
Improved Synthesis of Phosphoramidite-Protected N6-Methyladenosine via BOP-Mediated SNAr Reaction


1
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK
2
Department of Biochemistry, Medical College of Wisconsin, Milwaukee, WI 53226, USA
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(1), 147; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010147
Submission received: 10 November 2020
/
Revised: 16 December 2020
/
Accepted: 18 December 2020
/
Published: 31 December 2020
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:N6-methyladenosine(m6A) is the most abundant modification in mRNA. Studies on proteins that introduce and bind m6A require the efficient synthesis of oligonucleotides containing m6A. We report an improved five-step synthesis of the m6A phosphoramidite starting from inosine, utilising a 1-H-benzotriazol-1-yloxytris(dimethylamino)phosphoniumhexafluorophosphate (BOP)-mediated SNAr reaction in the key step. The route manifests a substantial increase in overall yield compared to reported routes, and is useful for the synthesis of phosphoramidites of other adenosine derivatives, such as ethanoadenosine, an RNA analogue of the DNA adduct formed by the important anticancer drug Carmustine.
1. Introduction
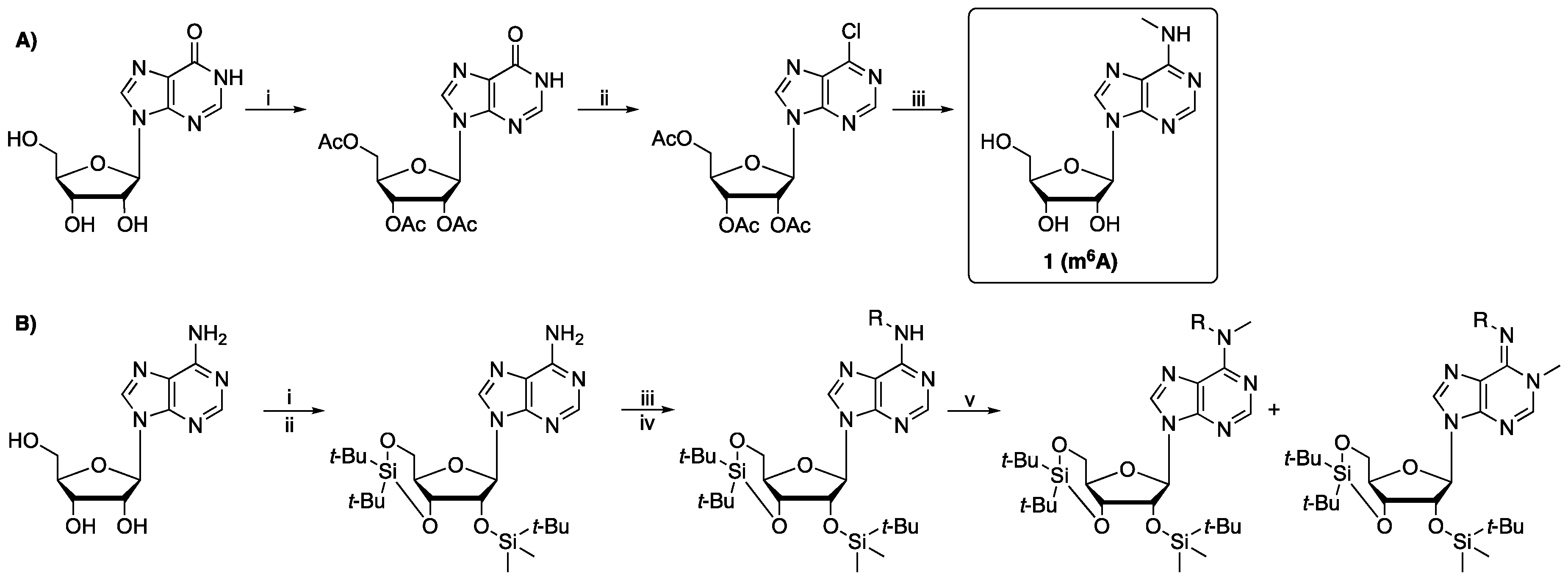
Modifications to mRNA are presently of interest from basic biological science and medicinal chemistry perspectives [1]. N6-methyladenosine(m6A) is the most abundant modification to mRNA, being reported to be present at an average of 2–3 sites/animal mRNA molecule [2]. Some of the methyltransferases, demethylases and reader domains that introduce, remove and bind m6A in mRNA, respectively, are linked to disease [3]. Studies on the roles of such enzymes/proteins and the medicinal chemistry targeting them requires the efficient synthesis of oligonucleotides containing m6A. Research on m6A has been hampered by its high cost, due to low yields for the reported seven to eight-step syntheses of phosphoramidite-protected m6A and the use of relatively expensive starting materials [4,5,6,7]. In one strategy, acetylation of the hydroxyls of inosine is followed by Vilsmeier-type aryl chlorination giving 2′,3′,5′-O-triacetyl-6-chloroinosine [8] (Scheme 1A). SNAr reaction with methylamine and cleavage of the acetyl groups gives m6A (1, Scheme 1A) which can be phosphoramidite protected. Alternatively, the hydroxyls of adenosine are first silyl protected using Beigelman’s strategy [9], then the exocyclic amine is protected by benzoylation or acetylation. Methylation is achieved using methyliodide, however, formation of N1-methyladenosine(m1A) significantly compromises the m6A yield [4] (Scheme 1B).
Here we describe a five-step synthesis of the m6A phosphoramidite from inosine employing BOP-mediated SNAr reaction. The lack of use of POCl3 or methyliodide enables a two to three-fold improvement in the m6A phosphoramidite yield compared to reported methods (Scheme 2) [4,5,6,7]. BOP- or PyBOP-mediated SNAr reaction is also useful for the synthesis of m6A analogues [10,11], e.g., methylethanoadenosine and ethylethanoadenosine.
2. Results
2.1. Synthesis of m6A Phosphoramidite
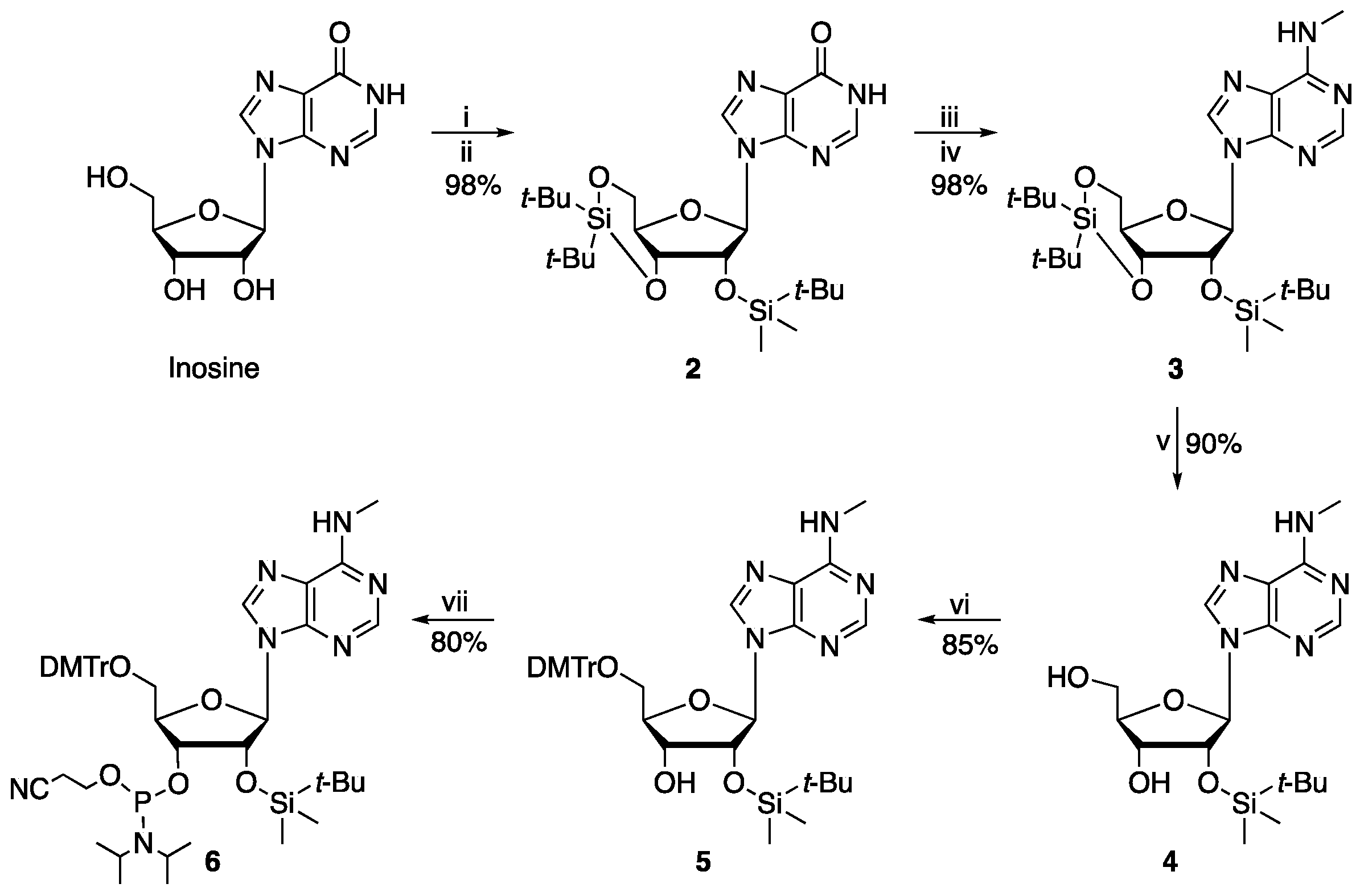
In our synthesis of the m6A phosphoramidite, the 5′- and 3′-hydroxyls of inosine were first selectively protected using di-tert-butylsilyl bis(trifluoromethanesulfonate) (DTBS ditriflate) (Scheme 3). The 2′-hydroxyl group was then protected using tert-butyldimethylsilyl chloride (TBDMSCl) in a one-pot procedure [9] (Beigelman’s strategy, Scheme 3). The resultant 3′,5′-O-(di-tert-butyl)silyl-2′-O-dimethyl(tert-butyl)silyl-inosine (2) was treated with BOP using 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) as a base, to give O6-benzotriazol-3′,5′-O-(di-tert-butyl)silyl-2′-O-dimethyl(tert-butyl)silyl-inosine (II, Figure 1), which was reacted in situ with excess methylamine to give 3′,5′-O-(di-tert-butyl)silyl-2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine [10] (3, 98% apparent yield, over two steps, Scheme 3). The DTBS group of 3 was selectively cleaved using a dilute solution of HF.pyridine, to give 2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine [4] (4, 90%, Scheme 3). The 5′-hydroxyl of 4 was selectively protected using 4,4′-dimethoxytritylchloride (DMTrCl) to give 5′-O-(4,4′-dimethoxytrityl)-2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine (5, 85%, Scheme 3) as reported [7]; portionwise addition of DMTrCl gave an improved yield. Note that TBDMS group migration from the 3′- to the 2′-hydroxyl and vice-versa occurs in protic solvents [12], hence use of such solvents, e.g., methanol, should be avoided for chromatography of 5.
