
Syntheses, Reactivities, Characterization, and Crystal Structures of Dipalladium Complexes Containing the 1,3-pyrimidinyl Ligand: Structures of [Pd(PPh3)(Br)]2(μ,η2-C4H3N2)2, [Pd(Br)]2(μ,η2-Hdppa)2, and [{Pd(PPh3)(CH3CN)}2(μ,η2-C4H3N2)2][BF4]2


Abstract
:1. Introduction
2. Results and Discussion
2.1. Syntheses
2.2. Infrared Spectroscopy and Mass Spectrometry
2.3. Nuclear Magnetic Resonance Spectroscopy
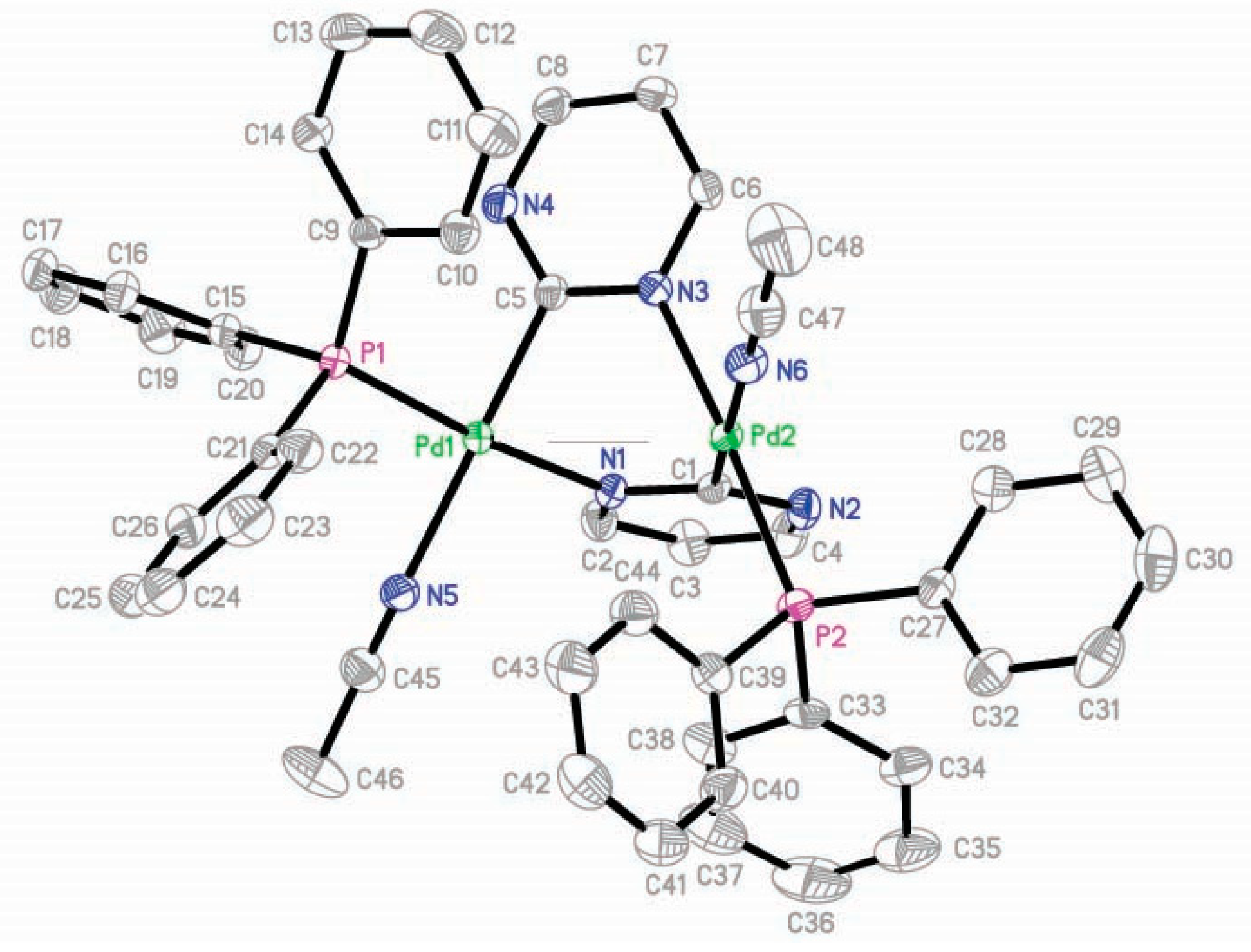
2.4. X-ray Single-Crystal Structures of 3, 4, and 8
3. Materials and Method
3.1. Materials
3.1.1. [Pd(PPh3)(Br)]2(μ,η2-C4H3N2)2, 3
3.1.2. [Pd(Br)]2(μ,η2-Hdppa)2, 4
3.1.3. [Pd(PPh3)(η1-C4H3N2)(η2-S2CNC4H8)], 5
3.1.4. [Pd(PPh3)(η1-C4H3N2)(η2-S2COEt)], 6
3.1.5. [Pd(PPh3)(η1-C4H3N2)(η2-Tp)], 7
3.1.6. [{Pd(PPh3)(CH3CN)}2(μ,η2-C4H3N2)2][BF4]2, 8
3.2. X-ray Crystallography
Single-Crystal X-ray Diffraction Analyses of 3, 4, and 8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steffen, A.; Sladek, M.I.; Braun, T.; Neumann, B.; Stammler, H.-G. Catalytic C−C Coupling Reactions at Nickel by C−F Activation of a Pyrimidine in the Presence of a C−Cl Bond: The Crucial Role of Highly Reactive Fluoro Complexes. Organometallics 2005, 24, 4057–4064. [Google Scholar] [CrossRef]
- Beeby, A.; Bettington, S.; Fairlamb, I.J.S.; Goeta, A.E.; Kapdi, A.R.; Niemela, E.H.; Dobrzynski, E.D.; Angelici, R.J. Synthesis of carbene complexes by reaction of platinum thiocarbamoyl and thio ester complexes with electrophiles. Inorg. Chem. 1975, 14, 1513–1518. [Google Scholar]
- Joshaghani, M.; Daryanavard, M.; Rafiee, E.; Nadri, S. Synthesis and applications of a new palladacycle as a high active catalyst in the Suzuki couplings. J. Organomet. Chem. 2008, 693, 3135–3140. [Google Scholar] [CrossRef]
- Yang, W.-H.; Lee, C.-S.; Pal, S.; Chen, Y.-N.; Hwang, W.-S.; Lin, I.J.; Wang, J.-C. Novel Ag(I), Pd(II), Ni(II) complexes of N,N′-bis-(2,2-diethoxyethyl)imidazole-2-ylidene: Synthesis, structures, and their catalytic activity towards Heck reaction. J. Organomet. Chem. 2008, 693, 3729–3740. [Google Scholar] [CrossRef]
