
A Review of Composite Phase Change Materials Based on Porous Silica Nanomaterials for Latent Heat Storage Applications

 , and
, and
Abstract
:1. Introduction
2. Thermal Energy Storage
2.1. Phase Change Materials
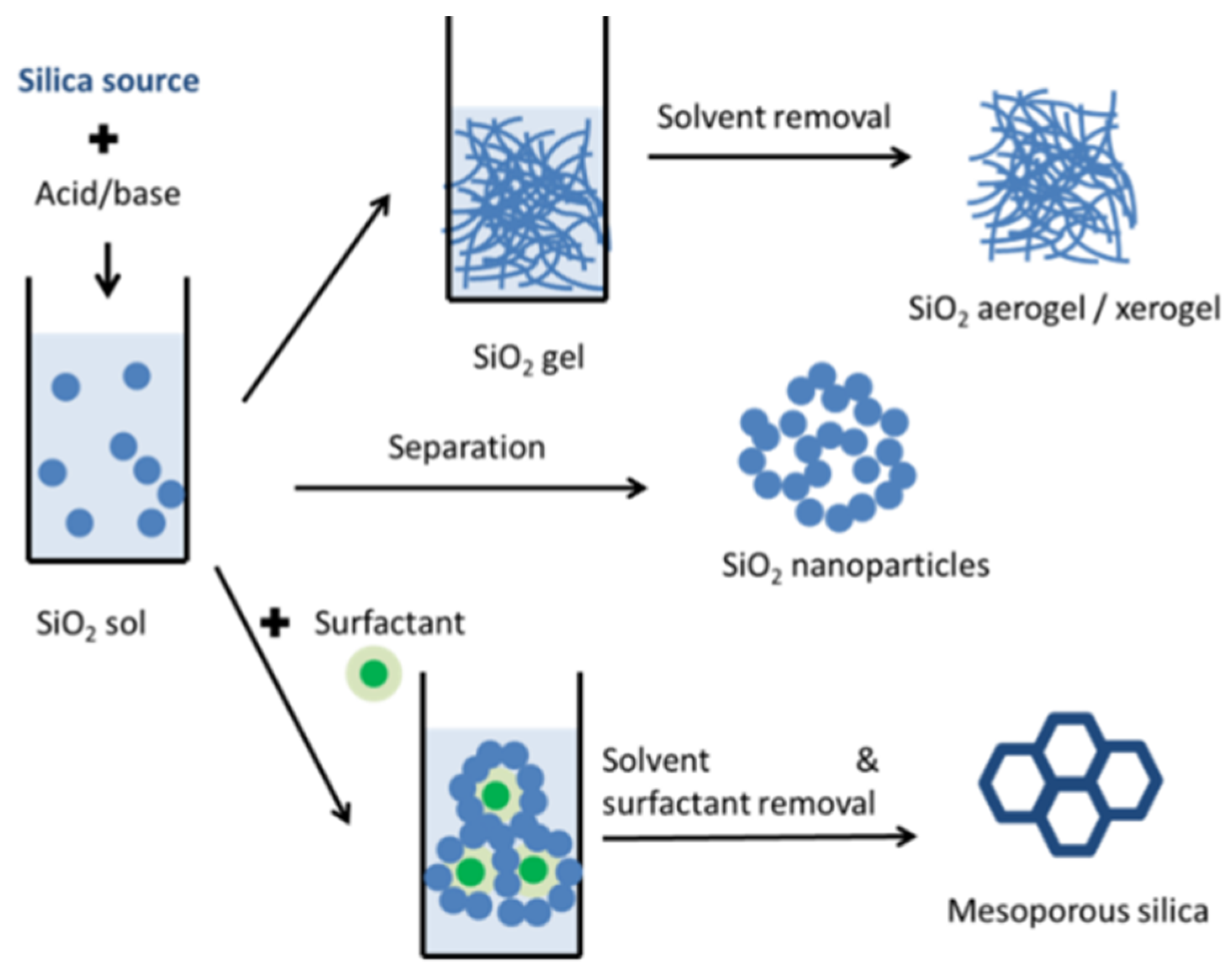
2.2. Porous Silica Matrices
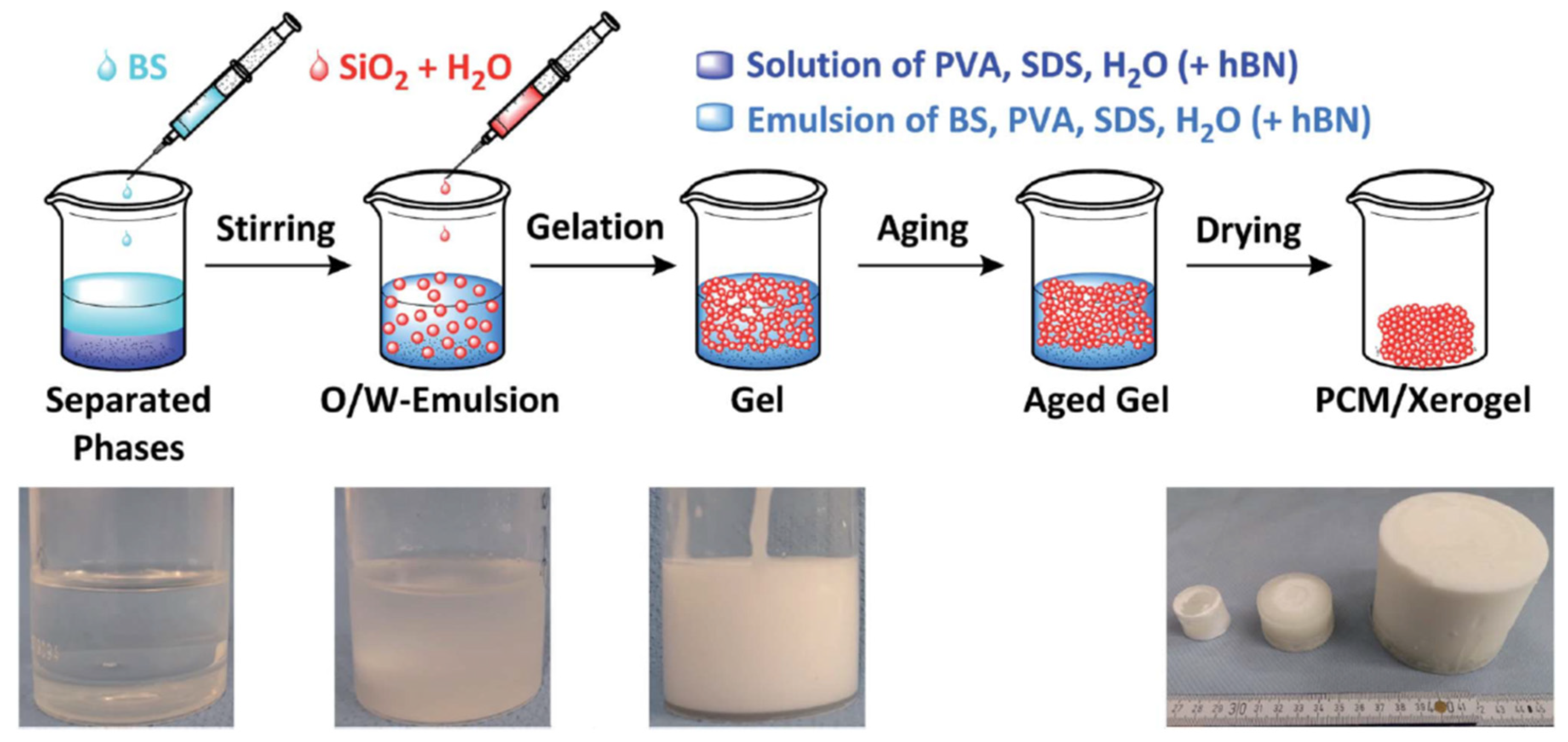
2.3. Porous Silica Nanocomposites: Desired Properties and Synthesis
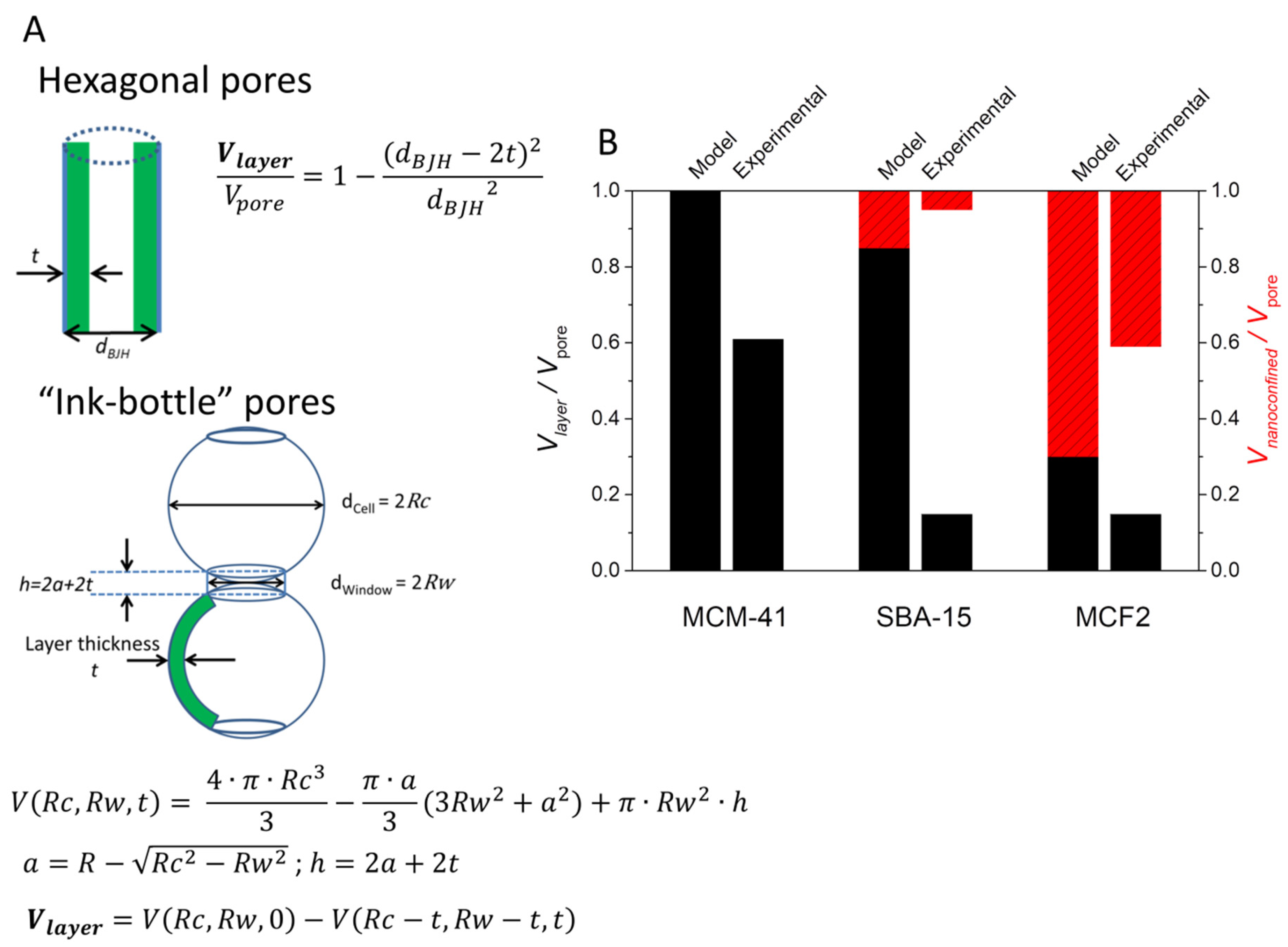
2.4. Porous Silica Nanocomposites: Structure and Physico-Chemical Properties under Nanoconfinement
3. Phase Change Materials Containing Porous Silica Matrices and Different Heat Storage Compounds
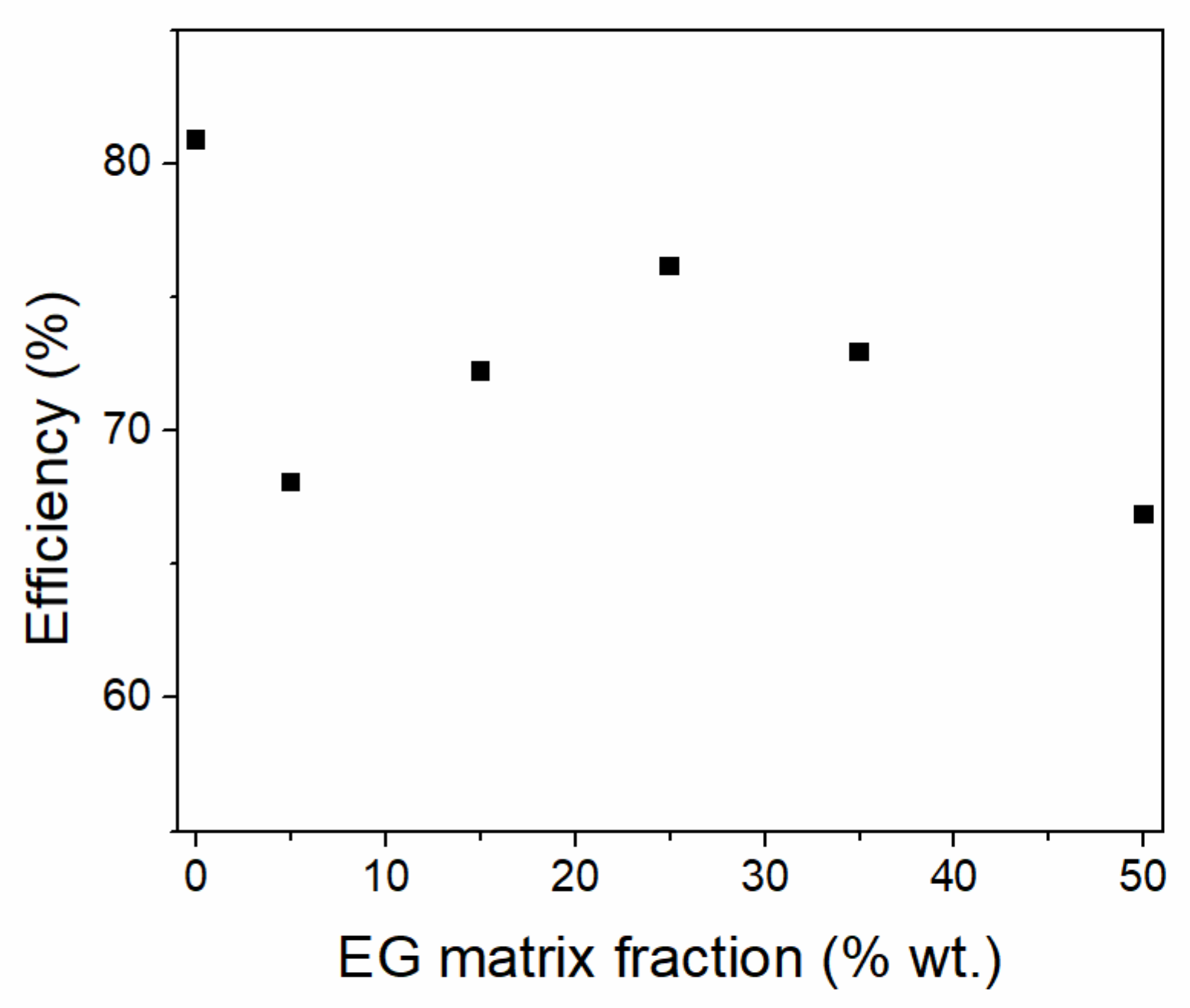
3.1. Paraffins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
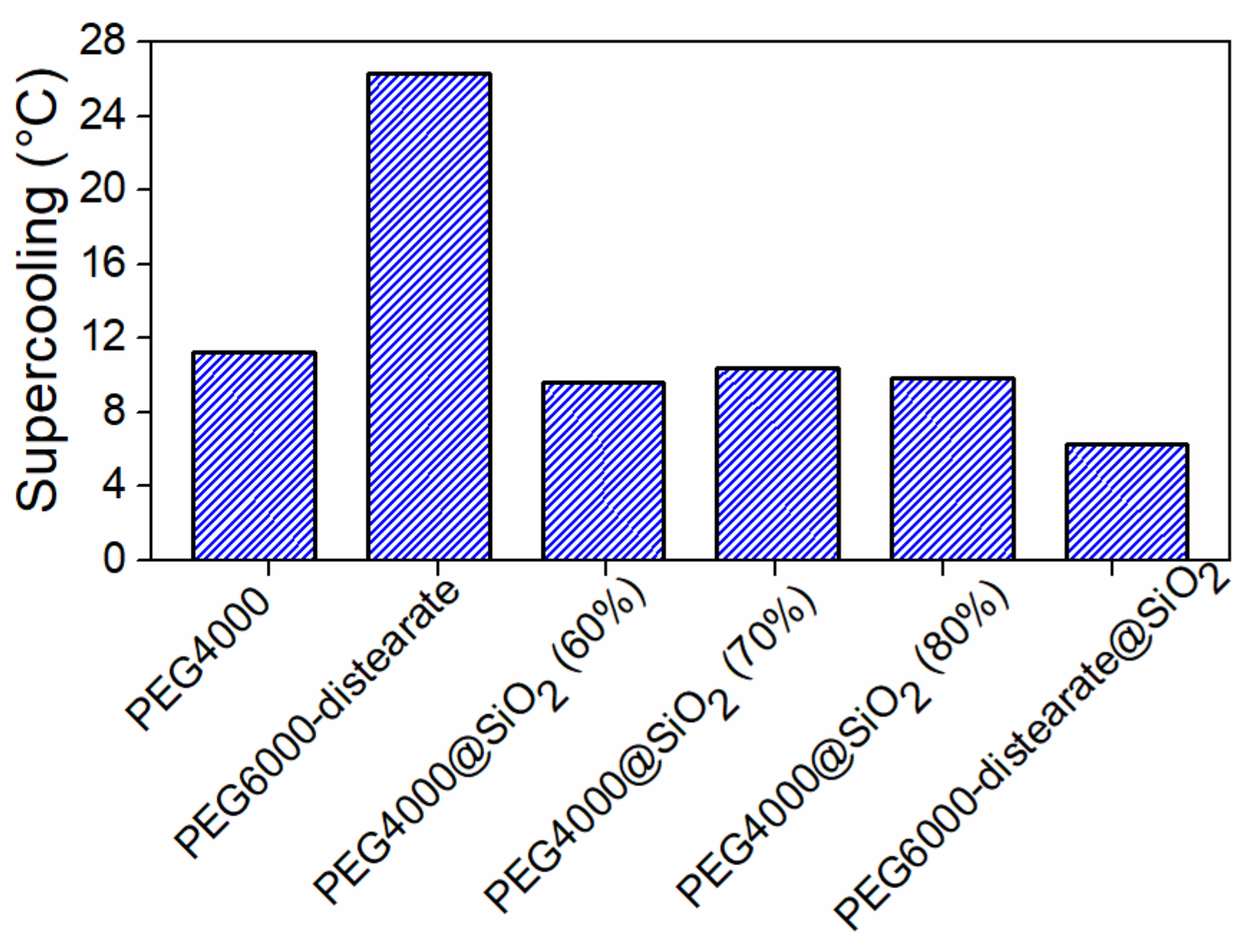{kind=link}
{kind=link}
{kind=link}
| PCM | Porous Silica Composite | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Sample/Synthesis | %PCM (wt.) * | m.p. (°C) | ΔHf (J g−1) | |
| Paraffin wax | 29.0 | 142.0 | Direct synthesis/CTAB + n-pentanol | - | 30.0 | 95.0 | [82] |
| Paraffin | 51.0 | 151.5 | Direct synthesis/TEOS/HCl | 92.1 | 50.2 | 139.6 | [83] |
| Paraffin | 57.0 | - | Aerogel/supercritical EtOH | 75 | - | - | [27] |
| Rubitherm RT 28 | - | - | TEOS/HCl, EG, paraffin | - | 27.7 | 104.4 | [111] |
| n-eicosane | 39.2 | 237.1 | n-C20H42,Fe3O4@SiO2 | 70 | 39.2 | 170.2 | [120] |
| PX25 | ~23 | 96 | 9% hydrophobic fumed silica vs. PCM; 30:70 wt. composite: cement | 27.3 | ~23 | 14.2 | [102] |
| Paraffin | 50.1 | 173.9 | Parafin@SiO2-GO/PVA, Span80, Tween80 | 49 | 49.7 | 87.1 | [118] |
| 25# Paraffin | 25.8 | 107.6 | Vacuum melt impregnation | - | 21.6 | 56.3 | [91] |
| n-eicosane | 37.13 | 249.0 | Aerogel melt impregnation | 84.3 | 36.8 | 198.4 | [101] |
| Paraffin | 57.7 | 161.4 | TEOS/HCl | 60 | 58.2 | 98.0 | [85] |
| Paraffin | 51.9 | 184.1 | 2% EG; 8% 20 nm SiO2; melt addition | 90 | 51.8 | 168.3 | [112] |
| Octadecane | 26.5 | 2300 | Gas transport/12.5 nm SBA-15 | - | 17.4 | 103.3 | [109] |
| Tetradecane | 6.2 | 216.0 | - | −7.4 | 124.2 | ||
| Paraffin | 49.7 | 200.4 | Solvent impregnation/SiO2 NPs | 80 | 52.0 | 156.6 | [100] |
| Paraffin | 28.0 | 168.0 | Melt impregnation/MCM-41 | 60 | 25.5 | 95.0 | [92] |
| Paraffin | 42.2 | 243.0 | Vacuum melt/SiO2-PTFE aerogel | 62.8 | 42.0 | 128.0 | [96] |
| Paraffin | 59.6 | 191.1 | Solution impregnation/CH3-fucntionalized aerogel | 70 | 59.6 | 112.9 | [104] |
| n-Eicosane | 36.9 | 243.3 | Solution impregnation/7% EG/70:30 Eicosane:20 nm SiO2 NP | 65.1 | 35.4 | 135.8 | [114] |
| Paraffin wax | 63.8 | 209.1 | Vacuum melt/CH3/HO-aerogel | 88 | 63.7 | 163.6 | [105] |
| Paraffin wax | 25.7 | 198.0 | Vacuum melt/SiO2-EG 1:1 | 80 | 26.7 | 105.9 | [115] |
| Paraffin | 56.8 | 182.2 | Melt impregnation/aerogel | 75 | 56.3 | 165.2 | [94] |
| Hexadecane | 17.7 | 220 | Vacuum melt/mesoporous silica/300 heat-cool cycles | 45 | 17.1 | 100.1 | [99] |
| Octadecane | 29.9 | 223 | Vacuum melt/mesoporous silica | 45.3 | 28.9 | 84.5 | [81] |
| Octadecane | 28.5 | 212.6 | Direct synthesis | - | 26.3 | 99.3 | [88] |
| Nonadecane | 29.4 | 201.0 | Direct synthesis | - | 26.2 | 80.8 | [89] |
| Octadecane | 28.2 | 232.5 | Vacuum melt/SiO2 NP | 70 | 27.7 | 85.0 | [122] |
3.2. Fatty Acids and Derivatives
| PCM | Porous Silica Composite | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Sample/Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf (J g−1) | |
| Stearic acid | 55.6 | 176.7 | Direct synthesis/TEOS/HCl | 60 | 54.9 | 109.4 | [85] |
| Dodecanoic acid | 44.6 | 169.0 | Gas transport/12.5 nm SBA-15 | - | 22.3 | 65.9 | [109] |
| Tetradecanol | 36.0 | 198.0 | - | 11.4 | 48.4 | ||
| Decanoic acid | 30.0 | 163.0 | - | 11.1 | 64.3 | ||
| Dodecanol | 24.0 | 196.0 | - | 0.2 | 69.5 | ||
| Quinary eutectic | 12.3 | 134.4 | Melt impregnation/electrospun SiO2 fibers | 80.2 | 13.4 | 107.8 | [132] |
| LA:PA:PAR eutectic | 33.1 | 140.6 | Melt impregnation/SiO2 NPs + HDPE | 75 | 31.5 | 104.4 | [133] |
| Lauric acid | 44.4 | 180.8 | Direct synthesis | 65 | 42.5 | 117.2 | [124] |
| Stearic acid | 59.9 | 177.8 | Solution impregnation/fumed silica | 46 | 58.8 | 82.5 | [137] |
| Octadecanol | - | 235 | Vacuum melt/CH3-aerogel | 86 | - | 153.7 | [144] |
| Lauric acid | 42.7 | 166.0 | Hexane solution/MCF | 83 | 34.0/ 41.2 | 123.7 | [67] |
| CA: LA:PA = 61.9:31.0:7.1 | 15.0 | 120.2 | Melt impregnation/electrospun SiO2 fibers | 81 | 13.7 | 100.9 | [131] |
| Stearic acid | 65.2 | 239.4 | Solution impregnation/Tannic acid templated SiO2 | 70 | 67.1 | 108.8 | [138] |
