
Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi


Abstract
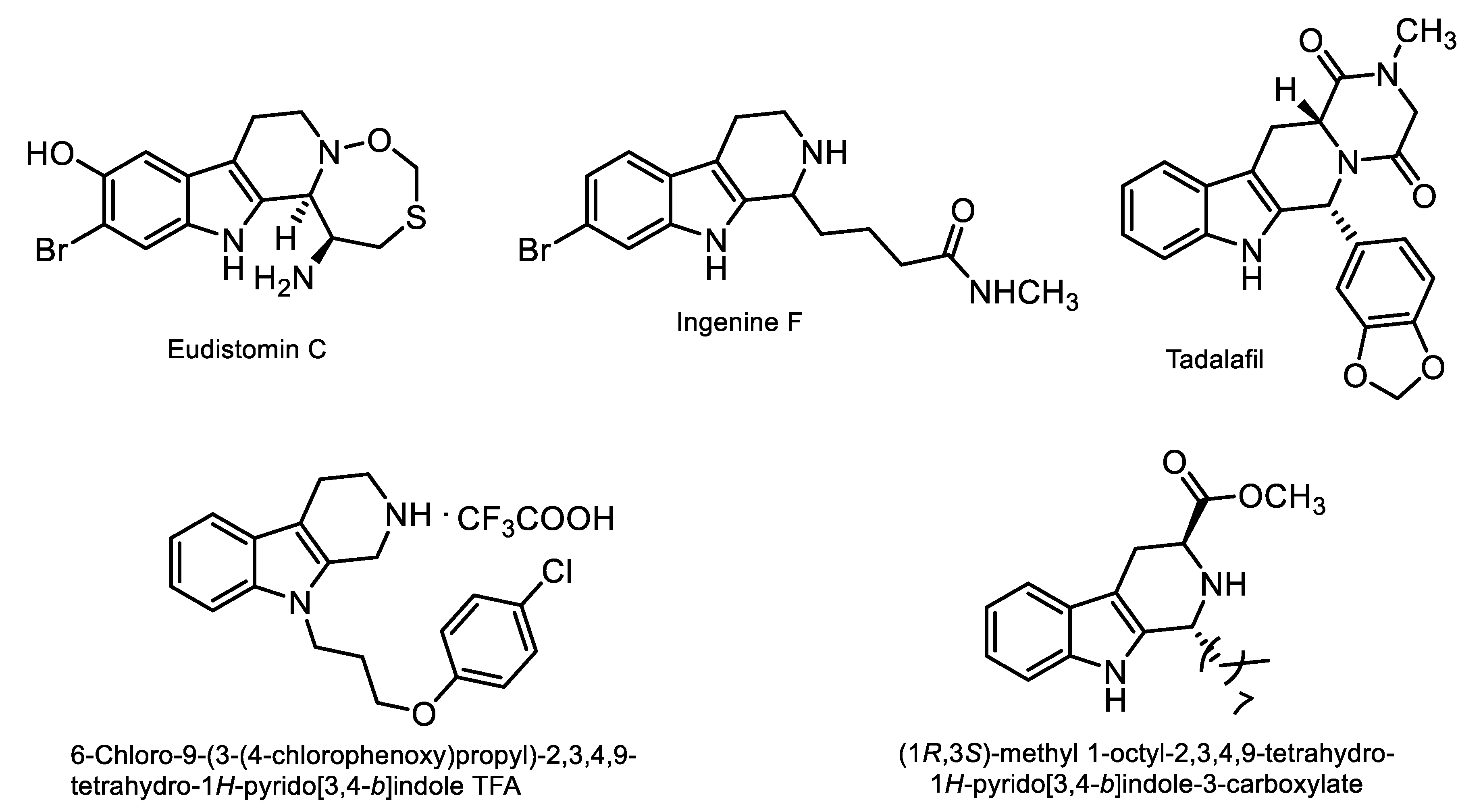
:1. Introduction
2. Results and Discussion
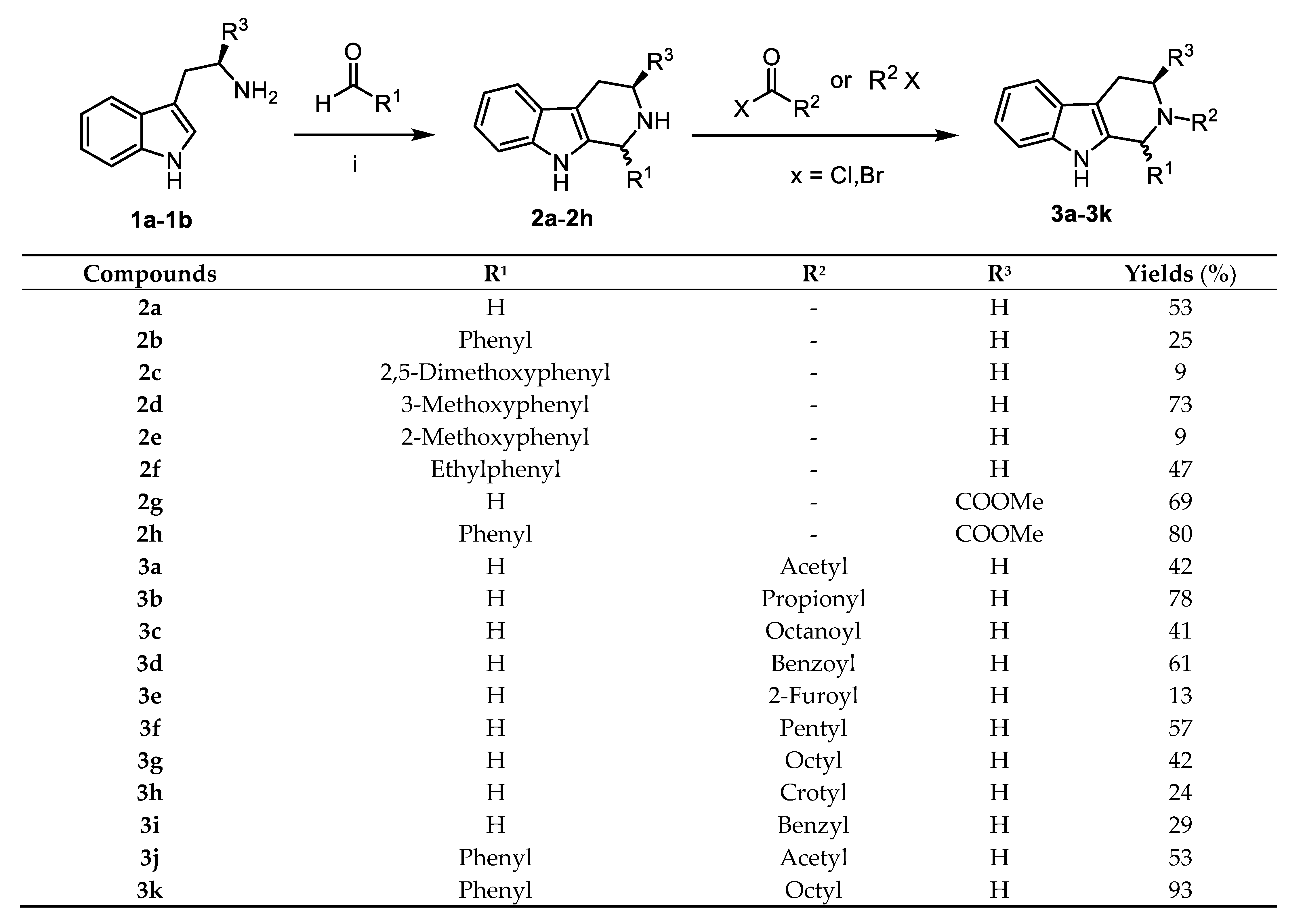
2.1. Chemistry
2.2. Antifungal Activity
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedure for the Synthesis of 1-substituted-tetrahydro-β-carbolines (2a–2h)