Phosphitylation of the 3′-hydroxyl of 5 was achieved using 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite to give 5′-O-(4,4′-dimethoxytrityl)-(3′-O-[(2-cyanoethyl)(N,N-diisopropylamino)phosphino]-2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine (6, 80%, Scheme 3), using 10 equivalents of base and 2.5 equivalents of the phosphitylating reagent with moisture-free purification under Ar. The optimised route comprises a five-step procedure proceeding to a 60% overall yield from inosine, a substantial advantage over reported routes (Scheme 2) [4,5,6,7].
We also investigated the direct reaction of BOP-activated inosine with methylamine to give m6A (1, 90%, Scheme 4); treatment of m6A (1) with DMTrCl gave 5′-O-(4,4′-dimethoxytrityl)-N6-methyladenosine (7) as the major crude product (Scheme 4). Treatment of the crude product mixture with TBDMSCl/imidazole, gave a mixture of the 2′- and 3′- TBDMS regioisomers of protected m6A (8 and 9, Scheme 4). However, this mixture was difficult to separate by flash column chromatography; a low yield (28–37%) of the desired isomer (8, Scheme 4) has been reported in the literature [7,8]. Thus, although this procedure is shorter, it is presently less efficient than our afore-described route.
2.2. Synthesis of m6A Analogues
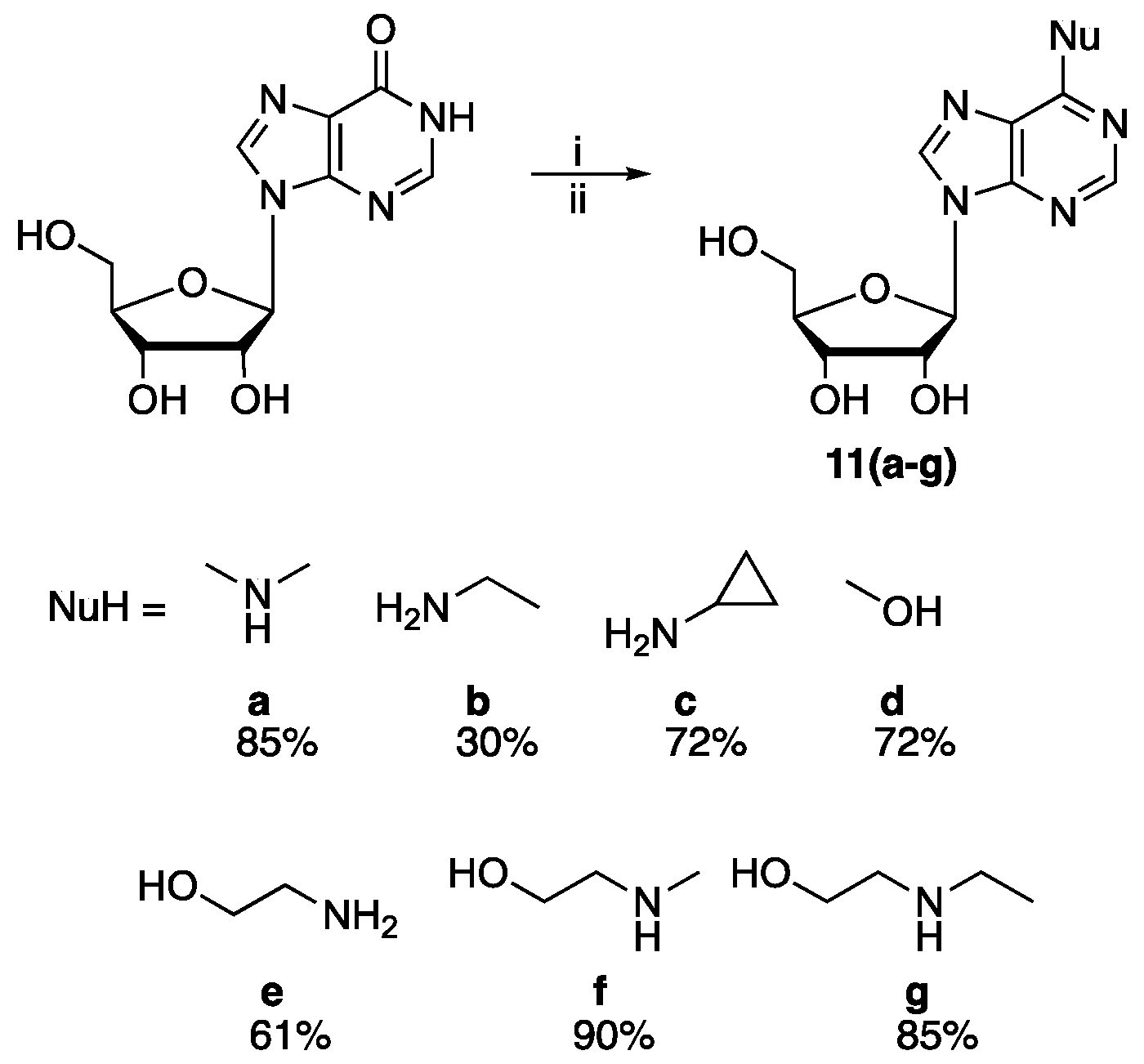
To test the generality of the SNAr reaction, we explored the synthesis of m6A analogues (11a–g, Scheme 5). Thus, treatment of inosine with BOP/DBU in DMF (40 min) was followed by addition of an amine and reaction overnight to give the desired products in generally good non-optimised yields.
2.3. Synthesis of N1,N6-Ethanoadenosine and N6, N1,N6-(m)Ethylethanodenosine Phosphoramidites
We also investigated the synthesis of N1,N6-ethanoadenosine and analogues using the BOP-mediated SNAr reaction. Following treatment of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]inosine (12, Scheme 6) with BOP/DBU, addition of excess ethanolamine, methylethanolamine, or ethylethanolamine resulted in the desired 6-substituted adenosine derivatives (13a–c, Scheme 6) in high yields (86–90%). 2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6-(2-hydroxyethyl)adenosine (13a, Scheme 6) was treated with (triphenoxy)phosphoniummethyl iodide to give intermediate 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6-(2-iodoethyl)adenosine (14a, Scheme 6) which spontaneously cyclises to give the desired 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N1,N6-ethanoadenosine (15a, 60%, Scheme 6). Cyclisation of 13 (b and c) using (triphenoxy)phosphonium-methyl iodide gives the positively charged cyclic products (15b (50%) and 15c (36%), Scheme 6). TBDMS deprotection using 3HF.Et3N [13] (15a–c, Scheme 6) gave N1,N6-ethanoadenosine, N6,N1,N6-methylethanoadenosine, or N6,N1,N6-ethylethanoadenosine (16a–c, 22–70%, Scheme 6).
3. Discussion
Overall, we have developed a modular and efficient five-step synthesis of phosphoramidite-protected m6A starting from inosine, which avoids the use of POCl3/the Vilsmeier reagent (Scheme 1A). The key step comprises the efficient (90% yield) BOP-mediated SNAr reaction of methylamine with a readily prepared protected inosine derivative, Although, there are reports of using BOP-mediated SNAr to prepare ribonucleoside analogues [10,14,15,16,17], use of the reaction to prepare phosphoramidite-protected materials suitable for oligonucleotide synthesis has been limited [18,19]. Investigations on the mechanism of BOP-mediated SNAr reactions are reported [11]. Thus, the oxide formed by base-mediated deprotonation of the hydroxyl group on the aromatic inosine ring reacts with the electrophilic phosphorus of BOP to form an (acyloxy)phosphonium intermediate (I, Figure 1), which undergoes SNAr reaction with the oxybenzotriazole to form an intermediate (II, Figure 1) which is subsequently replaced by a nucleophile in a second SNAr reaction to form the desired product (III, Figure 1). The method is also suited for the synthesis of phosphoramidites of other purines/pyrimidines modified at the C-6/C-4 positions, respectively, as shown by the preparation of N1,N6-ethanoadenosine and its analogues using BOP-mediated SNAr (Scheme 6); these reactions resulted in moderate (22%) to very good (90%) non-optimised yields ( (11b, Scheme 5) (in the case of N6-ethyladenosine, the yield was low due to a non-optimal work-up procedure), showing the generality of the reaction. The method also works with alcohol nucleophiles, though since MeOH is a poor nucleophile, after the formation of the proposed oxybenzotriazol intermediate (II, Figure 1), the crude mixture was evaporated and redissolved in excess methanol in the presence of a base (DBU) to give the desired O6-methylinosine (11d) product (72%) (Scheme 5). The absence of protection of the hydroxyl groups of inosine does not substantially impact on yields as evident from the formation of 11(e–g) vs. 13(a–c), although, more polar solvent is required to dissolve unprotected inosine.