| CA:PA: SA = 79.3:14.7:6.0 | 18.5 | 139.3 | Melt impregnation/SiO2 NPs | 75 | 17.2 | 99.4 | [134] |
| CA:MA = 72:28 | 21.7 | 139.2 | Direct synthesis | 40 | 21.15 | 55.6 | [126] |
| Stearic acid | - | 221.8 | Solution impregnation/MOS | 70 | - | 108.0 | [140] |
| Myristic acid | 57.7 | 184.3 | Solution impregnation/WMSN | 65 | 54.7 | 92.0 | [142] |
| Stearic acid | 52.5 | 172.7 | Vacuum melt/SiO2 NP | 70 | 52.1 | 77.6 | [122] |
| Octadecanol | 57.2 | 234.5 | 70 | 56.4 | 47.0 | ||
| Lauric acid | 42.7 | 176.1 | Vacuum melt/MCF | 84 | 31.5/41.7 | 128.1 | [146] |
| CA-PA (85:15) | 27.5 | 151.5 | Melt/Fumed silica+5% CNT | 30.4 | 25.2 | 41.2 | [147] |
| Stearic acid | 56.6 | 170.3 | Direct synthesis | 76 | 53.8 | 118.3 | [127] |
| Lauric acid | 44.2 | 165.8 | Direct synthesis; TEOS+MTES | 42.2 | 82.7 | [129] | |
| Stearic acid | 68.4 | 213.6 | Melt impregnation/MCF-COOH | 79 | 58.9/68.8 | 128.3 | [66] |
| Methyl laurate | 4.0 | 210.1 | Direct synthesis, VTES/PVA | 71 | 6.7 | 151.3 | [145] |
| Palmitic acid | 62.8 | 209.7 | Solution impregnation; 5% GNP | 70 | 60.6 | 128.4 | [148] |
| Octadecanol | 57.8 | 237.8 | Melt impregnation/2% GO- SiO2 aerogel | 75 | 53 | 129.6 | [149] |
| 1, 8-Cctanediol | 62.4 | 225.1 | Direct synthesis, TEOS/HCl | 70 | 61.3 | 157.7 | [128] |
| Caprylic acid | 12.0 | 139.9 | Melt impregnation/silica gel | 48 | 13.8 | 46.4 | [136] |
| Palmitic acid | 50.0 | 213.1 | Melt impregnation/C-SiO2 aerogel | 82 | 187.7 | 43.4 | [150] |
3.3. Polyethylene Glycol (PEG)-Based PCMs
| PCM | Porous Silica Composite | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf (J g−1) | |
| PEG 600 | 18.5 | 118.2 | solution impregnation | 62 | 19.6 | 71.6 | [171] |
| PEG 2000 | 52.5 | 153.0 | vacuum impregnation | 60 | 50.8 | 58.76 | [173] |
| PEG 4000 | 53.8 | 202.1 | vacuum impregnation | 80 | 50.8 | 136.6 | [167] |
| PEG 4000 | 59.1 | 183.4 | vacuum impregnation | 70 | 57.8 | 121.7 | [166] |
| PEG 4000 | 54.5 | 192.4 | vacuum impregnation | 70 | 53.0 | 73.8 | [168] |
| PEG 6000 | 61.7 | 178.6 | sol-gel | 97.3 | 60.4 | 164.9 | [172] |
| PEG 6000 distearate | 52.9 | 145.1 | sol-gel | 61.6 | 52.9 | 69.7 | [170] |
| PEG 6000 | 59 | 171 | sol-gel | 44.3 | 56.8 | 91.9 | [160] |
| PEG 6000 | 61.4 | 212.8 | sol-gel | 88.5 | 58.4 | 167.0 | [163] |
| PEG 1000 | 35.1 | 146.7 | sol-gel | 84.5 | 35.2 | 113.8 | [159] |
| PEG 1500 | 41.1 | 164.6 | sol-gel | 38.4 | 80.0 | 132.4 | [158] |
| PEG 2000 + PEG 10,000 | 51.7/62.4 | 180.6/ 170.9 | sol-gel co-crystallization | 36 | 56.5 | 108.6 | [165] |
3.4. Small Organic Compounds
3.5. Hydrated Salts
| PCM | Porous Silica Composite | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf (J g−1) | |
| Na2SO4·10H2O-Na2HPO4·12H2O | 36.7 | 226.9 | Melt impregnation/sol-gel SiO2 + PVP | 70 | 30.1 | 106.2 | [185] |
| Melt impregnation/sol-gel SiO2 | 70 | 28.5 | 67.5 | [186] | |||
| MgCl2.6H2O:Mg(NO3)2·6H2O (41.3:58.7) | 58.8 | 118.5 | Melt impregnation/fumed silica | 85 | 54.3 | 88.1 | [180] |
| Na2SO4·10H2O-Na2HPO4·12H2O | - | 221.4 | - | 70 | - | 64.1 | [191] |
| TBAB:H2O (4:6) | 11.8 | 211.9 | Melt impregnation/hydrophilic fumed silica | 70 | 8.3 | 134.0 | [188] |
| CaCl2·6H2O | 29.4 | 199.9 | Melt impregnation/SiO2 NPs | 75 | 25.1 | 148.2 | [181] |
3.6. Molten Salts
| PCM | Porous Silica Composite | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf (J g−1) | |
| Na2SO4 | 888.7 | 167.1 | Direct sol-gel synthesis | 50 | 886.0 | 82.3 | [192] |
| NaNO3 | 308.0 | 189.0 | Direct sol-gel synthesis | 60 | 302.0 | 108.0 | [193] |
| LiNO3 | 253.8 | 369.9 | Melt impregnation/KCC-1 | 70 | 251.3 | 292.2 | [166] |
| Na2CO3-K2CO3/MgO | 702.9 | 81.4 | Sintering/SiO2 NPs | 90 | 703.6 | 76.2 | [196] |
| NaCl-CaCl2 (1:1 mol) | 499.5 | 208.2 | Reactive melting/8.1 nm MSN | 95 | 499.1 | 60.8 | [197] |
| Na2SO4 | 886.7 | 167.1 | Direct sol-gel synthesis | 60 | 886.9 | 100.8 | [199] |
| LiNO3 | - | - | Direct sol-gel synthesis | 60 | 232.8 | 236.3 | [195] |
| NaNO3:KNO3 (1:1 mol) | 221.8 | 96.9 | Melt impregnation/MCF | 90 | 201.0 221.2 | 78.7 | [74] |
| Na(Cl, Br, MoO4) | 454.5 522.1 | 78.9 137.3 | Melt impregnation/MCM-41 | 80 | 450.4 514.4 | 52.8 111.2 | [198] |
3.7. Metals, Alloys and Elemental PCMs
4. Conclusions
5. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Minakshi, M.; Mitchell, D.R.G.; Jones, R.T.; Pramanik, N.C.; Jean-Fulcrand, A.; Garnweitner, G. A Hybrid Electrochemical Energy Storage Device Using Sustainable Electrode Materials. ChemistrySelect 2020, 5, 1597–1606. [Google Scholar] [CrossRef]