3.1.3. General Procedure for Preparation of 2-substituted-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (3a–3i)
3.1.4. General Procedure for the Preparation of 2-substituted-1-phenyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (3j–3k)
3.2. Antifungal Activity Assay
3.2.1. Agar Well Diffusion Method
3.2.2. Minimum Inhibitory Concentration Test
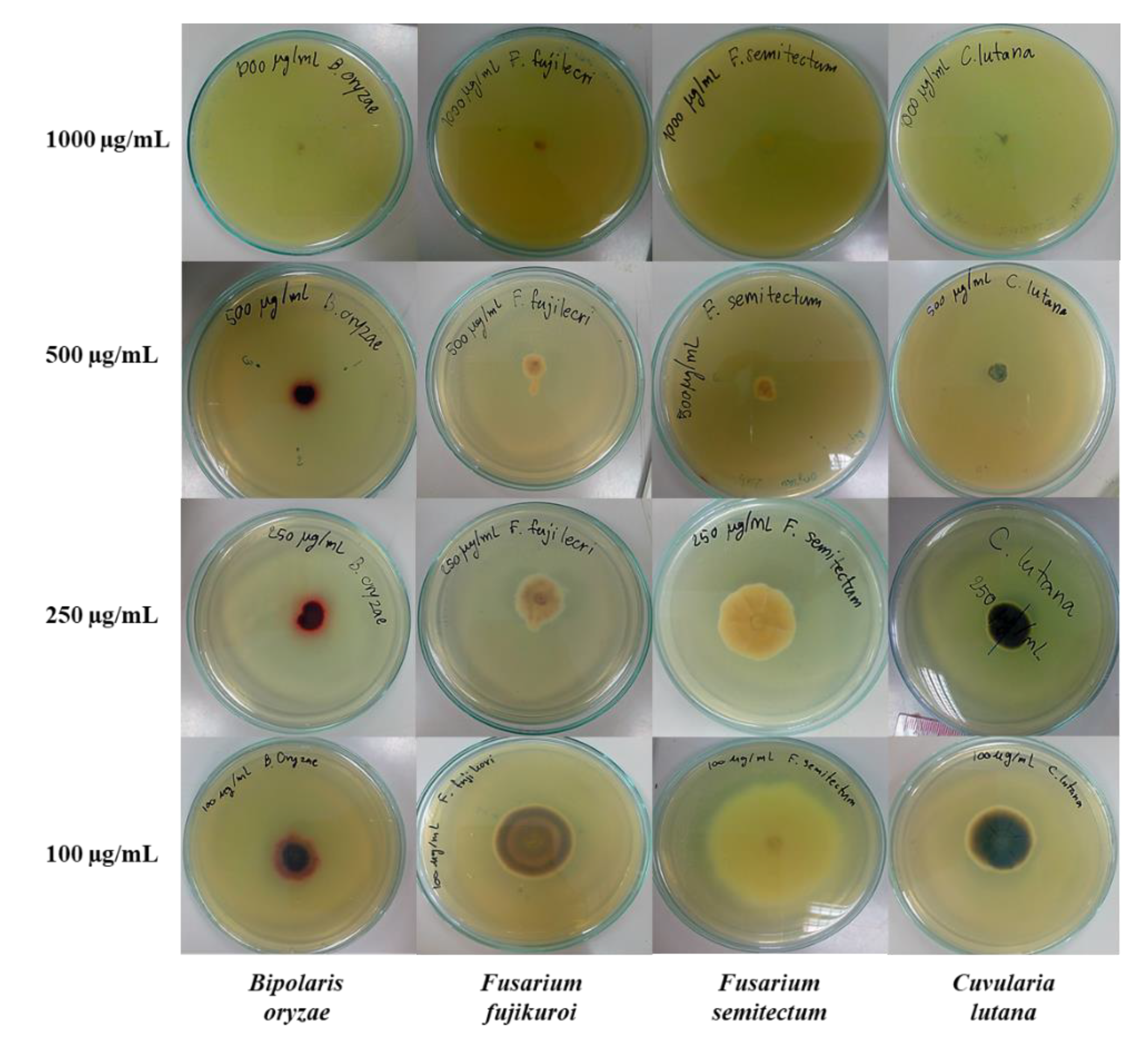
3.2.3. Test for Inhibitory Activity against Fungal Radial Growth
- IR = inhibitory activity against radial growth in percent.
- DC = diameter of fungal colony without compounds (control).