- Chin, C.C.H.; Yeo, J.S.L.; Loh, Z.-H.; Vittal, J.J.; Henderson, W.; Hor, A. Synthesis and electrospray mass spectrometry of palladium(II) diphosphine complexes from oxidative addition of 2-bromopyridine to Pd0. J. Chem. Soc. Dalton Trans. 1998, 3777–3784. [Google Scholar] [CrossRef]
- Beeby, A.; Bettington, S.; Fairlamb, I.J.S.; Goeta, A.E.; Kapdi, A.; Niemelä, E.; Thompson, A.L. A new precatalyst for the Suzuki reaction—a pyridyl-bridged dinuclear palladium complex as a source of mono-ligated palladium(0). New J. Chem. 2004, 28, 600–605. [Google Scholar] [CrossRef]
- Lin, Y.C.; Yih, K.-H.; Lee, G.-H.; Huang, S.-L.; Wang, Y. Synthesis, Dissociation Behavior and Crystal Structures of Palladium(II) Complexes with N,N-Dimethylthiocarbamoyl, Me2NC=S, Containing Ligand: Structures of [Pd(PPh3)2(η1-SCNMe2)(Cl)], [Pd(PPh3)(Cl)]2(μ,η2-SCNMe2)2, [Pd(PPh3)2{η1-C(SMe)(NMe2)}(η2-S2CO)]. J. Chin. Chem. Soc. 2004, 51, 279–290. [Google Scholar] [CrossRef]
- Yih, K.-H.; Lee, G.-H. Sulfur-assisted Chloride and Triphenylphosphine Dissociation of Palladium(II) Complex with Phenyloxythiocarbonyl, PhOC(S), Containing Ligand: X-ray Structure of [Pd(PPh3)2{η1-C(S)OPh}(Cl)]. J. Chin. Chem. Soc. 2004, 51, 265–270. [Google Scholar] [CrossRef]
- Yih, K.-H.; Wang, H.-F.; Lee, G.-H. The Nitrogen-Assisted Triphenylphosphine Displacement of Thiazoline Ligand in Palladium(II) Complex: Crystal Structures of [Pd(PPh3)2(η1-)(Br)] and [Pd(PPh3)(Br)]2(μ,η2-)2. J. Chin. Chem. Soc. 2007, 54, 553–558. [Google Scholar] [CrossRef]
- Yih, K.-H.; Lee, G.-H. The Nitrogen-Assisted Triphenylphosphine Displacement of Methylpyridine Ligand in Palladium(II) Complex: Crystal Structures of [Pd(PPh 3 ) 2 {?1-C 5 H 3 N(CH 3 )}(Br)] and [Pd(PPh 3 )Br] 2 {?,? 2 -C 5 H 3 N(CH 3 )} 2. J. Chin. Chem. Soc. 2008, 55, 109–114. [Google Scholar] [CrossRef]
- Yih, K.-H.; Lee, G.-H.; Wang, Y. Sulfur-assisted chloride and triphenylphosphine dissociation of palladium(II) complex [Pd(PPh3)2(η1-SCNMe2)(Cl)]. X-ray structures of [Pd(PPh3)2(η1-SCNMe2)(Cl)], [Pd(PPh3)(Cl)]2(μ,η2-SCNMe2)2, and [Pd(PPh3)2(η2-SCNMe2)][PF6]. Inorg. Chem. Commun. 2003, 6, 577–580. [Google Scholar] [CrossRef]