- Divakaran, A.M.; Hamilton, D.; Manjunatha, K.N.; Minakshi, M. Design, Development and Thermal Analysis of Reusable Li-Ion Battery Module for Future Mobile and Stationary Applications. Energies 2020, 13, 1477. [Google Scholar] [CrossRef] [Green Version]
- Ling, T.-C.; Poon, C.-S. Use of phase change materials for thermal energy storage in concrete: An overview. Constr. Build. Mater. 2013, 46, 55–62. [Google Scholar] [CrossRef]
- Pardo, P.; Deydier, A.; Anxionnaz-Minvielle, Z.; Rougé, S.; Cabassud, M.; Cognet, P. A review on high temperature thermochemical heat energy storage. Renew. Sustain. Energy Rev. 2014, 32, 591–610. [Google Scholar] [CrossRef] [Green Version]
- Abhat, A. Low temperature latent heat thermal energy storage: Heat storage materials. Sol. Energy 1983, 30, 313–332. [Google Scholar] [CrossRef]
- Zalba, B.; Marín, J.M.; Cabeza, L.F.; Mehling, H. Review on thermal energy storage with phase change: Materials, heat transfer analysis and applications. Appl. Eng. 2003, 23, 251–283. [Google Scholar] [CrossRef]
- Sharma, A.; Tyagi, V.V.; Chen, C.R.; Buddhi, D. Review on thermal energy storage with phase change materials and applications. Renew. Sustain. Energy Rev. 2009, 13, 318–345. [Google Scholar] [CrossRef]
- Cabeza, L.F.; Castell, A.; Barreneche, C.; de Gracia, A.; Fernández, A.I. Materials used as PCM in thermal energy storage in buildings: A review. Renew. Sustain. Energy Rev. 2011, 15, 1675–1695. [Google Scholar] [CrossRef]
- Oró, E.; de Gracia, A.; Castell, A.; Farid, M.M.; Cabeza, L.F. Review on phase change materials (PCMs) for cold thermal energy storage applications. Appl. Energy 2012, 99, 513–533. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas, B.; León, N. High temperature latent heat thermal energy storage: Phase change materials, design considerations and performance enhancement techniques. Renew. Sustain. Energy Rev. 2013, 27, 724–737. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, S. Medium- and high-temperature latent heat thermal energy storage: Material database, system review, and corrosivity assessment. Int. J. Energy Res. 2019, 43, 621–661. [Google Scholar] [CrossRef]
- Gerislioglu, B.; Ahmadivand, A.; Karabiyik, M.; Sinha, R.; Pala, N. VO2-Based Reconfigurable Antenna Platform with Addressable Microheater Matrix. Adv. Electron. Mater. 2017, 3, 1700170. [Google Scholar] [CrossRef]
- Gerislioglu, B.; Bakan, G.; Ahuja, R.; Adam, J.; Mishra, Y.K.; Ahmadivand, A. The role of Ge2Sb2Te5 in enhancing the performance of functional plasmonic devices. Mater. Today Phys. 2020, 12, 100178. [Google Scholar] [CrossRef]
- Bakan, G.; Gerislioglu, B.; Dirisaglik, F.; Jurado, Z.; Sullivan, L.; Dana, A.; Lam, C.; Gokirmak, A.; Silva, H. Extracting the temperature distribution on a phase-change memory cell during crystallization. J. Appl. Phys. 2016, 120, 164504. [Google Scholar] [CrossRef] [Green Version]
- Laraib Tariq, S.; Muhammad Ali, H.; Ammar Akram, M.; Mansoor Janjua, M.; Ahmadlouydarab, M. Nanoparticles enhanced Phase Change Materials (NePCMs)-A Recent Review. Appl. Eng. 2020, 176, 115305. [Google Scholar]
- Tauseef, u.R.; Ali, H.M.; Janjua, M.M.; Sajjad, U.; Yan, W.-M. A critical review on heat transfer augmentation of phase change materials embedded with porous materials/foams. Int. J. Heat Mass Transf. 2019, 135, 649–673. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Li, X.; Wu, X. Novel composite phase change materials with enhancement of light-thermal conversion, thermal conductivity and thermal storage capacity. Sol. Energy 2020, 196, 419–426. [Google Scholar] [CrossRef]
- Brown, R.C.; Rasberry, J.D.; Overmann, S.P. Microencapsulated phase-change materials as heat transfer media in gas-fluidized beds. Powder Technol. 1998, 98, 217–222. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, Z.; Yang, T.; Qin, D.; Li, S.; Zhang, G.; Haghighat, F.; Joybari, M.M. A review on macro-encapsulated phase change material for building envelope applications. Build. Environ. 2018, 144, 281–294. [Google Scholar] [CrossRef]
- Qi, H.; Zhang, T.; Zhang, D.; Wang, K.; Wang, Y. Paraffin/chitosan composite phase change materials fabricated by piercing-solidifying method for thermal energy storage. AIP Adv. 2020, 10, 035218. [Google Scholar] [CrossRef]
- Tao, Y.B.; He, Y.-L. A review of phase change material and performance enhancement method for latent heat storage system. Renew. Sustain. Energy Rev. 2018, 93, 245–259. [Google Scholar] [CrossRef]
- Hawes, D.W.; Feldman, D. Absorption of phase change materials in concrete. Sol. Energy Mater. Sol. Cells 1992, 27, 91–101. [Google Scholar] [CrossRef]