- DT = diameter of fungal colony treated with compound.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laine, A.E.; Lood, C.; Koskinen, A.M.P. Pharmacological importance of optically active tetrahydro-β-carbolines and synthetic approaches to create the C1 stereocenter. Molecules 2014, 19, 1544–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manda, S.; Khan, S.I.; Jain, S.K.; Mohammed, S.; Tekwani, B.L.; Khan, I.A.; Vishwakarma, R.A.; Bharate, S.B. Synthesis, antileishmanial and antitrypanosomal activities of N-substituted tetrahydro-β-carbolines. Bioorganic Med. Chem. Lett. 2014, 24, 3247–3250. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.R.; Wilson, J.; Emmerson, D.; Garcia, M.D.; Smith, M.R.; Gray, S.J.; Britton, R.G.; Mahale, S.; Chaudhuri, B. Design, synthesis and biological evaluation of new tryptamine and tetrahydro-β-carboline-based selective inhibitors of CDK4. Bioorg. Med. Chem. 2008, 16, 7728–7739. [Google Scholar] [CrossRef] [PubMed]
- Foderaro, T.A.; Barrows, L.R.; Lassota, P.; Ireland, C.M. Bengacarboline, a New β-Carboline from a Marine Ascidian Didemnum sp. J. Org. Chem. 1997, 62, 6064–6065. [Google Scholar] [CrossRef]
- Peng, J.; Hu, J.; Kazi, A.B.; Li, Z.; Avery, M.; Peraud, O.; Hill, R.T.; Franzblau, S.G.; Zhang, F.; Schinazi, R.F.; et al. Manadomanzamines A and B: A Novel Alkaloid Ring System with Potent Activity against Mycobacteria and HIV-1. J. Am. Chem. Soc. 2003, 125, 13382–13386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, A.D.; Freyer, A.J.; Carte, B.; Taylor, P.B.; Johnson, R.K.; Faulkner, D.J. Haploscleridamine, a Novel Tryptamine-Derived Alkaloid from a Sponge of the Order Haplosclerida: An Inhibitor of Cathepsin, K.J. Nat. Prod. 2002, 65, 628–629. [Google Scholar] [CrossRef]
- Davis, R.A.; Duffy, S.; Avery, V.M.; Camp, D.; Hooper, J.N.A.; Quinn, R.J. (+)-7-Bromotrypargine: An antimalarial β-carboline from the Australian marine sponge Ancorina sp. Tetrahedron Lett. 2010, 51, 583–585. [Google Scholar] [CrossRef]
- Maity, P.; Adhikari, D.; Jana, K.A. An Overview on Synthetic Entries to Tetrahydro-β-carbolines. Tetrahedron 2019, 75, 965–1028. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Mascal, M.; Holt, T.G.; Shield, L.S.; Lafargue, F. Eudistomins A-Q, beta.-carbolines from the antiviral Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1987, 109, 3378–3387. [Google Scholar] [CrossRef]
- Youssef, D.T.A. Hyrtioerectines A-C, Cytotoxic Alkaloids from the Red Sea Sponge Hyrtios erectus. J. Nat. Prod. 2005, 68, 1416–1419. [Google Scholar] [CrossRef]
- Shen, Y.C.; Chen, C.Y.; Hsieh, P.W.; Duh, C.Y.; Lin, Y.M.; Ko, C.L. The Preparation and Evaluation of 1-Substituted 1,2,3,4-Tetrahydro- and 3,4-Dihydro-β-carboline Derivatives as Potential Antitumor Agents. Chem. Pharm. Bull. 2005, 53, 32–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Kubota, T.; Kobayashi, J.I. Eudistomidins H–K, new β-carboline alkaloids from the Okinawan marine tunicate Eudistoma glaucus. Bioorganic Med. Chem. Lett. 2011, 21, 4220–4223. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Mohamed, G.; Al Haidari, R.; El-Kholy, A.; Zayed, M.; Ingenine, F. A New Cytotoxic Tetrahydro Carboline Alkaloid from the Indonesian Marine Sponge Acanthostrongylophora ingens. Pharmacogn. Mag. 2018, 14, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Zhu, L.F.; Yu, X.C.; Sun, M.Q.; Miao, F.; Zhou, L. Design, Synthesis, and Structure–Activity Relationship of New 2-Aryl-3,4-dihydro-β-carbolin-2-ium Salts as Antifungal Agents. J. Agric. Food Chem. 2016, 64, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.P.; Sarma, B.K.; Mishra, P.K.; Ray, A.B. Antifungal activity of venenatine, an indole alkaloid isolated from Alstonia venenata. Folia Microbiol. 2000, 45, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.M.F.; Queiroz, E.F.; Zeraik, M.L.; Marti, G.; Favre-Godal, Q.; Simões-Pires, C.; Marcourt, L.; Carrupt, P.A.; Cuendet, M.; Paulo, M.Q.; et al. Antifungals and acetylcholinesterase inhibitors from the stem bark of Croton heliotropiifolius. Phytochem. Lett. 2014, 10, lxxxviii–xciii. [Google Scholar] [CrossRef]
- Sheng, C.; Che, X.; Wang, W.; Wang, S.; Cao, Y.; Yao, J.; Miao, Z.; Zhang, W. Design and synthesis of antifungal benzoheterocyclic derivatives by scaffold hopping. Eur. J. Med. Chem. 2011, 46, 1706–1712. [Google Scholar] [CrossRef]
- Tu, J.; Li, Z.; Jiang, Y.; Ji, C.; Han, G.; Wang, Y.; Liu, N.; Sheng, C. Discovery of Carboline Derivatives as Potent Antifungal Agents for the Treatment of Cryptococcal Meningitis. J. Med. Chem. 2019, 62, 2376–2389. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Liu, W.; Liu, N.; Zhang, Y.; Dong, G.; Liu, Y.; Li, Z.; He, X.; Miao, Z.; et al. Novel Carboline Derivatives as Potent Antifungal Lead Compounds: Design, Synthesis, and Biological Evaluation. ACS Med. Chem. Lett. 2014, 5, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Jaisingh, A.; Maurya, I.K.; Salunke, D.B. Design, synthesis and bio-evaluation of C-1 alkylated tetrahydro-β-carboline derivatives as novel antifungal lead compounds. Bioorganic Med. Chem. Lett. 2020, 30, 126869. [Google Scholar] [CrossRef]
- Li, H.; Dang, H.T.; Li, J.; Sim, C.J.; Hong, J.; Kim, D.K.; Jung, J.H. Pyroglutamyl dipeptides and tetrahydro-β-carboline alkaloids from a marine sponge Asteropus sp. Biochem. Syst. Ecol. 2010, 38, 1049–1051. [Google Scholar] [CrossRef]
- Salmoun, M.; Devijver, C.; Daloze, D.; Braekman, J.C.; van Soest, R.W.M. 5-Hydroxytryptamine-Derived Alkaloids from Two Marine Sponges of the Genus Hyrtios. J. Nat. Prod. 2002, 65, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kubota, T.; Fromont, J.; Kobayashi, J.I. Zamamidines A and B, New Manzamine Alkaloids from the Sponge Amphimedon Species. Org. Lett. 2009, 11, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Pilli, R.A.; Rawal, V.H. Enantioselective Total Syntheses of (+)-Arborescidine A, (−)-Arborescidine B, and (−)-Arborescidine, C.J. Org. Chem. 2004, 69, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, R.M.; Jompa, J.; Tahir, A.; Ireland, C.M. Trypargine Alkaloids from a Previously Undescribed Eudistoma sp. Ascidian. J. Nat. Prod. 1999, 62, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Pénez, N.; Culioli, G.; Pérez, T.; Briand, J.F.; Thomas, O.P.; Blache, Y. Antifouling Properties of Simple Indole and Purine Alkaloids from the Mediterranean Gorgonian Paramuricea clavata. J. Nat. Prod. 2011, 74, 2304–2308. [Google Scholar]
- Ovenden, P.B.S.; Nielson, J.L.; Liptrol, H.C.; Willis, H.R.; Tapiolas, M.D.; Wright, D.A.; Motti, A.C. Callophycin A, a cytotoxic tetrahydro-β-carboline from the red alga Callophycus oppositifolius. Phytochem. Lett. 2011, 4, 69–71. [Google Scholar] [CrossRef]