- Yih, K.H.; Lee, G.H. Thiocarbarmoyl-Assisted Formation of a New Type of α- and β-Isomer Paddlewheel Pd24+ Compounds. Synthesis, Characterization, and Crystal Structures of [Pd(η1-SCNMe2)(η2-Tp)(PPh3)], [Pd2(μ-Hdppa)2(μ-SCNMe2)2][Cl]2, [Pd2(μ-dppa)2(μ-SCNMe2)2], and [Pd{η2-S2P(OEt)2}]2(μ-dppa)(μ-SCNMe2). Organometallics 2010, 29, 3397–3403. [Google Scholar]
- Lee, G.H.; Wang, H.F.; Yih, K.H. trans-Bromido(pyrimidinyl-kC2)- bis(triphenylphosphane-kP)palladium(II). Acta Crystallographica Section E 2010, E66, m1345. [Google Scholar] [CrossRef] [PubMed]
- Yih, K.H.; Lee, G.H.; Wang, Y. Novel palladium and platinum carbene-complexes containing dithiocarbonate ligand [M(PPh3){η2(S,S)-S2CO]{C(SR)(NMe2)}] formed viaalkyl migration of O-alkyldithiocarbonate to thiocarbamoyl ligand. Dalton Trans. 2003, 14, 2810–2812. [Google Scholar] [CrossRef]
- Yih, K.H.; Wang, H.F.; Huang, K.F.; Kwan, C.C.; Lee, G.H. The Nitrogen-Assisted Triphenylphosphine Displacement of 3-Hydroxypyridine and 3-Aminopyridine Ligands in Palladium(II) Complexes: Crystal Structures of [Pd(PPh3)Br]2{μ,η2-C5H3N(OH)}2 and [Pd(PPh3)Br]2{μ,η2-C5H3N(NH2)}2. J. Chin. Chem. Soc. 2009, 56, 718–724. [Google Scholar] [CrossRef]
- Lee, C.L.; Yang, Y.P.; Rettig, S.J.; James, B.R.; Nelson, D.A.; Lilga, M.A. Synthesis and characterization of dinuclear palladium(I) and mononuclear palladium(II) complexes containing 1,1-bis(diphenylphosphino)ethane (dpmMe) and related mixed-ligand complexes containing dpmMe with either bis(diphenylphosphino)methane (dpm) or 2-(diphenylphosphino)pyridine (Ph2Ppy): X-ray crystal structures of PdCl2(dpmMe) and Pd2Cl2(.mu.-dpmMe)2. Organometallics 1986, 5, 2220–2228. [Google Scholar] [CrossRef]
- Boehm, J.R.; Doonan, D.J.; Balch, A.L. Preparation and some reactions of dimeric isocyanide complexes of palladium(I) and platinum(I). J. Am. Chem. Soc. 1976, 98, 4845–4850. [Google Scholar] [CrossRef]