- Tang, J.; Yang, M.; Dong, W.; Yang, M.; Zhang, H.; Fan, S.; Wang, J.; Tan, L.; Wang, G. Highly porous carbons derived from MOFs for shape-stabilized phase change materials with high storage capacity and thermal conductivity. RSC Adv. 2016, 6, 40106–40114. [Google Scholar] [CrossRef]
- Sarı, A.; Karaipekli, A.; Alkan, C. Preparation, characterization and thermal properties of lauric acid/expanded perlite as novel form-stable composite phase change material. Chem. Eng. J. 2009, 155, 899–904. [Google Scholar] [CrossRef]
- Voronin, D.V.; Ivanov, E.; Gushchin, P.; Fakhrullin, R.; Vinokurov, V. Clay Composites for Thermal Energy Storage: A Review. Molecules 2020, 25, 1504. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Xia, W.; Zou, R. Nanoconfinement of phase change materials within carbon aerogels: Phase transition behaviours and photo-to-thermal energy storage. J. Mater. Chem. A 2014, 2, 19963–19968. [Google Scholar] [CrossRef]
- Xiangfa, Z.; Hanning, X.; Jian, F.; Changrui, Z.; Yonggang, J. Pore structure modification of silica matrix infiltrated with paraffin as phase change material. Chem. Eng. Res. Des. 2010, 88, 1013–1017. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, Y.; Yang, X.; Lin, J.; Zhu, Z. Encapsulation of metal-based phase change materials using ceramic shells prepared by spouted bed CVD method. Sol. Energy Mater. Sol. Cells 2017, 170, 137–142. [Google Scholar] [CrossRef]
- Gupta, R.; Kedia, S.; Saurakhiya, N.; Sharma, A.; Ranjan, A. Composite nanofibrous sheets of fatty acids and polymers as thermo-regulating enclosures. Sol. Energy Mater. Sol. Cells 2016, 157, 676–685. [Google Scholar] [CrossRef]
- Hölderich, W.; Hesse, M.; Näumann, F. Zeolites: Catalysts for Organic Syntheses. Angew. Chem. Int. Ed. Engl. 1988, 27, 226–246. [Google Scholar] [CrossRef]
- Koohsaryan, E.; Anbia, M. Nanosized and hierarchical zeolites: A short review. Chin. J. Catal. 2016, 37, 447–467. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Chen, J.; Ling, Z.; Fang, X.; Zhang, Z. Experimental and numerical investigation of form-stable dodecane/hydrophobic fumed silica composite phase change materials for cold energy storage. Energy Convers. Manag. 2015, 105, 817–825. [Google Scholar] [CrossRef]
- Kistler, S.S. Coherent Expanded Aerogels and Jellies. Nature 1931, 127, 741. [Google Scholar] [CrossRef]
- Soleimani Dorcheh, A.; Abbasi, M.H. Silica aerogel; synthesis, properties and characterization. J. Mater. Process. Technol. 2008, 199, 10–26. [Google Scholar] [CrossRef]
- Kistler, S.S.; Caldwell, A.G. Thermal Conductivity of Silica Aërogel. Ind. Eng. Chem. 1934, 26, 658–662. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Huo, Q.; Margolese, D.I.; Stucky, G.D. Surfactant Control of Phases in the Synthesis of Mesoporous Silica-Based Materials. Chem. Mater. 1996, 8, 1147–1160. [Google Scholar] [CrossRef]
- Fan, J.; Yu, C.; Gao, F.; Lei, J.; Tian, B.; Wang, L.; Luo, Q.; Tu, B.; Zhou, W.; Zhao, D. Cubic Mesoporous Silica with Large Controllable Entrance Sizes and Advanced Adsorption Properties. Angew. Chem. 2003, 115, 3254–3258. [Google Scholar] [CrossRef]
- Rao, A.P.; Rao, A.V.; Pajonk, G.M. Hydrophobic and Physical Properties of the Two Step Processed Ambient Pressure Dried Silica Aerogels with Various Exchanging Solvents. J. Sol-Gel Sci. Technol. 2005, 36, 285–292. [Google Scholar] [CrossRef]
- Deaconu, M.; Nicu, I.; Tincu, R.; Brezoiu, A.-M.; Mitran, R.-A.; Vasile, E.; Matei, C.; Berger, D. Tailored doxycycline delivery from MCM-41-type silica carriers. Chem. Zvesti 2018, 72, 1869–1880. [Google Scholar] [CrossRef]
- Iswar, S.; Malfait, W.J.; Balog, S.; Winnefeld, F.; Lattuada, M.; Koebel, M.M. Effect of aging on silica aerogel properties. Microporous Mesoporous Mater. 2017, 241, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Ramkumar, R.; Minakshi Sundaram, M. A biopolymer gel-decorated cobalt molybdate nanowafer: Effective graft polymer cross-linked with an organic acid for better energy storage. New J. Chem. 2016, 40, 2863–2877. [Google Scholar] [CrossRef]
- Barmi, M.J.; Minakshi, M. Tuning the Redox Properties of the Nanostructured CoMoO4 Electrode: Effects of Surfactant Content and Synthesis Temperature. ChemPlusChem 2016, 81, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhang, Z.; Zhang, W.; Wu, Y.; Zhang, J.; Imran, Z.; Zhang, D. Synthesis and Applications of Porous Glass. Shanghai Jiaotong Daxue Xuebao 2019, 24, 681–698. [Google Scholar] [CrossRef]
- Wu, H.; Tang, B.; Wu, P. Novel Hollow Mesoporous Silica Spheres/Polymer Hybrid Membrane for Ultrafiltration. J. Phys. Chem. C 2011, 116, 2246–2252. [Google Scholar] [CrossRef]