The modified nucleosides described here will have use in ongoing investigations to probe the selectivity and inhibition of RNA modifying enzymes, including m6A demethylases and m6A reader proteins. The BOP-mediated SNAr reaction may also have other applications, e.g., in improving the reported route to N1,N6-ethanodeoxyadenosine phosphoramidite [20] or because N1,N6-ethanoadenosine can be oxidised to N1,N6-ethenoadenosine using MnO2, to give a fluorescent analogue of adenosine [21].
4. Materials and Methods
4.1. General Experimental Considerations
Reagents for synthesis were from Sigma-Aldrich, Alfa Aesar, Cambridge Biotech, Fischer Scientific, or Link Technology, unless otherwise stated. Anhydrous solvents used in reactions were either analytical grade, as obtained commercially (Alfa Aesar), or were freshly distilled. HPLC grade solvents were employed for work-up and chromatography. For the chromatographic purification of phosphoramidites, solvents were dried over P2O5 prior to use. Reactions involving moisture-sensitive reagents were carried out under an argon atmosphere; glassware was oven dried and cooled under nitrogen before use. Reagents were used as supplied (analytical or HPLC grade) without prior purification. Anhydrous MgSO4 was used as a drying agent.
Thin layer chromatography was performed using aluminium plates coated with 60 F254 silica. Plates were visualised using UV light (254 nm), or 1% (m/v) aq. KMnO4 stain. Flash column chromatography was performed using Kieselgel 60 silica in a glass column, or on a Biotage SP4 flash column chromatography platform. Retention factors (Rf) are quoted to a precision of 0.05.
Deuterated solvents were from Sigma and Apollo Scientific Ltd. 1H-NMR and 13C-NMR spectra were recorded using Bruker AVIII400, AVII500, AVIII600 and AVIII700 NMR spectrometers (Bruker, Banner Lane, UK). Fields were locked by external referencing to the relevant residual deuterium resonance. Chemical shifts (δ) are reported in ppm; coupling constants (J) are recorded in Hz to the nearest 0.5 Hz; when peak multiplicities are reported, the following abbreviations are used: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broadened, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets. Spectra were recorded at room temperature unless otherwise stated. 1H and 13C-NMR spectra of compounds are available in the online Supplementary Materials.
Low-resolution mass spectra (m/z) and high-resolution mass spectra (HRMS) were recorded using an LCT Premier XE (Waters, Elstree, UK) or a microTOF machine (Bruker, Banner Lane, UK).
Melting points were recorded on a Gallenkamp Hot Stage apparatus (Gallenkamp, Loughborough, UK). IR spectra were recorded using a Bruker Tensor 27 FT-IR spectrometer (Bruker, Banner Lane, UK)as thin films. Selected characteristic peaks are reported in cm−1.
4.2. Experimental Details for Synthesis
General Procedure A: Synthesis of N-Alkyladenosine
DBU (1.5 mmol) was added dropwise to a stirred solution of inosine (1 mmol) and BOP (1.2 mmol) in DMF; the mixture was then heated at 40 °C. After the consumption of starting material (approximately 40 min, as assessed by TLC), the reaction was cooled to room temperature and the appropriate amine (5 mmol) was added dropwise and the reaction was stirred overnight. The crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under vacuum. The resulted solid was recrystallised twice from iso-propanol.
N6-Methyladenosine (1). The desired product was prepared according to General Procedure A; white solid (250 mg, 90%). m.p 214.5 °C; 1H-NMR (500 MHz, D2O) δ 2.93 (s, 3H), 3.74 (dd, J = 13.0, 3.5 Hz, 1H), 3.83 (dd, J = 13.0, 2.5 Hz, 1H), 4.20 (q, J = 3.0 Hz, 1H), 4.32 (dd, J = 5.0, 3.0 Hz, 1H), 4.66 (t, J = 5.5 Hz, 1H), 5.90 (d, J = 6.0 Hz, 1H), 8.02 (s, 1H), 8.11 (s, 1H); 13C-NMR (126 MHz, D2O) δ 27.5, 62.1, 71.1, 74.0, 88.36, 88.39, 120.0, 140.1, 152.9, 155.6. HRMS (ESI) m/z: calculated for C11H16O4N5 [M + H]+ 282.1197, observed: 282.1196.
3′,5′-O-(Di-tert-butyl)silyl-2′-O-dimethyl(tert-butyl)silylinosine (2). The desired compound was prepared according to a modified version of the reported procedure [22]. To a stirred suspension of inosine (2.12 g, 8 mmol) in 40 mL anhydrous DMF at 0 °C, di-t-butylsilyl ditrifluoromethanesulfonate (3.0 mL, 8.8 mmol) was added dropwise under an N2 atmosphere. After consumption of starting material (30 min, as assessed by TLC), the reaction was quenched immediately with imidazole (2.7 g, 40 mmol) at 0 °C. After 5 min, the reaction was warmed to room temperature. t-Butyldimethylsilyl chloride (1.5 g, 9.6 mmol) was then added portionwise and the reaction was refluxed at 60 °C for 12 h. The suspension was then cooled to room temperature, water was added, and the precipitate was collected by suction filtration. The filtrate was discarded, and the white precipitate was washed with cold methanol. The methanol layer was evaporated under reduced pressure and the product was crystallised from CH2Cl2 to give a white solid (4.0 g, 98%). m.p 191–193.4 °C. TLC Rf 0.45 (3:2 cyclohexane/ethyl acetate); 1H NMR (600 MHz, CDCl3) δ 0.17 (s, 3H), 0.18 (s, 3H), 0.96 (s, 9H), 1.07 (s, 9H), 1.10 (s, 9H), 4.02–4.09 (m, 1H), 4.25 (td, J = 10.0, 5.0 Hz, 1H), 4.38 (dd, J = 9.5, 4.5 Hz, 1H), 4.45–4.58 (m, 2H), 5.96 (s, 1H), 7.87 (s, 1H), 8.11 (s, 1H), 12.56 (s, 1H); 13C NMR (151 MHz, CDCl3) δ −5.0, −4.3, 18.3, 20.4, 22.8, 25.9, 27.0, 27.5, 67.8, 74.8, 75.89, 75.94, 92.3, 125.5, 138.3, 144.7, 148.1, 158.9; HRMS (ESI) m/z: calculated for C24H43O4N528Si2 [M + H]+ 523.2767, observed: 523.2756.
3′,5′-O-Bis(tert-butyl)silyl-2′-O-(tert-butyldimethyl)silyl-N6-methyladenosine (3). The desired compound was prepared according to a modified version of the reported procedure [10]. To a stirred solution of 3′,5′-O-Bis(tert-butylsilyl)-2′-O-(tert-butyldimethylsilyl)inosine (2; 663 mg, 1.2 mmol) and BOP (0.64 g, 1.44 mmol) in 20 mL of THF, DBU (0.3 mL, 1.8 mmol) was added dropwise and the mixture was heated at 40 °C. After the consumption of the starting material (40 min, as assessed by TLC), the reaction was cooled to room temperature and methylamine (0.3 mL, 6.0 mmol) was added dropwise and the reaction was stirred overnight. The crude product mixture was concentrated under reduced pressure and diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under vacuum. The residue was purified by column chromatography (9:1 to 3:2 cyclohexane/ethyl acetate) which resulted in an oil (665 mg, 98%). TLC Rf 0.20 (7:3 cyclohexane/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 0.00 (s, 3H) 0.02 (s, 3H) 0.78 (s, 9H) 0.90 (s, 9H) 0.94 (s, 9H) 3.05 (d, J = 1.0 Hz, 3H) 3.86–3.90 (m, 1H) 4.02–4.10 (m, 1H) 4.34 (dd, J = 9.0, 5.0 Hz, 1 H) 4.38–4.44 (m, 1 H) 4.47 (d, J = 4.5 Hz, 1 H) 5.76 (b.s, 2 H) 7.62 (s, 1 H) 8.22 (s, 1 H); 13C NMR (101 MHz, CDCl3) δ −5.0, −4.3, 18.3, 20.4, 22.8, 25.9, 27.1, 27.5, 27.6, 67.9, 74.6, 75.5, 75.8, 92.4, 120.5, 125.0, 138.0, 153.4, 155.5; HRMS (ESI) m/z: calculated for C25H46O4N528Si2 [M + H]+ 536.3082, observed: 536.3078. Analytical data are consistent with those reported [4].
2′-O-(tert-Butyldimethyl)silyl-N6-methyladenosine (4). The desired compound was prepared according to the reported procedure [4]. To a stirred solution of 3′,5′-O-Bis(tert-butylsilyl)-2′-O-(tert-butyldimethylsilyl)-N6-methyladenosine (3; 240 mg, 0.45 mmol) in 4 mL of CH2Cl2 at −15 °C, a cooled solution of (HF)x·pyridine (0.06 mL, 2.3 mmol) in 365 μL pyridine was added. The reaction temperature was maintained at -15 °C and stirred for 12 h. The reaction was diluted with CH2Cl2, then washed first with sat. aq. NaHCO3 solution, then with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (9:1 to 3:2 cyclohexane/ethyl acetate) which resulted in oil (160 mg, 90%). TLC Rf 0.15 (2:3 hexane/ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ 0.00 (s, 3H), 0.02 (s, 3H), 0.94 (s, 9H), 3.42 (d, J = 1.0 Hz, 3H), 3.89 (dd, J = 10.5, 9.0 Hz, 1H), 4.01–4.11 (m, 1H), 4.34 (dd, J = 9.0, 5.0 Hz, 1H), 4.41 (dd, J = 9.0, 5.0 Hz, 1H), 4.47 (d, J = 5.0 Hz, 1H), 5.76 (s, 2H), 7.62 (s, 1H), 8.22 (s, 1H); 13C NMR (101 MHz, CDCl3) δ −5.4, −5.3, 17.9, 25.6, 25.8, 27.5, 63.5, 73.1, 74.4, 87.8, 91.3, 119.7, 140.0, 140.1, 152.9, 155.8; HRMS (ESI) m/z: calculated for C17H30O4N528Si [M + H]+ 396.2062, observed: 396.2068. Analytical data are consistent with those reported [4].