- Gabe, E.J.; Lee, F.L.; Lepage, Y.; Shhelldrick, E.G.M.; Kruger, C.; Goddard, R. Crystallographic Computing 3; Clarendon Press: Oxford, UK, 1985; Volume 167. [Google Scholar]
- Birmingham, K.P. International Tables for X-ray Crystallography; Reidel: Dordrecht, The Netherlands, 1974; Volume IV. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3·CH2Cl2 | 4·CH3COCH3 | 8·2CH3CN | |
|---|---|---|---|
| Chemical formula | C45H38Br2Cl2N4P2Pd2 | C51H48Br2N2OP4Pd2 | C52H48B2F8N8P2Pd2 |
| Formula weight | 1140.25 | 1201.41 | 1233.34 |
| Crystal system | triclinic | monoclinic | triclinic |
| Space group | P-1 | P2(1)/c | P-1 |
| a, Å | 10.4218(5) | 13.6382(8) | 13.3314(6) |
| b, Å | 13.0314(7) | 17.0076(10) | 13.7552(6) |
| c, Å | 17.6646(9) | 21.1326(12) | 16.7500(7) |
| α, deg | 76.958(1) | 90 | 71.6169(9) |
| β, deg | 77.213(1) | 105.145(2) | 80.4954(10) |
| γ, deg | 71.316(1) | 90 | 65.0198(9) |
| V, Å3 | 2184.93(19) | 4731.5(5) | 2640.3(2) |
| Z | 2 | 4 | 2 |
| ρcalcd, g cm−3 | 1.733 | 1.687 | 1.551 |
| μ,(Mo Kα), mm−1 | 2.885 | 2.625 | 0.814 |
| λ, Å | 0.71073 | 0.71073 | 0.71073 |
| T, K | 150(2) | 150(2) | 150(2) |
| θ range, deg | 1.89–27.50 | 1.56–27.50 | 1.83–27.50 |
| Independent rflns | 10003 | 10865 | 12109 |
| no. of variables | 514 | 561 | 690 |
| Ra | 0.041 | 0.055 | 0.039 |
| Rwb | 0.084 | 0.116 | 0.087 |
| Sc | 1.027 | 1.044 | 1.034 |
| Bond Lengths | Bond Angles | ||
|---|---|---|---|
| Complex 3 | |||
| Pd(1)-C(5) | 1.981(4) | C(5)-Pd(1)-Br(1) | 173.84(10) |
| Pd(1)-N(1) | 2.083(3) | C(1)-Pd(2)-Br(2) | 173.39(10) |
| Pd(1)-P(1) | 2.2702(9) | N(1)-Pd(1)-P(1) | 176.00(8) |
| Pd(1)-Br(1) | 2.4974(5) | N(3)-Pd(2)-P(2) | 172.13(9) |
| Pd(1)-Pd(2) | 3.2008(4) | C(5)-Pd(1)-N(1) | 86.39(13) |
| Pd(2)-C(1) | 1.976(4) | C(1)-Pd(2)-N(3) | 86.83(13) |
| Pd(2)-N(3) | 2.097(3) | P(1)-Pd(1)-Br(1) | 94.56(3) |
| Pd(2)-P(2) | 2.2716(10) | P(2)-Pd(2)-Br(2) | 95.26(3) |
| Pd(2)-Br(2) | 2.5099(5) | P(1)-Pd(1)-Pd(2) | 118.19(3) |
| Complex 4 | |||
| Pd(1)-P(3) | 2.2830(13) | P(3)-Pd(1)-P(1) | 176.23(5) |
| Pd(1)-P(1) | 2.2945(14) | P(3)-Pd(1)-Br(1) | 90.08(4) |
| Pd(1)-Br(1) | 2.5168(7) | P(1)-Pd(1)-Br(1) | 92.11(4) |