- Diacon, A.; Rusen, E.; Trifan, A.; Șomoghi, R.; Tutunaru, O.; Crăciun, G.; Busuioc, C.; Voicu, G. Preparation of metal and metal oxide doped silica hollow spheres and the evaluation of their catalytic performance. Colloid Polym. Sci. 2020, 298, 1401–1410. [Google Scholar] [CrossRef]
- Feng, X.; Fryxell, G.E.; Wang, L.-Q.; Kim, A.Y.; Liu, J.; Kemner, K.M. Functionalized Monolayers on Ordered Mesoporous Supports. Science 1997, 276, 923–926. [Google Scholar] [CrossRef]
- Mitran, R.-A.; Nastase, S.; Matei, C.; Berger, D. Tailoring the dissolution rate enhancement of aminoglutethimide by functionalization of MCM-41 silica: A hydrogen bonding propensity approach. RSC Adv. 2015, 5, 2592–2601. [Google Scholar] [CrossRef]
- Kecht, J.; Schlossbauer, A.; Bein, T. Selective Functionalization of the Outer and Inner Surfaces in Mesoporous Silica Nanoparticles. Chem. Mater. 2008, 20, 7207–7214. [Google Scholar] [CrossRef]
- Tao, Z. Mesoporous silica-based nanodevices for biological applications. RSC Adv. 2014, 4, 18961–18980. [Google Scholar] [CrossRef]
- Mitran, R.-A.; Berger, D.; Băjenaru, L.; Năstase, S.; Andronescu, C.; Matei, C. Azobenzene functionalized mesoporous AlMCM-41-type support for drug release applications. Cent. Eur. J. Chem. 2014, 12, 788–795. [Google Scholar] [CrossRef]
- Lin, S.; Shi, L.; Ribeiro Carrott, M.M.L.; Carrott, P.J.M.; Rocha, J.; Li, M.R.; Zou, X.D. Direct synthesis without addition of acid of Al-SBA-15 with controllable porosity and high hydrothermal stability. Microporous Mesoporous Mater. 2011, 142, 526–534. [Google Scholar] [CrossRef]
- Berger, D.; Nastase, S.; Mitran, R.A.; Petrescu, M.; Vasile, E.; Matei, C.; Negreanu-Pirjol, T. Mesostructured silica and aluminosilicate carriers for oxytetracycline delivery systems. Int. J. Pharm. 2016, 510, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Chaikriangkrai, A.; Takeshita, Y.; Shibata, M. Synthesis of iron-containing mesoporous silica aiming to use as a new sunscreen ultraviolet absorber. Chem. Lett. 2011, 40, 693–695. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q. A simple method to directly synthesize Al-SBA-15 mesoporous materials with different Al contents. Solid State Commun. 2008, 148, 529–533. [Google Scholar] [CrossRef]
- Souza, K.C.; Mohallem, N.D.S.; Sousa, E.M.B. Mesoporous silica-magnetite nanocomposite: Facile synthesis route for application in hyperthermia. J. Sol-Gel Sci. Technol. 2009, 53, 418–427. [Google Scholar] [CrossRef]
- Hurley, K.R.; Lin, Y.-S.; Zhang, J.; Egger, S.M.; Haynes, C.L. Effects of Mesoporous Silica Coating and Postsynthetic Treatment on the Transverse Relaxivity of Iron Oxide Nanoparticles. Chem. Mater. 2013, 25, 1968–1978. [Google Scholar] [CrossRef] [Green Version]
- Mitran, R.-A.; Matei, C.; Berger, D.; Băjenaru, L.; Moisescu, M.G. Controlling drug release from mesoporous silica through an amorphous, nanoconfined 1-tetradecanol layer. Eur. J. Pharm. Biopharm. 2018, 127, 318–325. [Google Scholar] [CrossRef]
- Baeza, A.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337. [Google Scholar] [CrossRef]
- Mitran, R.-A.; Culita, D.C.; Atkinson, I. Thermal stability enhancement of mesoporous SBA-15 silica through nanoconfinement of ceria nanoparticles. Microporous Mesoporous Mater. 2020, 306, 110484. [Google Scholar] [CrossRef]
- Doukeh, R.; Popovici, D.; Trifoi, A.; Bombos, M.; Banu, I. A study on the alkylation of m-cresol with 1-decene over mesoporous silica supported tungstophosphoric acid (HPW). React. Kinet. Mech. Catal. 2020, 131, 793–804. [Google Scholar] [CrossRef]
- Neffati, R.; Judeinstein, P.; Rault, J. Freezing, melting and dynamics of supercooled water confined in porous glass. J. Phys. Condens. Matter 2020, 32, 465101. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, Z.; Chang, Y.; Gao, H.; Cheng, P.; Tao, Z.; Lv, J. Toward Tailoring Chemistry of Silica-Based Phase Change Materials for Thermal Energy Storage. iScience 2020, 23, 101606. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, C.; Li, W.; Fan, X.; Wu, Z.; Zheng, J.; Li, X. Novel phase change behavior of n-eicosane in nanoporous silica: Emulsion template preparation and structure characterization using small angle X-ray scattering. Phys. Chem. Chem. Phys. 2013, 15, 14390–14395. [Google Scholar] [CrossRef]
- Matei, C.; Buhǎlţeanu, L.; Berger, D.; Mitran, R.-A. Functionalized mesoporous silica as matrix for shape-stabilized phase change materials. Int. J. Heat Mass Transf. 2019, 144, 118699. [Google Scholar] [CrossRef]