5′-O-(4,4′-Dimethoxytrityl)−2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine (5). The desired compound was prepared according to the reported procedure [4]. To a stirred solution of 2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine (4) (2.6 g, 6.6 mmol) in 4 mL anhydrous pyridine at 0 °C, DMTrCl (2.7 g, 8.0 mmol) was added portionwise at regular intervals for 12 h. The reaction was quenched by addition of an excess of anhydrous methanol (0.5 mL) at room temperature. After 1 h, the solution was concentrated under vacuum. The crude solid was first dissolved and fractioned between aqueous NaHCO3 and ethyl acetate; the organic layer was then washed with water (3 × 10 mL). The organic layer was dried (MgSO4) and concentrated under vacuum. The residue was purified by column chromatography (9:1 to 3:2 cyclohexane/ethyl acetate) resulted in a green oil (3.9 g, 85%). TLC Rf 0.45 (2:3 cyclohexane/ethyl acetate); 1H-NMR (400 MHz, CDCl3) δ −0.13 (s, 3H) 0.00 (s, 3H) 0.86 (s, 9H) 2.77 (d, J = 4.0 Hz, 1H) 3.17 (s, 3H) 3.36–3.43 (m, 1H) 3.54 (dd, J = 10.5, 3.5 Hz, 1H) 3.80 (s, 6 H) 4.27 (d, J = 3.5 Hz, 1H) 4.33–4.37 (m, 1H) 5.02 (t, J = 5.5 Hz, 1H) 5.85 (d, J = 4.5 Hz, 1H) 6.04 (b.s, 2H) 6.83 (d, J = 9.0 Hz, 4H) 7.18–7.28 (m, 3H) 7.36 (d, J = 8.0 Hz, 4H) 7.47 (dd, J = 8.5 Hz, 1.5, 2H) 7.98 (s, 1H) 8.35 (s, 1H); 13C NMR (101 MHz, CDCl3) δ −5.6, −5.5, 18.3, 25.8, 25.9, 55.2, 60.4, 63.0, 73.6, 75.5, 85.0, 87.5, 89.2, 113.4, 120.0, 127.3, 128.1, 128.3, 130.39, 130.45, 135.9, 138.0, 145.0, 153.0, 155.4, 158.89, 158.91; HRMS (ESI) m/z: calculated for C38H48O6N528Si [M + H]+ 698.3368, observed: 698.3359. Analytical data are consistent with those reported [4].
5′-O-(4,4′-Dimethoxytrityl)-(3′-O-[(2cyanoethyl)(N,N-diisopropylamino)phosphino]−2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine (6). The desired compound was prepared according to the reported procedure [4]. To a stirred solution of 5′-O-(4,4′-dimethoxytrityl)−2′-O-dimethyl(tert-butyl)silyl-N6-methyladenosine (5, 500 mg, 0.7 mmol) in anhydrous CH2Cl2 in an over-dried flask under argon, DIPEA (1.3 mL, 7.2 mmol) was added dropwise and the reaction mixture was allowed to stir at 0 °C for 10 min. (2-Cyanoethyl)-N,N-diisopropylchlorophosphoramidite (0.40 mL, 1.8 mmol) was added to the reaction mixture dropwise at 0 °C under an argon atmosphere. The reaction was stirred at 0 °C for 30 min, then gradually (about 30 min) warmed to room temperature. After another five hours under an inert atmosphere, the reaction mixture was treated with a saturated aq. KCl solution, then evaporated by rotary evaporation. The desired product was separated by silica gel column chromatography (1:1:0.01 hexane/ethyl acetate/pyridine) resulting in a colourless oil (520 mg, 80%) yield. TLC Rf 0.40 (1:1:0.01 hexane/ethyl acetate/pyridine); 1H NMR (700 MHz, CD2Cl2-d2) Major peaks are listed. δ −0.15 (s, 3H), −0.01 (s, 3H), 0.82 (s, 9H), 1.10 (s,3H), 1.11 (s, 3H), 1.22 (s, 3H), 1.22 (s, 3H), 1.65 (s, 2H), 2.62–2.74 (m, 2H), 3.19 (s, 3H), 3.36 (dd, J = 10.5, 4.5 Hz, 1H), 3.54 (dd, J = 10.5, 4.0 Hz, 1H), 3.82 (s, 6H), 3.85–3.93 (m, 1H), 3.95–4.10 (m, 1H), 4.41–4.49 (m, 1H), 5.12 (dd, J = 6.1, 4.4 Hz, 1H), 5.33–5.40 (m, 2H), 5.79 (s, 1H), 5.99 (d, J = 6.0 Hz, 1H), 6.78–6.90 (m, 4H), 7.23–7.29 (m, 1H), 7.28–7.34 (m, 2H), 7.34–7.40 (m, 4H), 7.47–7.52 (m, 2H), 7.94 (s, 1H), 8.25 (s, 1H); 13C NMR (176 MHz, CD2Cl2) Major peaks are listed. δ −5.4, −5.0, 0.8, 17.8, 20.4, 20.44, 21.1, 24.37, 24.4, 25.4, 25.44, 42.9, 43.0, 55.2, 58.8, 58.9, 63.5, 72.8, 72.9, 74.7, 74.7, 83.46, 83.48, 86.5, 88.4, 113.1, 117.8, 125.2, 126.8, 127.8, 128.1, 128.2, 129.0, 130.10, 130.14, 135.7, 139.0, 144.9, 153.0, 155.5, 158.6, 158.7; 31P-NMR (202 MHz, CD2Cl2) δ 148.0, 150.8.
N6,N6-Dimethyladenosine (11a). The desired product was prepared according to General Procedure A; white solid (250 mg, 85%). 1H NMR (600 MHz, D2O) δ 3.06 (s, 6H), 3.75 (dd, J = 13.0, 3.5 Hz, 1H), 3.85 (dd, J = 13.0, 2.5 Hz, 1H), 4.19 (q, J = 3.0 Hz, 1H), 4.29–4.33 (t, J = 4.5 Hz, 1H), 4.59 (t, J = 5.5 Hz, 1H), 5.82 (d, J = 6.0 Hz, 1H), 7.78 (b.s, 1H), 8.00 (s, 1H); 13C-NMR (151 MHz, D2O) δ 38.7, 61.4, 70.5, 73.7, 85.5, 88.2, 118.9, 138.2, 148.2, 151.3, 153.6; HRMS (ESI) m/z: calculated for C12H18O4N5 [M + H]+ 296.1353, observed: 296.1352.
N6-Ethyladenosine (11b). The desired product was prepared according to General Procedure A; Ethylamine was prepared in-situ. To a stirred solution of ethylamine hydrochloride (1.1 g, 13.7 mmol) in 10 mL of ethanol in 50 mL round bottom flask, Ag2O (3.8 g, 16.4 mmol) was added and the mixture was stirred at room temperature under N2 for 1 h. The precipitate was collected by suction filtration; the resultant solution was then added to the stirred mixture of inosine, BOP and DBU. Yellowish solid (90 mg, 30%). 1H NMR (600 MHz, D2O + DMSO-d6) δ 2.63 (t, J = 2.0 Hz, 3H), 3.53 (b.s, 2H), 3.77 (dd, J = 13.0, 3.5 Hz, 1H), 3.85 (dd, J = 13.0, 3.0 Hz, 1H), 4.23 (q, J = 3.0 Hz, 1H), 4.35 (dd, J = 5.0, 3.0 Hz, 1H), 5.98 (d, J = 6.5 Hz, 1H), 8.18 (s, 1H), 8.24 (s, 1H), (1H under solvent peak); 13C-NMR (151 MHz, D2O + DMSO-d6) δ 13.9, 61.6, 70.8, 73.7, 86.0, 88.3, 117.6, 130.0, 140.1, 152.7, 154.6, (1C under solvent peak); HRMS (ESI) m/z: calculated for C12H18O4N5 [M + H]+ 296.1308, observed: 296.1353.
N6-Cyclopropyladenosine (11c). The desired product was prepared according to General Procedure A; white solid (222 mg, 72%). 1H-NMR (700 MHz, DMSO-d6 + D2O) δ 0.62–0.71 (m, 2H), 0.88 (dd, J = 7.0, 2.0 Hz, 2H), 3.00 (bs, 1H), 3.67 (dd, J = 12.0, 3.5 Hz, 1H), 3.77 (dd, J = 12.0, 3.5 Hz, 1H), 4.21–4.28 (m, 1H), 4.67 (t, J = 5.5 Hz, 1H), 5.97 (d, J = 6.0 Hz, 1H), 8.32 (s, 1H), 8.41 (s, 1H), (1 proton under solvent); 13C NMR (176 MHz, DMSO-d6 + D2O) δ 7.0, 61.9, 70.8, 70.9, 73.9, 86.2, 86.3, 88.4, 119.8, 140.3, 140.4, 152.7, 155.9; HRMS (ESI) m/z: calculated for C13H18O4N5 [M + H]+ 308.1353, observed: 308.1351.
