-H), 6.56–6.53 (m, 2H, 4-H + 3′-H), 5.78 (s, 1H, OH), 4.86 (s, 1H, 3-H3), 3.88 (s, 3H, OMe(4′)), 3.82 (s, 3H, OMe(2′)), 3.79 (s, 3H, OMe(5)), 3.70 (s, 3H, OMe(5′)) ppm; 13C-NMR (126 MHz, CDCl3): δ = 176.57 (C-2), 151.72 (C-2′), 149.96 (C-4′), 148.03 (C-7a), 146.24 (C-6), 143.61 (C-5), 143.36 (C-5′), 118.20 (C-3a), 116.33(C-1′), 114.11 (C-6‘), 106.85 (C-4), 98.63 (C-3‘), 98.44 (C-7), 56.83 (OMe(5′)), 56.79 (OMe(2′)), 56.62 (OMe(5)), 56.18 (OMe(4′)), 46.19 (C-3) ppm; MS (ESI, MeOH): m/z (%) = 345.6 ([M − H]−, 100); analysis calcd for C18H18O7 (346.33): C 62.44, H 5.24; found: C 62.26, H 4.98.
4.12. 5-Methoxy-3-(2-methoxyphenyl)benzofuran-2,6-dione (18)
Compound 2 (240 mg, 1.0 mmol) and anisole (440 mg, 4 mmol) were suspended in hydrochloric acid (25 mL) and the mixture was stirred for 4 h at 40 °C, diluted with water (10 mL), extracted with CHCl3 (3 × 25 mL). The combined organic extracts were washed with brine, dried (MgSO4), the solvent was evaporated and the residue re-crystallized from EtOAc to yield 18 (211 mg, 74%) as a red crystalline solid; m.p. 194–196 °C; RF = 0.54 (SiO2, CHCl3); 1H-NMR (500 MHz, CDCl3): δ = 7.52–7.45 (m, 2H, 6′-H + 4‘-H), 7.11 (td, J = 7.6, 1.0 Hz, 1H, 5′-H), 7.05 (dd, J = 8.3, 1.0 Hz, 1H, 3′-H), 6.27 (s, 1H, 4-H), 6.09 (s, 1H, 7-H), 3.89 (s, 3H, OMe(2′)), 3.82 (s, 3H, OMe(5)) ppm; 13C-NMR (126 MHz CDCl3): δ = 181.29 (C6), 160.53 (C2), 157.38 (C2′), 154.42 (C5), 140.69 (C7a), 132.40 (C4‘), 131.67 (C6‘), 126.18 (C3a), 121.29 (C5‘), 117.65 (C1′), 111.88 (C3‘), 104.49 (C7), 99.83 (C4), 56.41 (OMe(2′)), 55.77 (OMe(5′)) ppm; MS (ESI, MeOH): m/z (%) = 285.4 ([M + H]+, 100), 307.3 ([M + Na]+, 38); analysis calcd for C16H12O5 (284.26): C 67.60, H 4.26; found: C 67.41, H 4.39.
4.13. 3-(4-Aminophenyl)-5-methoxybenzofuran-2,6-dione (19)
To a suspension of 2 (500 mg, 2.08 mmol) in hydrochloric acid (25 mL) Boc-aniline (781 mg, 4 mmol) was added and the mixture was stirred for 4 h at 40 °C. Usual aqueous workup followed by column chromatography (SiO2, CHCl3/MeOH, 95:5) and recrystallization from EtOAc gave 19 (243 mg, 43%) as black crystalline solid; m.p. 243–246 °C; RF = 0.14 (SiO2, CHCl3); IR (ATR): ν = 3456w, 3331m, 3219w, 1780m, 1662m, 1643m, 1628m, 1600s, 1575s, 1551s, 1515s, 1506m, 1470w, 1445m, 1439m, 1415s, 1355m, 1331m, 1227s, 1181s, 1157s, 995m, 945m, 850m, 840s, 832s, 821m, 789m, 773m, 687m, 679m, 602m cm−1; UV–Vis (MeOH): λmax (log ε) 251 (3.67), 317 (3.80), 507 (3.92) nm; 1H-NMR (500 MHz, DMSO-d6): δ = 7.71 (d, J = 8.7 Hz, 2H, 2′-H), 6.75 (s, 1H, 4-H), 6.72 (d, J = 8.8 Hz, 1H, 3′-H), 6.26 (s, 2H, NH), 6.12 (s, 1H, 7-H), 3.85 (s, 3H, OMe) ppm; 13C NMR (126 MHz, DMSO-d6): δ = 179.71 (C-6), 167.28 (C-2), 159.86 (C-7a), 153.98 (C-5), 152.18(C-3), 131.58 (C-2′), 130.82 (C-3a), 126.93 (C-1′), 116.23 (C-4′), 114.06 (C-3′), 103.10 (C-7), 99.84 (C-4), 56.03 (OMe) ppm; MS (ESI, MeOH): m/z (%) = 270.2 ([M + H]+, 100), 292.0 ([M + Na]+, 14), 302.1 ([M + H + MeOH]+, 20), 423.5 ([3M + Ca]2+, 18), 560.8 ([2M + Na]+, 90); analysis calcd for C15H11NO4 (269.25): C 66.91, H 4.12, N 5.20; found: C 66.72, H 4.34, N 5.02.