| Pd(1)-Pd(2) | 2.6414(6) | P(3)-Pd(1)-Pd(2) | 88.50(4) |
| Pd(2)-P(2) | 2.2665(14) | P(1)-Pd(1)-Pd(2) | 89.43(4) |
| Pd(2)-P(4) | 2.2794(14) | Br(1)-Pd(1)-Pd(2) | 177.52(2) |
| Pd(2)-Br(2) | 2.5171(7) | P(2)-Pd(2)-P(4) | 171.63(5) |
| P(1)-N(1) | 1.693(4) | P(2)-Pd(2)-Br(2) | 94.05(4) |
| P(3)-N(2) | 1.682(4) | P(4)-Pd(2)-Br(2) | 93.81(4) |
| Complex 8 | |||
| Pd(1)-C(5) | 1.965(3) | C(5)-Pd(1)-N(5) | 173.45(11) |
| Pd(1)-N(1) | 2.094(3) | N(1)-Pd(1)-P(1) | 174.17(7) |
| Pd(1)-N(5) | 2.117(3) | C(1)-Pd(2)-N(6) | 173.39(11) |
| Pd(1)-P(1) | 2.2813(9) | N(3)-Pd(2)-P(2) | 176.05(7) |
| Pd(1)-Pd(2) | 3.2276(4) | N(1)-Pd(1)-N(5) | 89.76(9) |
| Pd(2)-C(1) | 1.961(3) | N(3)-Pd(2)-N(6) | 88.83(10) |
| Pd(2)-N(3) | 2.0823) | N(1)-Pd(1)-Pd(2) | 59.98(6) |
| Pd(2)-N(6) | 2.104(3) | P(1)-Pd(1)-Pd(2) | 120.09(2) |
| Pd(2)-P(2) | 2.2741(8) | N(6)-Pd(2)-P(2) | 94.35(7) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.-F.; Yih, K.-H.; Lee, G.-H. Syntheses, Reactivities, Characterization, and Crystal Structures of Dipalladium Complexes Containing the 1,3-pyrimidinyl Ligand: Structures of [Pd(PPh3)(Br)]2(μ,η2-C4H3N2)2, [Pd(Br)]2(μ,η2-Hdppa)2, and [{Pd(PPh3)(CH3CN)}2(μ,η2-C4H3N2)2][BF4]2. Molecules 2020, 25, 2035. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092035
Wang H-F, Yih K-H, Lee G-H. Syntheses, Reactivities, Characterization, and Crystal Structures of Dipalladium Complexes Containing the 1,3-pyrimidinyl Ligand: Structures of [Pd(PPh3)(Br)]2(μ,η2-C4H3N2)2, [Pd(Br)]2(μ,η2-Hdppa)2, and [{Pd(PPh3)(CH3CN)}2(μ,η2-C4H3N2)2][BF4]2. Molecules. 2020; 25(9):2035. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092035
Chicago/Turabian StyleWang, Hsiao-Fen, Kuang-Hway Yih, and Gene-Hsiang Lee. 2020. "Syntheses, Reactivities, Characterization, and Crystal Structures of Dipalladium Complexes Containing the 1,3-pyrimidinyl Ligand: Structures of [Pd(PPh3)(Br)]2(μ,η2-C4H3N2)2, [Pd(Br)]2(μ,η2-Hdppa)2, and [{Pd(PPh3)(CH3CN)}2(μ,η2-C4H3N2)2][BF4]2" Molecules 25, no. 9: 2035. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092035