O6-Methylinosine (11d). DBU (1.5 mmol) was added dropwise to a stirred solution of inosine (1 mmol), BOP (1.2 mmol) in THF; the mixture was heated at 40 °C. After the consumption of starting material (40 min, as assessed by TLC), the reaction mixture was concentrated under reduced pressure and an excess of MeOH was added to the flask and the reaction was stirred at 40 °C overnight. The crude product mixture was concentrated under reduced pressure and diluted with ethyl acetate, then washed with water (3 × 10 mL). The organic layer was dried (MgSO4) and concentrated under vacuum. The crude mixture was purified (99:1 to 9:1 ethyl acetate/methanol) by column chromatography which resulted in a white solid (0.2 g, 72%). TLC Rf 0.3 (9:1 CH2Cl2/MeOH); 1H-NMR (600 MHz, DMSO-d6) δ 3.58 (ddd, J = 12.0, 6.0, 4.0 Hz, 1H), 3.69 (dt, J = 12.0, 4.5 Hz, 1H), 3.98 (q, J = 4.0 Hz, 1H), 4.11 (s, 3H), 4.17 (q, J = 4.5 Hz, 1H), 4.60 (q, J = 5.5 Hz, 1H), 5.13 (t, J = 5.5 Hz, 1H), 5.22 (d, J = 5.0 Hz, 1H), 5.50 (d, J = 6.0 Hz, 1H), 6.00 (d, J = 5.5 Hz, 1H), 8.57 (s, 1H), 8.63 (s, 1H); 13C-NMR (151 MHz, DMSO-d6) δ 54.5, 61.8, 70.8, 74.2, 86.2, 88.2, 121.6, 142.9, 152.2, 152.24, 160.9; calculated for C11H13O5N4 [M + H]+ 283.0934, observed: 283.0932.
N6-(2-Hydroxyethyl)adenosine (11e). The desired product was prepared according to General Procedure A; white solid (190 mg, 61%). 1H-NMR (600 MHz, DMSO-d6) δ 3.57 (dd, J = 12.0, 4.0 Hz, 1H), 3.67 (dd, J = 12.0, 3.5 Hz, 1H), 4.13–4.21 (m, 1H), 4.51–4.64 (m, 3H), 5.96 (d, J = 6.0 Hz, 1H), 8.49 (s, 1H), 8.52 (s, 1H) (4 protons under the residual water peak); 13C NMR (151 MHz, DMSO-d6) δ 61.6, 63.4, 70.7, 74.0, 86.1, 88.2, 121.5, 142.7, 152.0, 152.2, 160.6; HRMS (ESI) m/z: calculated for C12H18O5N5 [M + H]+312.1302, observed: 312.1297.
N6,N6-Methyl(2-hydroxyethyl)adenosine (11f). The desired product was prepared according to General Procedure A; white solid (293 mg, 90%). 1H-NMR (700 MHz, DMSO-d6) δ 3.56 (ddd, J = 12.0, 7.0, 3.5 Hz, 1H), 3.61–4.43 (m, 6H), 4.59 (q, J = 6.0 Hz, 1H), 4.75 (t, J = 5.5 Hz, 1H), 5.18 (d, J = 5.0 Hz, 1H), 5.37 (dd, J = 7.0, 4.5 Hz, 1H), 5.45 (d, J = 6.0 Hz, 1H), 5.91 (d, J = 6.0 Hz, 1H), 8.22 (s, 1H), 8.37 (s, 1H) (3 methyl protons and one hydroxyl group under the residual water peak in DMSO); 13C NMR (176 MHz, DMSO-d6) δ 37.3, 52.7, 60.0, 62.0, 71.0, 73.9, 86.2, 88.3, 120.1, 139.2, 150.4, 152.2, 154.6; HRMS (ESI) m/z: calculated for C13H20O5N5 [M + H]+ 326.1459, observed: 326.1459.
N6,N6-Ethyl(2-hydroxyethyl)adenosine (11g). The desired product was prepared according to General Procedure A; white solid (275 mg, 85%). 1H-NMR (700 MHz, D2O) δ 1.18 (t, J = 7.0 Hz, 3H), 2.53 (d, J = 9 Hz, 1H)3.75 (dd, J = 13.0, 3.5 Hz, 1H), 3.78–4.09 (m, 7H), 4.21 (q, J = 3.5 Hz, 1H), 4.34 (dd, J = 5.0, 3.5 Hz, 1H), 5.98 (d, J = 6.0 Hz, 1H), 8.13 (s, 1H), 8.17 (s, 1H) 13C-NMR (176 MHz, D2O) δ 15.4, 44.6, 59.6, 61.1, 61.5, 70.6, 73.5, 85.8, 88.1, 119.3, 138.7, 149.2, 152.0, 154.1; HRMS (ESI) m/z: calculated for C14H22O5N5 [M + H]+ 340.1617, observed: 340.1615.
2′,3′,5′-O-Tris(tert-butyldimethyl)silylinosine (12). To a stirred solution of inosine (3.75 g, 13.24 mmol) and imidazole (3.6 g, 53.0 mmol) in anhydrous DMF in a 50 mL round bottom flask, TBDMSCl (6.6 g, 43.7 mmol) was added portionwise. The reaction was heated at 60 °C for 12 h. The suspension was cooled to room temperature, water was added and the precipitate was collected by suction filtration. The filtrate was discarded, and the white precipitate was washed with cold methanol. The methanol layer was evaporated under vacuum; the product was crystallised as a white solid from CH2C12 (7.8 g, 94%). TLC Rf 0.6 (1:9 MeOH/CH2Cl2); 1H-NMR (400 MHz, CDCl3) δ −0.31 (s, 3H), −0.16 (s, 3H), −0.04 (s, 3H), −0.03 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.68 (s, 9H), 0.79 (s, 9H), 0.82 (s, 9H), 3.66 (dd, J = 11.5, 2.5 Hz, 1H), 3.86 (dd, J = 11.5, 4.0 Hz, 1H), 4.00 (q, J = 3.5 Hz, 1H), 4.17 (t, J = 4.0 Hz, 1H), 4.38 (t, J = 4.5 Hz, 1H), 5.88 (d, J = 5.0 Hz, 1H), 7.97 (s, 1H), 8.09 (s, 1H), 12.83 (s, 1H); 13C NMR (101 MHz, CDCl3) δ −5.4, −5.0, −4.70, −4.66, −4.4, 17.9, 18.1, 18.6, 25.7, 25.8, 26.1, 62.4, 71.8, 85.5, 88.3, 125.0, 139.1, 144.5, 148.9. 159.2; HRMS (ESI) m/z: calculated for C28H55O5N428Si3 [M + H]+ 611.3474, observed: 611.3468.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6-(2-hydroxyethyl)adenosine (13a). To a stirred solution of 2′,3′,5′-O-tris(tert-butyldimethyl)silylinosine (12; 0.1 g, 0.16 mmol) and PyBOP (0.1 g, 0.2 mmol) in 10 mL of THF in a 50 mL round bottom flask, DIPEA (42 μL, 0.24 mmol) was added dropwise and the mixture was heated at 40 °C. After the consumption of the starting material (40 min, as assessed by TLC), the reaction was cooled to room temperature and ethanolamine (0.2 mL, 0.35 mmol) was added dropwise; the reaction was then stirred overnight. The crude product mixture was concentrated under reduced pressure and then diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 94:6 CH2Cl2/MeOH) which resulted in oil (95 mg, 90%). 1H NMR (500 MHz, CD3OD) δ −0.28 (s, 3H), −0.02 (s, 3H), 0.17 (s, 6H), 0.18 (s, 6H), 0.79 (s, 9H), 0.98 (s, 9H), 0.99 (s, 9H), 3.71–3.84 (m, 4H), 3.85 (dd, J = 11.5, 3.0 Hz, 1H), 4.06 (dd, J = 11.0, 4.5 Hz, 1H), 4.15 (dt, J = 5.0, 3.0 Hz, 1H), 4.40 (dd, J = 4.5, 2.5 Hz, 1H), 4.83 (dd, J = 6.0, 4.5 Hz, 1H), 4.89 (s, 5H), 6.07 (d, J = 6.0 Hz, 1H), 8.26 (s, 1H), 8.31 (s, 1H), (-OH peak under solvent peak); 13C-NMR (126 MHz, CD3OD) δ −6.6, −6.5, −6.3, −5.6, −5.6, −5.5, 17.4, 17.6, 18.0, 24.9, 25.1, 25.2, 45.97, 46.0, 60.4, 62.7, 72.7, 76.0, 86.2, 87.7, 119.5, 139.3, 148.6, 152.5, 155.0; HRMS (ESI) m/z: calculated for C30H60O5N528Si3 [M + H]+ 635.3713, observed: 635.3797.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N6-methyl(2-hydroxyethyl)adenosine (13b). To a stirred solution of 2′,3′,5′-O-tris(tert-butyldimethyl)silylinosine (12; 1 g, 1.64 mmol) and BOP (0.9 g, 1.96 mmol) in 25 mL of EtOH in a 50 mL round bottom flask, DBU (0.3 mL, 1.97 mmol) was added dropwise; the mixture was heated at 40 °C. After the consumption of the starting material (40 min, TLC), the reaction was cooled to room temperature and methylethanolamine (0.65 mL, 8.2 mmol) was added dropwise; the reaction was then stirred overnight. The crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and was washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 94:6 CH2Cl2/MeOH) which resulted in oil (0.82 g, 80%). TLC Rf 0.6 (6:94 MeOH/CH2Cl2). 1H-NMR (600 MHz, CDCl3) δ −0.29 (s, 3H), −0.14 (s, 3H), −0.01 (s, 3H), 0.00 (s, 3H), 0.02 (s, 3H), 0.03 (s, 3H), 0.71 (s, 9H), 0.83 (s, 9H), 0.85 (s, 9H), 3.42 (s, 3H), 3.67 (dd, J = 11.5, 3.0 Hz, 1H), 3.87 (t, J = 5.0 Hz, 2H), 3.92 (dd, J = 11.4, 4.0 Hz, 1H), 3.94–4.09 (m, 4H), 4.21 (t, J = 4.0 Hz, 1H), 4.58 (t, J = 4.5 Hz, 1H), 5.92 (d, J = 5.0 Hz, 1H), 7.97 (s, 1H), 8.20 (s, 1H); 13C-NMR (151 MHz, CDCl3) δ −5.38, −5.36, −5.0, −4.72, −4.70, −4.4, 17.9, 18.1, 18.5, 25.7, 25.9, 26.1, 53.8, 61.6, 62.5, 71.9, 75.6, 85.3, 88.3, 120.4, 137.7, 150.5, 152.2, 155.6; HRMS (ESI) m/z: calculated for C31H62O5N528Si3 [M + H]+ 668.4053, observed: 668.4042.