- Badre, A.; Boulanger, A.; Abou-Mansour, E.; Banaigs, B.; Combaut, G.; Francisco, C. Eudistomin U and Isoeudistomin U, New Alkaloids from the Carribean Ascidian Lissoclinum fragile. J. Nat. Prod. 1994, 57, 528–533. [Google Scholar] [CrossRef]
- Lake, R.J.; Blunt, J.W.; Munro, M.H.G. Eudistomins from the New Zealand Ascidian Ritterella sigillinoides. Aust. J. Chem. 1989, 42, 1201–1206. [Google Scholar] [CrossRef]
- Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Rinehart, K.L., Jr. Eudistomins A, D, G, H, I, J, M, N, O, P, and Q, bromo, hydroxy, pyrrolyl and iminoazepino beta-carbolines from the antiviral Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1526–1528. [Google Scholar] [CrossRef]
- Schupp, P.; Poehner, T.; Edrada, R.; Ebel, R.; Berg, A.; Wray, V.; Proksch, P. Eudistomins W and X, Two New β-Carbolines from the Micronesian Tunicate Eudistoma sp. J. Nat. Prod. 2003, 66, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Chinen, T.; Yoshida, K.; Kudo, S.; Nagumo, Y.; Shiwa, Y.; Yamada, R.; Umihara, H.; Iwasaki, K.; Masumoto, H.; et al. Eudistomin C, an Antitumor and Antiviral Natural Product, Targets 40S Ribosome and Inhibits Protein Translation. Chembiochem 2016, 17, 1616–1620. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.; Mohamed, G.A. Ingenine E, a new cytotoxic beta-carboline alkaloid from the Indonesian sponge Acanthostrongylophora ingens. J. Asian. Nat. Prod. Res. 2017, 19, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Mohamed, G.A.; Zayed, M.F.; Sayed, H.M. Ingenines A and B, Two New Alkaloids from the Indonesian Sponge Acanthostrongylophora ingens. Drug Res. 2015, 65, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Gorki, V.; Singh, R.; Walter, N.S.; Bagai, U.; Salunke, D.B. Synthesis and Evaluation of Antiplasmodial Efficacy of β-Carboline Derivatives against Murine Malaria. ACS Omega 2018, 3, 13200–13210. [Google Scholar] [CrossRef] [Green Version]
- Eardley, I.; Cartledge, J. Tadalafil (Cialis) for men with erectile dysfunction. Int. J. Clin. Pract. 2002, 56, 300–304. [Google Scholar]
- Levin, Y.D.; White, R.J. Novel therapeutic approaches in pulmonary arterial hypertension: Focus on tadalafil. Drugs Today 2011, 47, 145–156. [Google Scholar] [CrossRef]
- Shrestha, S.K.; Garzan, A.; Garneau-Tsodikova, S. Novel alkylated azoles as potent antifungals. Eur. J. Med. Chem. 2017, 133, 309–318. [Google Scholar] [CrossRef]
- Ji, H.; Zhang, W.; Zhang, M.; Kudo, M.; Aoyama, Y.; Yoshida, Y.; Sheng, C.; Song, Y.; Yang, S.; Zhou, Y.; et al. Structure-Based de Novo Design, Synthesis, and Biological Evaluation of Non-Azole Inhibitors Specific for Lanosterol 14α-Demethylase of Fungi. J. Med. Chem. 2003, 46, 474–485. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, J.; Zhou, Y.; Li, Y.; Cheng, J.; Zheng, C. Design, synthesis, and antifungal activities in vitro of novel tetrahydroisoquinoline compounds based on the structure of lanosterol 14α-demethylase (CYP51) of fungi. Bioorganic Med. Chem. Lett. 2006, 16, 5285–5289. [Google Scholar] [CrossRef]
- Yao, B.; Ji, H.; Cao, Y.; Zhou, Y.; Zhu, J.; Lü, J.; Li, Y.; Chen, J.; Zheng, C.; Jiang, Y.; et al. Synthesis and Antifungal Activities of Novel 2-Aminotetralin Derivatives. J. Med. Chem. 2007, 50, 5293–5300. [Google Scholar] [CrossRef] [PubMed]