4.14. N-(4-(5-Methoxy-2,6-dioxo-2,6-dihydrobenzofuran-3-yl)phenyl)benzamide (20)
To a solution of 19 (200 mg, 0.74 mmol) in DCM (50 mL) and triethylamine (0.11 mL, 0.8 mmol) at 0 °C benzoylchloride (0.93 mL, 0.8 mmol) was added and the reaction was stirred for 6 h. Usual aqueous workup followed by column chromatography (SiO2, CHCl3/MeOH, 9:1) gave 20 (191 mg, 69%) as a red solid; m.p. 259–264 °C; RF = 0.11 (SiO2, CHCl3); IR (ATR): ν = 3377w, 3021w, 2973w, 2934w, 1811w, 1782m, 1749w, 1669m, 1629s, 1587s, 1572s, 1512s, 1489s, 1452m, 1421m, 1408s, 1351m, 1321s, 1303m, 1250s, 1232s, 1181s, 1158s, 1124m, 1105m, 1079w, 1066w, 1020w, 991m, 934m, 904m, 854m, 847s, 837s, 827s, 799m, 789m, 720s, 699m, 690m, 669m, 625s, 610s, 546s cm−1; UV–Vis (methanol): λmax (log ε) = 229 (3.89), 307 (3.97), 452 (3.92) nm; 1H-NMR (500 MHz, pyridine-d5): δ = 11.36 (s, 1H, NH), 8.37–8.32 (m, 2H, 2 × 2′-H), 8.27–8.21 (m, 2H, 2 × 2′’-H), 8.09–8.05 (m, 2H, 2 × 3′-H), 7.50–7.46 (m, 1H, 4′’-H), 7.45–7.38 (m, 2H, 2 × 3′’-H), 6.71 (s, 1H, 4-H), 6.32 (s, 1H, 7-H), 3.77 (s, 3H, OMe) ppm; 13C-NMR (126 MHz, pyridine-d5): δ = 180.98 (C-6), 167.90 (C=O), 167.75 (C-2), 160.91(C-7a), 156.36 (C-5), 143.12 (C-4′), 138.18 (C-3a), 136.42 (C-1′’), 132.47 (C-4′’), 131.38 (2x C-2′), 129.19 (2x C-3′’), 128.86 (2 × C-2′’), 127.09 (C-3), 125.27 (C-1′), 121.63 (2 × C-3′), 105.16 (C-7), 99.25 (C-4), 56.70 (OMe) ppm; MS (ESI, MeOH): m/z (%) = 374.20 ([M + H]+, 46), 396.07 ([M + Na]+, 18), 406.0 ([M + MeOH + H]+, 100), 428.1 ([M + MeOH + Na]+, 32), 766.7 ([2M + Na]+, 55), 800.7 ([2M + MeOH + Na]+, 70), 832.9 ([2M + 2MeOH + Na]+, 64); analysis calcd for C22H15NO5 (373.36): C 70.77, H 4.05, N 3.75; found: C 70.51, H 4.35, N 3.52.
4.15. 6-Hydroxy-5-methoxy-3-phenyl-3-(2,4,5-trimethoxyphenyl)benzofuran-2(3H)-one (21)
To a solution of 2 (240 mg, 1.0 mmol) and benzene (45 µL, 0.5 mmol) hydrochloric acid (25 mL) was added and the mixture was stirred for 2 h. Usual aqueous work-up followed by column chromatography (SiO2, hexanes/CHCl3, 1:1) gave 21 (74 mg, 18%) as a pale yellow crystalline solid; m.p. 112–117 °C; RF = 0.63 (SiO2, CHCl3); IR (KBr): ν = 3432br, 3081w, 3062w, 3008w, 2961w, 2941w, 2842w, 1763w, 1638s, 1586s, 1527m, 1516m, 1461m, 1427w, 1382w, 1412m, 1388m, 1348s, 1322w, 1233s, 1190s, 1140m, 1043m, 991m, 936m, 852s, 824m, 781m, 689m cm−1; UV-Vis (MeOH): λmax (log ε) = 239 (3.05), 284 (3.01), 331 (3.37) nm1H NMR (500 MHz, CDCl3): δ = 7.45 (d, J = 8.0 Hz, 2H, 2′’Ha + 2′’-Hb), 7.38–7.31 (m, 3H, 3”-Ha + 3′’-Hb + 4′’-H), 6.81 (s, 1H, 7-H), 6.54 (s, 1H, 4-H), 6.50 (s, 1H, 3′-H), 6.41 (s, 1H, 6′-H), 5.85 (s, 1H, OH), 3.86 (s, 3H, OMe(2′)), 3.81 (s, 3H, OMe(5)), 3.62 (s, 3H, OMe(5′)), 3.61 (s, 3H, OMe(4′)) ppm; 13C NMR (126 MHz, CDCl3): δ = 178.27 (C-2), 151.58 (C-2′), 149.83 (C-4), 147.79 (C-7a), 146.46 (C-6), 143.63 (C-5), 143.13 (C-5′), 138.09 (C-1′’), 129.27 (2 × C-2′’), 128.53 (2 × C-3′’), 128.32 (C-4′’), 123.36 (C-1′), 120.65 (C-3b), 114.05 (C-6′), 108.18 (C-4), 99.07 (C-3′), 98.57 (C-7), 57.96 (C-3), 56.9 (OMe(4′)), 56.9 (OMe(5′)), 56.8 (OMe(5)), 56.2 (OMe(2′)) ppm; MS (ESI): m/z (%) = 421.2 ([M − H]−, 100); analysis calcd for C24H22O7 (422.43): C 68.24, H 5.25; found: C 68.10, H 5.37.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