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N6-ethyl(2-hydroxyethyl)adenosine (13c). To a stirred solution of 2′,3′,5′-O-tris(tert-butyldimethyl)silylinosine (12; 1 g, 1.64 mmol) and BOP (0.87 g, 1.96 mmol) in 25 mL of EtOH in a 50 mL round bottom flask, DBU (0.3 mL, 2.0 mmol) was added dropwise and the mixture was heated at 40 °C. After the consumption of the starting material (40 min, as assessed by TLC), the reaction was cooled to room temperature and ethylethanolamine (0.65 mL, 8.2 mmol) was added dropwise; the reaction was then stirred overnight. The crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and washed with water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 94:6 CH2Cl2/MeOH) which resulted in an oil (960 mg, 86%). TLC Rf 0.5 (6:94 MeOH/CH2Cl2); 1H NMR (600 MHz, CDCl3) δ −0.30 (s, 3H), −0.15 (s, 3H), −0.02 (s, 3H), −0.01 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.69 (s, 9H), 0.82 (s, 9H), 0.83 (s, 9H), 1.18 (t, J = 7.0 Hz, 3H), 3.66 (dd, J = 11.0, 3.0 Hz, 1H), 3.72–4.15 (m, 8H), 4.21 (t, J = 4.0 Hz, 1H), 4.59 (t, J = 5.0 Hz, 1H), 4.67–5.17 (m, 1H), 5.89 (d, J = 5.0 Hz, 1H), 8.05 (s, 1H), 8.16 (s, 1H); 13C NMR (151 MHz, CDCl3) δ −5.39, −5.38, −5.0, −4.72, −4.70, −4.4, 13.3, 17.9, 18.1, 18.5, 25.7, 25.8, 26.0, 44.6, 51.6, 62.5, 62.6, 71.9, 75.5, 85.2, 88.3, 120.1, 137.8, 150.5, 152.1, 155.0. HRMS (ESI) m/z: calculated for C32H64O5N528Si3 [M + H]+ 682.4209, observed: 682.4201.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N1,N6-ethanoadenosine (15a). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N1,N6-(2-hydroxyethyl)adenosine (13a; 1.3 g, 2 mmol) and Et3N (1.4 mL, 10 mmol) in 30 mL of anhydrous DMF in a 50 mL round bottom flask, methyltriphenoxyphosphonium iodide (1 g, 2.4 mmol) was added and the mixture was stirred at room temperature for 1 h. Anhydrous methanol was added and the crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and washed with NaHCO3 and water (3 × 10 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 to 85:15 CH2Cl2/MeOH) resulting in an oil (0.75 g, 60%). 1H-NMR (600 MHz, CDCl3) δ −0.18 (s, 3H), −0.07 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.05 (s, 3H), 0.06 (s, 3H), 0.75 (s, 9H), 0.83 (s, 9H), 0.87 (s, 9H), 3.71 (dd, J = 11.5, 2.5 Hz, 1H), 3.93 (dd, J = 11.5, 3.0 Hz, 1H), 4.06 (dt, J = 5.5, 3.0 Hz, 1H), 4.18 (t, J = 4.5 Hz, 1H), 4.26–4.40 (m, 3H), 4.90–4.95 (m, 2H), 5.92 (d, J = 4.0 Hz, 1H), 8.38 (s, 1H), 8.51 (s, 1H); 13C NMR (151 MHz, CDCl3) δ −5.3, −5.27, −4.8, −4.6, −4.4, −4.2, 17.9, 18.0, 18.5, 25.7, 25.8, 26.1, 26.4, 26.5, 46.28, 46.33, 46.4, 49.1, 62.0, 71.1, 76.7, 85.2, 89.0, 117.6, 142.3, 143.4, 149.0, 151.3; HRMS (ESI) m/z: calculated for C30H58O4N528Si3 [M + H]+ 636.3767, observed: 636.3791.
N1,N6-Ethanoadenosine (16a). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6-ethanoadenosine (15a, 470 mg, 0.74 mmol) in CH2Cl2, 3HF.Et3N (361 µL, 2.2 mmol) was added dropwise. The solution was left to stir for 48 h. The mixture was reduced under pressure and was purified by column chromatography (99:1 to 9:1 ethyl acetate/methanol) which resulted in a white solid (150 mg, 70%). 1H NMR (700 MHz, DMSO-d6) δ 3.49–3.57 (m, 2H), 3.60–3.68 (m, 1H), 3.86–3.97 (m, 3H), 4.08–4.16 (m, 3H), 4.44 (t, J = 5.0 Hz, 1H), 5.11–5.21 (m, 2H), 5.46 (s, 1H), 5.78 (d, J = 6.0 Hz, 1H), 8.09 (s, 1H), 8.17 (s, 1H); 13C-NMR (176 MHz, DMSO-d6) δ 46.5, 53.3, 61.9, 70.8, 74.5, 86.1, 88.0, 120.0, 138.5, 145.2, 145.4, 150.2; HRMS (ESI) m/z: calculated for C12H16O4N5 [M + H]+ 294.1197, observed: 294.1191.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N1,N6-methylethanoadenosine (15b). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-methyl(2-hydroxyethyl)adenosine (13b; 0.5 g, 0.75 mmol) and Et3N (0.54 mL, 3.75 mmol) in 30 mL of anhydrous DMF in a 50 mL round bottom flask, methyltriphenoxyphosphonium iodide (0.85 g, 1.9 mmol) was added and the mixture was stirred at room temperature for 1 h. Anhydrous methanol was added and the crude product mixture was concentrated under reduced pressure, then diluted with ethyl acetate and washed with NaHCO3 and water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4), then concentrated under reduced pressure. The residue was purified by alumina column chromatography (99:1 to 90:10 CH3Cl/MeOH) which resulted in an oil. 1H NMR (600 MHz, CDCl3) δ −0.20 (s, 3H), −0.12 (s, 3H), −0.08 (s, 3H), −0.07 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.70 (s, 9H), 0.75 (s, 9H), 0.81 (s, 9H), 3.57 (s, 3H), 3.65 (dd, J = 11.5, 2.0 Hz, 1H), 3.88 (dd, J = 11.5, 3.0 Hz, 1H), 4.00 (dt, J = 5.0, 2.5 Hz, 1H), 4.12 (dd, J = 5.5, 4.0 Hz, 1H), 4.17–4.26 (m, 2H), 4.35–4.42 (m, 1H), 5.05–5.3 (m, 2H), 5.87 (d, J = 3.5 Hz, 1H), 8.43 (s, 1H), 8.50 (s, 1H); 13C NMR (151 MHz, CDCl3) δ −5.4, −5.2, −4.8, −4.6, −4.5, −4.2, 17.9, 18.1, 18.6, 25.7, 25.8, 26.2, 34.6, 45.9, 49.1, 51.6, 61.7, 70.7, 85.0, 89.3, 115.4, 117.0, 129.5, 142.8, 143.7, 149.9, 150.1; HRMS (ESI) m/z: calculated for C31H60O4N528Si3 [M] 650.3947, observed: 650.3924.
2′,3′,5′-O-Tris[dimethyl(tert-butyl)silyl]-N6,N1,N6-ethylethanoadenosine (15c). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-ethyl(2-hydroxyethyl)adenosine (15c; 0.4 g, 0.6 mmol) and Et3N ( 0.4 mL, 3 mmol) in 20 mL of anhydrous DMF in a 50 mL round bottom flask, methyltriphenoxyphosphonium iodide (0.6 g, 1.2 mmol) was added; the mixture was stirred at room temperature for 1 h. Anhydrous methanol was added and the crude product mixture was concentrated under reduced pressure and then diluted with ethyl acetate and was washed subsequently with NaHCO3 and water (3 × 10 mL). The organic layer was dried (anhydrous MgSO4) and concentrated under reduced pressure. The residue was purified by alumina column chromatography (99:1 to 95:5 CH2Cl2/MeOH) which resulted in a solid (0.26 g, 36%). mp 195 °C; 1H-NMR (600 MHz, CDCl3) δ −0.25 (s, 3H), −0.18 (s, 3H), −0.03 (s, 3H), −0.01 (s, 3H), 0.00 (s, 3H), 0.01 (s, 3H), 0.70 (s, 9H), 0.74 (s, 9H), 0.83 (s, 9H), 1.33 (t, J = 7.0 Hz, 3H), 3.2 (q, J = 7.0 Hz, 2H), 3.65 (dd, J = 12.0, 2.0 Hz, 1H), 3.88 (dd, J = 12.0, 3.0 Hz, 1H), 4.00 (dt, J = 5.0, 2.5 Hz, 1H), 4.23–4.28 (m, 2H), 4.59 (t, J = 5.0 Hz, 1H), 4.69 (t, J = 5.0 Hz, 1H) 5.0–5.18 (m, 2H), 5.83 (d, J = 5.0 Hz, 1H), 8.34 (s, 1H), 8.46 (s, 1H); 13C-NMR (151 MHz, CDCl3) δ −5.3, −5.0, −5.0, −4.6, −4.6, −4.2, 13.0, 17.9, 18.1, 18.5, 25.7, 25.8, 26.2, 35.6, 48.4, 51.6, 61.7, 70.7, 73.4, 85.0, 89.3, 115.4, 144.8, 149.9, 150.0, 152.0; HRMS (ESI) m/z: calculated for C31H60O4N528Si3 [M] 664.3886, observed: 664.3896.