- Steinbuch, K.B.; Benhamou, R.I.; Levin, L.; Stein, R.; Fridman, M. Increased Degree of Unsaturation in the Lipid of Antifungal Cationic Amphiphiles Facilitates Selective Fungal Cell Disruption. ACS Infect. Dis. 2018, 4, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Neto, V.; Voisin, A.; Héroguez, V.; Grelier, S.; Coma, V. Influence of the Variation of the Alkyl Chain Length of N-Alkyl-β-d-glycosylamine Derivatives on Antifungal Properties. J. Agric. Food Chem. 2012, 60, 10516–10522. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, M.; Rai, N.N.; Sawant, D. Mild and efficient cyanuric chloride catalyzed Pictet–Spengler reaction. Beilstein J. Org. Chem. 2013, 9, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dóra, L.; Clauder, O. The reduction of 1,2,3,4-tetrahydro-1-oxo-beta-carboline. Acta Pharm. Hung. 1968, 38, 78–83. [Google Scholar]
- Qi, S.H.; Zhang, S.; Yang, L.H.; Qian, P.Y. Antifouling and antibacterial compounds from the gorgonians Subergorgia suberosa and Scripearia gracillis. Nat. Prod. Res. 2008, 22, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Sharma, S.; Sawant, D.; Kundu, B. Water as an efficient medium for the synthesis of tetrahydro-β-carbolines via Pictet-Spengler reactions. Tetrahedron Lett. 2007, 48, 1379–1383. [Google Scholar] [CrossRef]
- Cheve, G.; Duriez, P.; Fruchart, J.; Teissier, E.; Poupaert, J.; Lesieur, D. Antioxidant activity of pinoline analogues in the LDL oxidation model. J. Mad. Chem. 2002, 11, 361–379. [Google Scholar]
- Barbero, M.; Bazzi, S.; Cadamuro, S.; Dughera, S. o-Benzenedisulfonimide as a reusable acid catalyst for an easy, efficient, and green synthesis of tetrahydroisoquinolines and tetrahydro-β-carbolines through Pictet–Spengler reaction. Tetrahedron Lett. 2010, 51, 6356–6359. [Google Scholar] [CrossRef]
- Ikeda, R.; Iwaki, T.; Iidaa, T.; Okabayashi, T.; Nishi, E.; Kurosawa, M.; Sakai, N.; Konakahara, T. 3-Benzylamino-ß-carboline derivatives induce apoptosis through G2/M arrest in human carcinoma cells HeLa S-3. Eur. J. Med. Chem. 2011, 46, 636–646. [Google Scholar] [CrossRef]
- Ding, L.; Dahse, H.-M.; Hertweck, C. Cytotoxic Alkaloids from Fusarium incarnatum Associated with the Mangrove Tree Aegiceras corniculatum. J. Nat. Prod. 2012, 75, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.B.; Mclsaac, W.M.; Walker, E.K. Inhibitors of monoamine oxidase II. Syntheses of some N-2(9)-Substituted tetrahydro-β-carbolines and evaluation of their inhibitory a ctivities. J. Pharm. Sci. 1968, 57, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Aubry, C.; Wilson, A.J.; Jenkins, R.P.; Mahale, S.; Chaudhuri, B.; Maréchalc, J.-D.; Sutcliffe, J.M. Design, synthesis and biological activity of new CDK4-specific inhibitors, based on fascaplysin. Org. Biomol. Chem. 2006, 4, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, F.; Stephensen, H. (Cyanomethyl)trialkylphosphonium Iodides: Efficient Reagents for the Intermolecular Alkylation of Amines with Alcohols in Solution and on Solid Phase. J. Org. Chem. 2001, 66, 2518–2521. [Google Scholar] [CrossRef] [PubMed]
- Freter, K.; HugoHübner, H.; Metz, H.; Schroeder, H.D.; Zeile, K. Reactions of 1-aryl-1,2,3,4-tetrahydro-β-carbolines in acid solution. Justus Liebigs Ann. Chem. 1965, 684, 159–187. [Google Scholar] [CrossRef]