- Mitran, R.A.; Berger, D.; Munteanu, C.; Matei, C. Evaluation of Different Mesoporous Silica Supports for Energy Storage in Shape-Stabilized Phase Change Materials with Dual Thermal Responses. J. Phys. Chem. C 2015, 119, 15177–15184. [Google Scholar] [CrossRef]
- Riikonen, J.; Salonen, J.; Lehto, V.-P. Utilising thermoporometry to obtain new insights into nanostructured materials. J. Anal. Calorim. 2011, 105, 811–821. [Google Scholar] [CrossRef]
- Juras, B.; Martynas, K.; Jan, M.; Georg, V.; Winfried, B.; Venkatesan, U.; Martin, H.; Andreas, P. Broadband dielectric spectroscopy of water confined in MCM-41 molecular sieve materials—low-temperature freezing phenomena. J. Phys. Condens. Matter 2005, 17, 2843. [Google Scholar]
- Petrov, O.V.; Vargas-Florencia, D.; Furó, I. Surface Melting of Octamethylcyclotetrasiloxane Confined in Controlled Pore Glasses: Curvature Effects Observed by 1H NMR. J. Phys. Chem. B 2007, 111, 1574–1581. [Google Scholar] [CrossRef]
- Liu, E.; Dore, J.C.; Webber, J.B.W.; Khushalani, D.; Jähnert, S.; Findenegg, G.H.; Hansen, T. Neutron diffraction and NMR relaxation studies of structural variation and phase transformations for water/ice in SBA-15 silica: I. The over-filled case. J. Phys. Condens. Matter 2006, 18, 10009. [Google Scholar] [CrossRef]
- Wallacher, D.; Knorr, K. Melting and freezing of Ar in nanopores. Phys. Rev. B 2001, 63, 104202. [Google Scholar] [CrossRef]
- Husár, B.; Commereuc, S.; Lukáč, I.; Chmela, Š.; Nedelec, J.M.; Baba, M. Carbon Tetrachloride as a Thermoporometry Liquid Probe To Study the Cross-Linking of Styrene Copolymer Networks. J. Phys. Chem. B 2006, 110, 5315–5320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitran, R.-A.; Lincu, D.; Buhǎlţeanu, L.; Berger, D.; Matei, C. Shape-stabilized phase change materials using molten NaNO3–KNO3 eutectic and mesoporous silica matrices. Sol. Energy Mater. Sol. Cells 2020, 215, 110644. [Google Scholar] [CrossRef]
- Mitran, R.A.; Berger, D.; Matei, C. Phase Change Materials Based on Mesoporous Silica. Curr. Org. Chem. 2018, 22, 2644–2663. [Google Scholar] [CrossRef]
- Liu, S.; Ma, G.; Xie, S.; Jia, Y.; Sun, J.; Jing, Y. Diverting the phase transition behaviour of adipic acid via mesoporous silica confinement. RSC Adv. 2016, 6, 111787–111796. [Google Scholar] [CrossRef]
- Sterczyńska, A.; Deryło-Marczewska, A.; Śliwińska-Bartkowiak, M.; Piotrowska, J.Z.; Jarek, M.; Domin, K. Phase transitions of octamethylcyclotetrasiloxane confined inside aluminosilicate and silicate nanoporous matrices. J. Anal. Calorim. 2014, 118, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tian, S.; Xiao, D. Experimental study on the phase change behavior of phase change material confined in pores. Sol. Energy 2007, 81, 653–660. [Google Scholar] [CrossRef]
- Majda, D.; Korzeniowska, A.; Makowski, W.; Michalik-Zym, A.; Napruszewska, B.D.; Zimowska, M.; Serwicka, E.M. Thermoporosimetry of n-alkanes for characterization of mesoporous SBA-15 silicas–Refinement of methodology. Microporous Mesoporous Mater. 2016, 222, 33–43. [Google Scholar] [CrossRef]
- Lee, J.A.; Rösner, H.; Corrigan, J.F.; Huang, Y. Phase Transitions of Naphthalene and Its Derivatives Confined in Mesoporous Silicas. J. Phys. Chem. C 2011, 115, 4738–4748. [Google Scholar] [CrossRef]
- Nomura, T.; Zhu, C.; Sheng, N.; Tabuchi, K.; Sagara, A.; Akiyama, T. Shape-stabilized phase change composite by impregnation of octadecane into mesoporous SiO2. Sol. Energy Mater. Sol. Cells 2015, 143, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Miao, C.; Lü, G.; Yao, Y.; Tang, G.; Weng, D. Preparation of shape-stabilized phase change materials as temperature-adjusting powder. Front. Mater. Sci. 2007, 1, 284–287. [Google Scholar] [CrossRef]
- Li, H.; Fang, G.; Liu, X. Synthesis of shape-stabilized paraffin/silicon dioxide composites as phase change material for thermal energy storage. J. Mater. Sci. 2010, 45, 1672–1676. [Google Scholar] [CrossRef]
- Zhang, M.; Hong, Y.; Ding, S.; Hu, J.; Fan, Y.; Voevodin, A.A.; Su, M. Encapsulated nano-heat-sinks for thermal management of heterogeneous chemical reactions. Nanoscale 2010, 2, 2790–2797. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Su, L.; Liu, M.; Li, J.; Zhang, Z.; Li, B. Confinement effect of silica mesopores on thermal behavior of phase change composites. J. Sol-Gel Sci. Technol. 2016, 80, 180–188. [Google Scholar] [CrossRef]
- Hu, M.; Yan, Z.; Peng, L.; Guo, N.; Liu, Z. Optimization of preparation and analysis of Paraffin/SiO2 composite PCMs via sol-gel method. IOP Conf. Ser. Earth Environ. Sci. 2019, 242, 032005. [Google Scholar] [CrossRef]