N6,N1,N6-Methylethanoadenosine (16b). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-methylethanoadenosine (15b; 500 mg, 0.8 mmol) in CH2Cl2, 3HF.Et3N (0.4 mL, 2.3 mmol) was added dropwise. The solution was stirred for 48 h. The mixture was reduced under pressure, then purified by alumina column chromatography (99:1 to 9:1 ethyl acetate/methanol) which resulted in a white solid (54 mg, 22%). mp 202.5 °C; 1H-NMR (600 MHz, DMSO-d6) δ 3.01 (s, 3H), 3.29 (m, 1H), 3.38 (dt, J = 12.5, 4.5 Hz, 1H), 3.68 (q, J = 4.0 Hz, 1H), 3.76–3.84 (m, 2H), 3.87 (b.s, 1H), 4.18 (q, J = 5.0 Hz, 1H), 4.43 (t, J = 9.5 Hz, 1H), 4.79 (t, J = 5.5 Hz, 1H), 5.01 (s, 1H), 5.28–5.37 (m, 2H), 5.68 (d, J = 5.0 Hz, 1H), 8.50 (s, 1H), 8.52 (s, 1H); 13C-NMR (151 MHz, D2O) δ 36.4, 50.5, 53.4, 63.6, 72.6, 77.1, 88.6, 90.8, 119.0, 145.7, 148.0, 152.2, 152.3; HRMS (ESI) m/z: calculated for C13H18O4N5 [M]+ 308.1353, observed: 308.1353.
N6,N1,N6-Ethylethanoadenosine (16c). To a stirred solution of 2′,3′,5′-O-tris[dimethyl(tert-butyl)silyl]-N6,N6-methylethanoadenosine (15c; 470 mg, 0.74 mmol) in CH2Cl2, 3HF.Et3N (360 µL, 2.2 mmol)was added dropwise. The solution was left to stir for 48 h. The mixture was reduced under pressure and was purified by alumina column chromatography (99:1 to 9:1 ethyl acetate/methanol) which resulted in a white solid (65 mg, 30%). mp 210 °C; 1H-NMR (600 MHz, DMSO-d6) δ 1.15 (t, J = 7.0 Hz, 3H), 2.88 (q, J = 7.0 Hz, 2H), δ 3.49–3.57 (m, 2H), 3.36 (dt, J = 12.5 Hz, 4.0 Hz, 1H), 3.54 (q, J = 4.0 Hz, 1H), 3.76–3.84 (m, 2H), 3.95 (b.s, 1H), 4.15–4.27 (m, 1H), 4.34–4.43 (m, 1H), 5.1 (b.s, 1H), 5.43–5.57 (m, 2H), 6.0 (d, J = 5.0 Hz, 1H), 8.48 (s, 1H), 8.63 (s, 1H); 13C-NMR (151 MHz, DMSO-d6) δ 13.0, 32.4, 48.3, 52.3, 62.4, 70.4, 75.3, 85.4, 90.8, 119.0, 146.4, 148.2, 151.9, 152.0; HRMS (ESI) m/z: calculated for C13H18O4N5 [M]+ 322.1518, observed: 322.1515.
5. Conclusions
Overall, we have developed an improved the synthesis of the m6A phosphoramidite starting from inosine. Following alcohol group protection, a BOP-mediated SNAr reaction was employed to introduce the desired N6-methylamino group in a suitably protected form to be efficiently converted to the phosphoramidite for incorporation into oligonucleotides. The BOP-mediated SNAr reaction can be employed to prepare other N6-alylamino substituted adenosine derivatives, including ethanoadenosine, an RNA analogue of the DNA adduct formed by the important anticancer drug Carmustine.
Supplementary Materials
The following are available online. 1H and 13C-NMR spectra of compounds are available.
Author Contributions
Conceptualization, S.S. and C.J.S.; writing—original draft preparation, S.S.; writing—review and editing, S.S and C.J.S.; supervision, C.J.S.; funding acquisition, C.J.S. Both authors have read and agreed to the published version of the manuscript.
Funding
C.J.S. thanks the Biotechnology and Biological Research Council, Cancer Research UK and the Wellcome Trust for funding. S.S. thanks Felix Scholarship Foundation and Vice-Chancellors’ Fund for support.
Acknowledgments
S.S. thanks Anna Dysko, currently working at Abcam, for her help in the phosphoramidite reaction procedure.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors, if available.
References
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, S.; Liu, J.; Huang, Y.; Gong, C.; Liu, J.; Xiao, Y.; Yang, S. New sights in cancer: Component and function of N6-methyladenosine modification. Biomed. Pharm. 2020, 122, 109694. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Zhang, D.; El-Sagheer, A.H.; Brown, T.; Claridge, D.W.T.; Schofield, J.C.; Hopkinson, J.R. NMR analyses on N-hydroxymethylated nucleobases—Implications for formaldehyde toxicity and nucleic acid demethylases. Org. Biomol. Chem. 2018, 16, 4021–4032. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.H.; Hayashi, G.; Okamoto, A. Diazirine photocrosslinking recruits activated FTO demethylase complexes for specific N6-methyladenosine recognition. ACS Chem. Biol. 2015, 10, 1450–1455. [Google Scholar] [CrossRef]
- Dai, Q.; Fong, R.; Saikia, M.; Stephenson, D.; Yu, Y.T.; Pan, T.; Piccirilli, J.A. Identification of recognition residues for ligation-based detection and quantitation of pseudouridine and N6-methyladenosine. Nucleic Acids Res. 2007, 35, 6322–6329. [Google Scholar] [CrossRef]
- Véliz, E.A.; Easterwood, L.M.; Beal, P.A. Substrate analogues for an RNA-editing adenosine deaminase: Mechanistic investigation and inhibitor design. J. Am. Chem. Soc. 2003, 125, 10867–10876. [Google Scholar] [CrossRef] [PubMed]
- Höbartner, C.; Kreutz, C.; Flecker, E.; Ottenschläger, E.; Pils, W.; Grubmayr, K.; Micura, R. The synthesis of 2′-O-[(Triisopropylsilyl)oxy] methyl (TOM) phosphoramidites of methylated ribonucleosides (m 1 G, m 2 G, m 2 2 G, m 1 I, m 3 U, m 4 C, m 6 A, m 6 2 A) for use in automated RNA solid-phase synthesis. Mon. Chem. 2003, 134, 851–873. [Google Scholar] [CrossRef]
- Serebryany, V.; Beigelman, L. An efficient preparation of protected ribonucleosides for phosphoramidite RNA synthesis. Tetrahedron Lett. 2002, 43, 1983–1985. [Google Scholar] [CrossRef]
- Wan, Z.K.; Binnun, E.; Wilson, D.P.; Lee, J. A highly facile and efficient one-step synthesis of N6-adenosine and N6-2’-deoxyadenosine derivatives. Org. Lett. 2005, 7, 5877–5880. [Google Scholar] [CrossRef]
- Wan, Z.K.; Wacharasindhu, S.; Levins, C.G.; Lin, M.; Tabei, K.; Mansour, T.S. The scope and mechanism of phosphonium-mediated SNAr reactions in heterocyclic amides and ureas. J. Org. Chem. 2007, 72, 10194–10210. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.S.; Reese, C.B. Migration of t-butyldimethylsilyl protecting groups. J. Chem. Soc. Perkin Trans. 1979, 1, 2762–2764. [Google Scholar] [CrossRef]
- Pirrung, C.M.; Shuey, W.S.; Lever, C.D.; Fallon, A.L. Convenient procedure for the deprotection of silylated nucleosides and nucleotides using triethylamine trihydrofluoride. Bioorg. Med. Chem. Lett. 1994, 4, 1345–1346. [Google Scholar] [CrossRef]
- Lakshman, M.K.; Frank, J. A simple method for C-6 modification of guanine nucleosides. Org. Biomol. Chem. 2009, 7, 2933–2940. [Google Scholar] [CrossRef] [PubMed]
- Satish, K.S.; Vuram, P.; Relangi, S.; Gurram, V.; Zhou, H.; Kreitman, R.; Montemayor, M.; Yang, L.; Kaliyaperumal, M.; Sharma, S.; et al. Cladribine analogues via O6-(benzotriazolyl) derivatives of guanine nucleosides. Molecules 2015, 20, 18437–18463. [Google Scholar] [CrossRef] [Green Version]
- Kokatla, H.P.; Lakshman, M.K. One-pot etherification of purine nucleosides and pyrimidines. Org. Lett. 2010, 12, 4478–4481. [Google Scholar] [CrossRef] [Green Version]
- Devine, S.M.; Scammells, P.J. Synthesis and utility of 2-halo-O6-(benzotriazol-1-yl)-functionalized purine nucleosides. Eur. J. Org. Chem. 2011, 6, 1092–1098. [Google Scholar] [CrossRef]
- Bae, S.; Lakshman, M.K. O6-(benzotriazol-1-yl)inosine derivatives: Easily synthesized, reactive nucleosides. J. Am. Chem. Soc. 2007, 129, 782–789. [Google Scholar] [CrossRef]
- Neuner, S.; Kreutz, C.; Micura, R. The synthesis of 15N(7)-hoogsteen face-labeled adenosine phosphoramidite for solid-phase RNA synthesis. Mon. Chem. 2017, 148, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Maruenda, H.; Chenna, A.; Liem, L.K.; Singer, B. Synthesis of 1,N6-Ethano-2′-deoxyadenosine, a metabolic product of 1,3-Bis(2-chloroethyl)nitrosourea, and its incorporation into oligomeric DNA. J. Org. Chem. 1998, 63, 4385–4389. [Google Scholar] [CrossRef]
- Saito, T.; Murakami, M.; Inada, T.; Hayashibara, H.; Fujii, T. Purines. LIV. Intramolecular cyclization of 9-ethyl-1-(2-hydroxyethyl)adenine caused by nucleophiles: Formation of N6, 1-ethanoadenine derivatives. Chem. Pharm. Bull. 1993, 41, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Ghanty, U.; Fostvedt, E.; Valenzuela, R.; Beal, P.A.; Burrows, C.J. Promiscuous 8 alkoxyadenosines in the guide strand of an siRNA: Modulation of silencing efficacy and off-pathway protein binding. J. Am. Chem. Soc. 2012, 134, 17643–17652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Scheme 1.