- Taechowisan, T.; Chaiseang, S.; Phutdhawong, S.W. Antifungal activity of biphenyls from Streptomyces sp. bo07 against selective phytopathogenic fungi of rice. Int. J. Curr. Res. 2018, 10, 66239–66244. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Tests for Filamentous Fungi; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2002. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Compounds | B. oryzae | F. fujikuroi | F. semitectum | C. lutana | ||||
|---|---|---|---|---|---|---|---|---|
| Zone of Inhibition | MIC 1 | Zone of Inhibition | MIC | Zone of Inhibition | MIC | Zone of Inhibition | MIC | |
| 2a | 0.00 | NT 2 | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2b | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.10 ± 0.0 | NT |
| 2c | 0.10 ± 0.1 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2d | 0.60 ± 0.1 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2e | 0.25 ± 0.3 | NT | 0.00 | NT | 0.00 | NT | 0.40 ± 0.0 | NT |
| 2f | 0.40 ± 0.0 | NT | 0.10 ± 0.0 | NT | 0.00 | NT | 0.20 ± 0.0 | NT |
| 2g | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 2h | 0.15 ± 0.1 | NT | 0.00 | NT | 0.00 | NT | 0.20 ± 0.1 | NT |
| 3a | 0.00 | NT | 0.00 | NT | 0.10 ± 0.0 | 870 ± 0.0 | 0.00 | NT |
| 3b | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3c | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3d | 0.30 ± 0.1 | 200 ± 0.1 | 0.1 ± 0.0 | >512 | 0.20 ± 0.0 | >512 | 0.30 ± 0.0 | >512 |
| 3e | 0.60 ± 0.0 | 400 ± 0.0 | 0.20 ± 0.1 | >512 | 0.25 ± 0.1 | >512 | 0.25 ± 0.2 | 250 ± 0.0 |
| 3f | 0.10 ± 0.0 | 110 ± 0.0 | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3g | 0.10 ± 0.0 | 28 ± 0.0 | 0.1 ± 0.0 | >512 | 0.1 ± 0.1 | >512 | 0.15 ± 0.1 | 200 ± 0.0 |
| 3h | 0.10 ± 0.0 | 220 ± 0.1 | 0.00 | NT | 0.00 | NT | 0.00 | NT |
| 3i | 0.00 | NT | 0.00 | NT | 0.00 | NT | 0.10 ± 0.0 | 250 ± 0.0 |
| 3j | 0.00 | NT | 0.00 | NT | 0.20 ± 0.1 | >512 | 0.00 | NT |
| 3k | 0.35 ± 0.1 | 210 ± 0.0 | 0.00 | NT | 0.20 ± 0.0 | 1700 ± 0.0 | 0.10 ± 0.0 | 270 ± 0.0 |
| Amphotericin B | 0.21 ± 0.1 | 0.78 ± 0.0 | 0.00 | >512 | 0.13 ± 0.1 | >512 | 0.15 ± 0.1 | 0.33 ± 0.1 |
| Compound | Concentration (µg/mL) | Diameter of Colony (cm) of Tested Fungi | |||
|---|---|---|---|---|---|
| B. oryzae | F. fujikuroi | F. semitectum | C. lutana | ||
| None (control) | - | 7.77 ± 0.15 | 5.48 ± 0.08 | 5.17 ± 0.23 | 5.00 ± 0.27 |
| 3g | 1000 | NG 1 (100%) | NG (100%) | NG (100%) | NG (100%) |
| 500 | 0.85 ± 0.05 (89.1%) | 1.30 ± 0.35 (76.3%) | 0.90 ± 0.07 (82.6%) | 0.90 ± 0.12 (82.0%) | |
| 250 | 1.43 ± 0.11 (81.7%) | 2.23 ± 0.08 (59.4%) | 3.08 ± 0.04 (40.6%) | 1.98 ± 0.11 (60.5%) | |
| 100 | 2.48 ± 0.13 (68.2%) | 3.43 ± 0.04 (37.4%) | 5.90 ± 0.21 (−14.0%) | 3.00 ± 0.07 (40%) | |
| Amphotericin B | 0.6 | 3.30 ± 0.14 (57.6%) | 5.68 ± 0.04 (−3.7%) | 2.45 ± 0.17 (52.7%) | 4.03 ± 0.08 (19.5%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buaban, K.; Phutdhawong, W.; Taechowisan, T.; Phutdhawong, W.S. Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi. Molecules 2021, 26, 207. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010207
Buaban K, Phutdhawong W, Taechowisan T, Phutdhawong WS. Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi. Molecules. 2021; 26(1):207. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010207
Chicago/Turabian StyleBuaban, Koonchira, Weerachai Phutdhawong, Thongchai Taechowisan, and Waya S. Phutdhawong. 2021. "Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of Plant Pathogenic Fungi" Molecules 26, no. 1: 207. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010207