- Hu, M.; Guo, N.; Wang, L. Preparation and assessment of paraffin/SiO2composite phase change material based on the efficacy coefficient method. Heat Mass Transf. 2020, 56, 1921–1929. [Google Scholar] [CrossRef]
- Liang, S.; Li, Q.; Zhu, Y.; Chen, K.; Tian, C.; Wang, J.; Bai, R. Nanoencapsulation of n-octadecane phase change material with silica shell through interfacial hydrolysis and polycondensation in miniemulsion. Energy 2015, 93, 1684–1692. [Google Scholar] [CrossRef]
- He, F.; Wang, X.; Wu, D. Phase-change characteristics and thermal performance of form-stable n-alkanes/silica composite phase change materials fabricated by sodium silicate precursor. Renew. Energy 2015, 74, 689–698. [Google Scholar] [CrossRef]
- Liu, H.; Niu, J.; Wang, X.; Wu, D. Design and construction of mesoporous silica/n-eicosane phase-change nanocomposites for supercooling depression and heat transfer enhancement. Energy 2019, 188, 116075. [Google Scholar] [CrossRef]
- Kong, X.; Zhong, Y.; Rong, X.; Min, C.; Qi, C. Building energy storage panel based on paraffin/expanded perlite: Preparation and thermal performance study. Materials 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zheng, S.; Zhu, S.; Ma, J.; Sun, Z.; Farid, M. Evaluation of paraffin infiltrated in various porous silica matrices as shape-stabilized phase change materials for thermal energy storage. Energy Convers. Manag. 2018, 171, 361–370. [Google Scholar] [CrossRef]
- Qu, Y.; Chen, J.; Liu, L.; Xu, T.; Wu, H.; Zhou, X. Study on properties of phase change foam concrete block mixed with paraffin / fumed silica composite phase change material. Renew. Energy 2020, 150, 1127–1135. [Google Scholar] [CrossRef]
- Zhou, X.; Xiao, H.; Feng, J.; Zhang, C.; Jiang, Y. Preparation and thermal properties of paraffin/porous silica ceramic composite. Compos. Sci. Technol. 2009, 69, 1246–1249. [Google Scholar] [CrossRef]
- Li, X.; Chen, H.; Liu, L.; Lu, Z.; Sanjayan, J.G.; Duan, W.H. Development of granular expanded perlite/paraffin phase change material composites and prevention of leakage. Sol. Energy 2016, 137, 179–188. [Google Scholar] [CrossRef]
- Lyu, J.; Li, G.; Liu, M.; Zhang, X. Aerogel-Directed Energy-Storage Films with Thermally Stimulant Multiresponsiveness. Langmuir 2019, 35, 943–949. [Google Scholar] [CrossRef]
- Snehal, K.; Das, B.B.; Kumar, S. Influence of Integration of Phase Change Materials on Hydration and Microstructure Properties of Nanosilica Admixed Cementitious Mortar. J. Mater. Civ. Eng. 2020, 32, 04020108. [Google Scholar] [CrossRef]
- Chung, O.; Jeong, S.G.; Yu, S.; Kim, S. Thermal performance of organic PCMs/micronized silica composite for latent heat thermal energy storage. Energy Build. 2014, 70, 180–185. [Google Scholar] [CrossRef]
- Goitandia, A.M.; Beobide, G.; Aranzabe, E.; Aranzabe, A. Development of content-stable phase change composites by infiltration into inorganic porous supports. Sol. Energy Mater. Sol. Cells 2015, 134, 318–328. [Google Scholar] [CrossRef]
- Belessiotis, G.V.; Papadokostaki, K.G.; Favvas, E.P.; Efthimiadou, E.K.; Karellas, S. Preparation and investigation of distinct and shape stable paraffin/SiO2 composite PCM nanospheres. Energy Convers. Manag. 2018, 168, 382–394. [Google Scholar] [CrossRef]
- Shaid, A.; Wang, L.; Islam, S.; Cai, J.Y.; Padhye, R. Preparation of aerogel-eicosane microparticles for thermoregulatory coating on textile. Appl. Eng. 2016, 107, 602–611. [Google Scholar] [CrossRef]
- Li, H.; Chen, H.; Li, X.; Sanjayan, J.G. Development of thermal energy storage composites and prevention of PCM leakage. Appl. Energy 2014, 135, 225–233. [Google Scholar] [CrossRef]
- Li, X.; Chen, H.; Li, H.; Liu, L.; Lu, Z.; Zhang, T.; Duan, W.H. Integration of form-stable paraffin/nanosilica phase change material composites into vacuum insulation panels for thermal energy storage. Appl. Energy 2015, 159, 601–609. [Google Scholar] [CrossRef]
- Gao, H.; Bo, L.; Liu, P.; Chen, D.; Li, A.; Ou, Y.; Dong, C.; Wang, J.; Chen, X.; Hou, C.; et al. Ambient pressure dried flexible silica aerogel for construction of monolithic shape-stabilized phase change materials. Sol. Energy Mater. Sol. Cells 2019, 201, 110122. [Google Scholar] [CrossRef]
- Yu, Y.; Xu, J.; Wang, G.; Zhang, R.; Peng, X. Preparation of paraffin/SiO2 aerogel stable-stabilized phase change composites for high-humidity environment. J. Mater. Sci. 2020, 55, 1511–1524. [Google Scholar] [CrossRef]