Previous strategies towards synthesis of (protected) m6A. (A) Route involving the Vilsmeier reagent [8]. Reaction conditions: (i) acetic anhydride (5.7 eq), pyridine, dimethylformamide (DMF), 75 °C, 0.5 h; (ii) Vilsmeier reagent (2 eq), CH2Cl2, 40 °C, 24 h; (iii) MeNH2 (33% in EtOH, 7 eq), EtOH, rt, 18 h. (B) Route involving methyl iodide [4,5]. Reaction conditions: (i) (t-Bu)2Si(OTf)2, imidazole, DMF, 0 °C, 1 h; (ii) TBDMSCl, imidazole, DMF, 60 °C, 12 h; (iii) RCl, pyridine, 0–20 °C, 3 days; (iv) NH3/MeOH, 23 °C; (v) CH3I, NaOH, CH2Cl2, tetrabutylammonium iodide, 24 h.
Scheme 1.
Previous strategies towards synthesis of (protected) m6A. (A) Route involving the Vilsmeier reagent [8]. Reaction conditions: (i) acetic anhydride (5.7 eq), pyridine, dimethylformamide (DMF), 75 °C, 0.5 h; (ii) Vilsmeier reagent (2 eq), CH2Cl2, 40 °C, 24 h; (iii) MeNH2 (33% in EtOH, 7 eq), EtOH, rt, 18 h. (B) Route involving methyl iodide [4,5]. Reaction conditions: (i) (t-Bu)2Si(OTf)2, imidazole, DMF, 0 °C, 1 h; (ii) TBDMSCl, imidazole, DMF, 60 °C, 12 h; (iii) RCl, pyridine, 0–20 °C, 3 days; (iv) NH3/MeOH, 23 °C; (v) CH3I, NaOH, CH2Cl2, tetrabutylammonium iodide, 24 h.

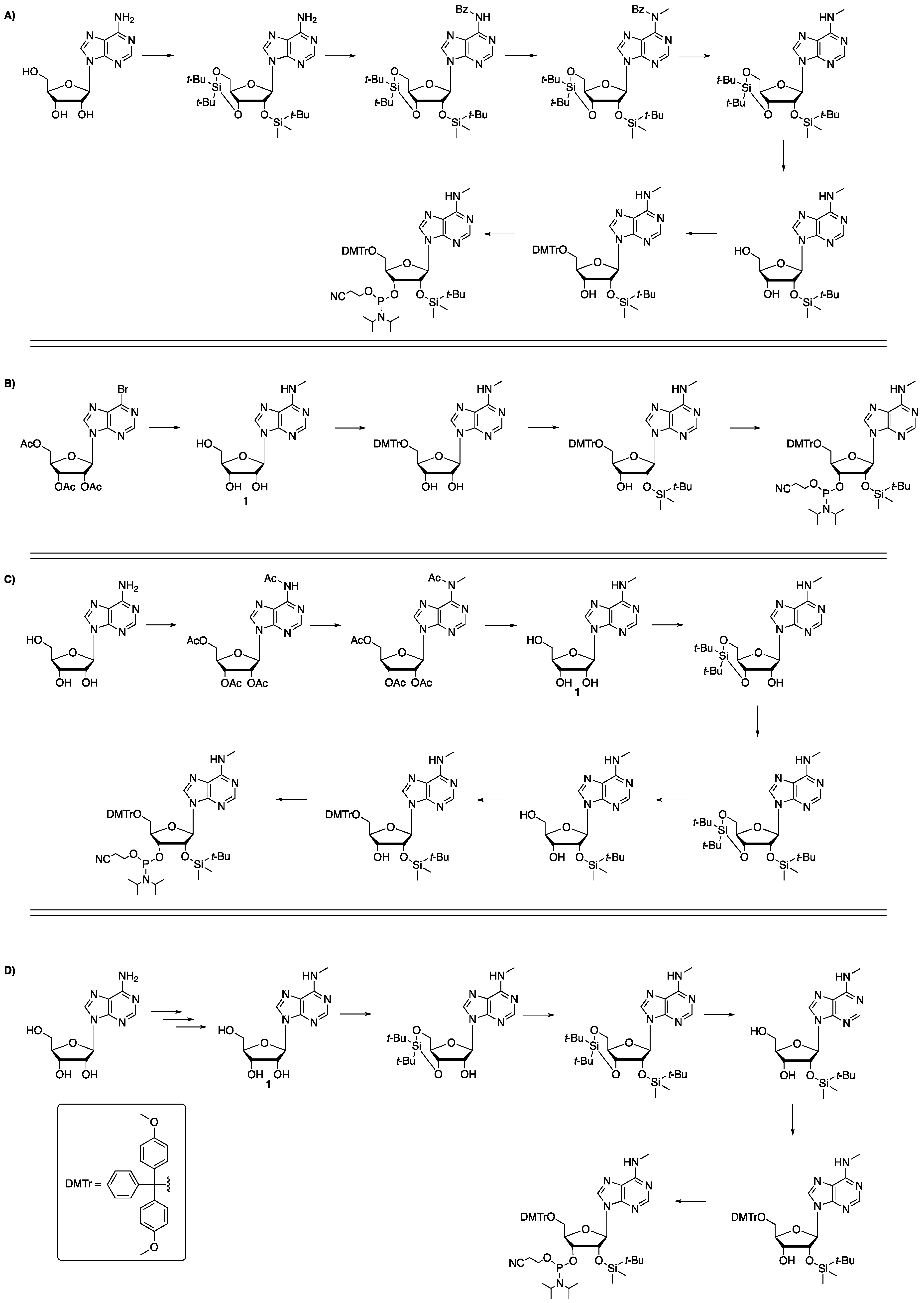
Scheme 2.
Overall yields from the shown starting materials of previously reported routes to protected m6A. (A) <8% [4]; (B) <18% [7]; (C) <20% [5]; (D) <30% [6].

Scheme 3.
Reaction conditions: (i) (t-Bu)2Si(OTf)2, imidazole, DMF, 0 °C, 1 h; (ii) TBDMSCl, imidazole, DMF, 60 °C, 12 h; (iii) DBU (1.5 eq), BOP (1.2 eq), THF, 40 °C, 40 min; (iv) methylamine (5 eq), rt, 12 h; (v) HF.pyridine (3 eq), pyridine, 0 °C, 5 h; (vi) DMTrCl (1.2 eq), pyridine, 0 °C, 12 h; (vii) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (2.5 eq), diisopropylethylamine (DIPEA, 10 eq), THF, 0 °C-rt, 6 h.
Scheme 3.
Reaction conditions: (i) (t-Bu)2Si(OTf)2, imidazole, DMF, 0 °C, 1 h; (ii) TBDMSCl, imidazole, DMF, 60 °C, 12 h; (iii) DBU (1.5 eq), BOP (1.2 eq), THF, 40 °C, 40 min; (iv) methylamine (5 eq), rt, 12 h; (v) HF.pyridine (3 eq), pyridine, 0 °C, 5 h; (vi) DMTrCl (1.2 eq), pyridine, 0 °C, 12 h; (vii) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (2.5 eq), diisopropylethylamine (DIPEA, 10 eq), THF, 0 °C-rt, 6 h.

Figure 1.
Proposed mechanism for BOP-mediated SNAr [11].
Figure 1.
Proposed mechanism for BOP-mediated SNAr [11].

Scheme 4.
Reaction conditions: (i) DBU (1.5 eq), BOP (1.2 eq), DMF, 40 °C, 40 min; (ii) methylamine (5 eq), rt, 12 h; (iii) DMTrCl (1.2 eq), pyridine, AgNO3, THF, 0 °C, 12 h; (iv) TBDMSCl, TEA, AgNO3, DMF, 60 °C, 12 h.
Scheme 4.
Reaction conditions: (i) DBU (1.5 eq), BOP (1.2 eq), DMF, 40 °C, 40 min; (ii) methylamine (5 eq), rt, 12 h; (iii) DMTrCl (1.2 eq), pyridine, AgNO3, THF, 0 °C, 12 h; (iv) TBDMSCl, TEA, AgNO3, DMF, 60 °C, 12 h.

Scheme 5.
Reaction conditions: (i) DBU (1.5 eq), BOP (1.2 eq), DMF, 40 °C, 40 min; (ii) NuH (5 eq), rt, 12 h (all yields non-optimised).
Scheme 5.
Reaction conditions: (i) DBU (1.5 eq), BOP (1.2 eq), DMF, 40 °C, 40 min; (ii) NuH (5 eq), rt, 12 h (all yields non-optimised).

Scheme 6.
Reaction conditions: (i) DBU (1.5 eq), BOP (1.2 eq), THF, 40 °C, 40 min; (ii) RNH2 (5 eq), rt, 12 h; (iii) (triphenoxy)phosphoniummethyliodide (1.2 eq), Et3N (5 eq), DMF, rt, 30 min; (iv) 3HF.Et3N (4 eq), CH2Cl2, rt, 24 h.
Scheme 6.
Reaction conditions: (i) DBU (1.5 eq), BOP (1.2 eq), THF, 40 °C, 40 min; (ii) RNH2 (5 eq), rt, 12 h; (iii) (triphenoxy)phosphoniummethyliodide (1.2 eq), Et3N (5 eq), DMF, rt, 30 min; (iv) 3HF.Et3N (4 eq), CH2Cl2, rt, 24 h.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shishodia, S.; Schofield, C.J. Improved Synthesis of Phosphoramidite-Protected N6-Methyladenosine via BOP-Mediated SNAr Reaction. Molecules 2021, 26, 147. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010147
AMA Style
Shishodia S, Schofield CJ. Improved Synthesis of Phosphoramidite-Protected N6-Methyladenosine via BOP-Mediated SNAr Reaction. Molecules. 2021; 26(1):147. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010147
Chicago/Turabian StyleShishodia, Shifali, and Christopher J. Schofield. 2021. "Improved Synthesis of Phosphoramidite-Protected N6-Methyladenosine via BOP-Mediated SNAr Reaction" Molecules 26, no. 1: 147. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010147