- Wang, F.; Gao, S.; Pan, J.; Li, X.; Liu, J. Short-chain modified SiO2 with high absorption of organic PCM for thermal protection. Nanomaterials 2019, 9, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, L.; Tao, S.; An, Y.; Meng, C.; Hu, T. Phase change in modified hierarchically porous monolith: An extra energy increase. Microporous Mesoporous Mater. 2014, 193, 69–76. [Google Scholar] [CrossRef]
- Wen, X.Y.; Ling, Z.Y.; Fang, X.M.; Zhang, Z.G. Preparation of RT28/ hydrophobic-fumed-silica composite phase change materials and their thermal insulation application in lithium ion battery. J. Chem. Eng. Chin. Univ. 2016, 30, 1178–1183. [Google Scholar]
- Choi, J.; Fujita, H.; Ogura, M.; Sakoda, A. Confinement effect on enthalpy of fusion and melting point of organic phase change materials in cylindrical nanospace of mesoporous silica and carbon. Adsorption 2018, 24, 345–355. [Google Scholar] [CrossRef]
- Choi, J.; Yoshie, K.; Moteki, T.; Ogura, M. Theoretical Evaluation of an Organic Phase Change Material (PCM)-Inserted Dual-Functional Adsorbent for the Recovery of Heat of Adsorption. Ind. Eng. Chem. Res. 2019, 58, 10114–10118. [Google Scholar] [CrossRef]
- Li, M.; Wu, Z.; Tan, J. Properties of form-stable paraffin/silicon dioxide/expanded graphite phase change composites prepared by sol-gel method. Appl. Energy 2012, 92, 456–461. [Google Scholar] [CrossRef]
- Han, X.; Zhao, T.; Gao, X.; Li, H. Preparation and characterization of high-temperature non-flowing SiO2/EG/paraffin composites by high-temperature refining. Colloids Surf. A 2018, 542, 1–7. [Google Scholar] [CrossRef]
- Lv, Y.; Situ, W.; Yang, X.; Zhang, G.; Wang, Z. A novel nanosilica-enhanced phase change material with anti-leakage and anti-volume-changes properties for battery thermal management. Energy Convers. Manag. 2018, 163, 250–259. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, C.; Fang, G. Preparation and thermal properties of n-eicosane/nano-SiO2/expanded graphite composite phase-change material for thermal energy storage. Mater. Chem. Phys. 2020, 240, 122178. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, K.; Kou, Y.; Wang, S.; Shi, Q. A facile strategy of constructing composite form-stable phase change materials with superior high thermal conductivity using silicagel industrial wastes. Sol. Energy 2020, 207, 51–58. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, Y.; Yuan, H.; Feng, D.; Zhang, X.; Wang, G. Thermal properties of C17H36/MCM-41 composite phase change materials. Comput. Mater. Sci. 2015, 109, 300–307. [Google Scholar] [CrossRef]
- Afgan, S.; Khushnood, R.A.; Memon, S.A.; Iqbal, N. Development of structural thermal energy storage concrete using paraffin intruded lightweight aggregate with nano-refined modified encapsulation paste layer. Constr. Build. Mater. 2019, 228, 116768. [Google Scholar] [CrossRef]
- Yuan, K.; Wang, H.; Liu, J.; Fang, X.; Zhang, Z. Novel slurry containing graphene oxide-grafted microencapsulated phase change material with enhanced thermo-physical properties and photo-thermal performance. Sol. Energy Mater. Sol. Cells 2015, 143, 29–37. [Google Scholar] [CrossRef]
- Amaral, C.; Gama, N.V.; Mohseni, F.; Amaral, J.S.; Amaral, V.S.; Marques, P.A.A.P.; Barros-Timmons, A.; Vicente, R. Development of structural layers PVC incorporating phase change materials for thermal energy storage. Appl. Eng. 2020, 179, 115707. [Google Scholar] [CrossRef]
- Jiang, F.; Wang, X.; Wu, D. Design and synthesis of magnetic microcapsules based on n-eicosane core and Fe3O4/SiO2 hybrid shell for dual-functional phase change materials. Appl. Energy 2014, 134, 456–468. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Li, J.; Ma, C.; Guo, L.; Meng, X. Solar-driven phase change microencapsulation with efficient Ti4O7 nanoconverter for latent heat storage. Nano Energy 2018, 53, 579–586. [Google Scholar] [CrossRef]
- Qian, T.; Li, J.; Min, X.; Fan, B. Integration of Pore Confinement and Hydrogen-Bond Influence on the Crystallization Behavior of C18 PCMs in Mesoporous Silica for Form-Stable Phase Change Materials. ACS Sustain. Chem. Eng. 2018, 6, 897–908. [Google Scholar] [CrossRef]
- Tan, N.; Xie, T.; Hu, P.; Feng, Y.; Li, Q.; Zhao, S.; Zhou, H.-N.; Zeng, W.-B.; Zeng, J.-L. Preparation and characterization of capric-palmitic acids eutectics/silica xerogel/exfoliated graphite nanoplatelets form-stable phase change materials. J. Energy Storage 2020, 102016. [Google Scholar] [CrossRef]
- Fang, G.; Li, H.; Liu, X. Preparation and properties of lauric acid/silicon dioxide composites as form-stable phase change materials for thermal energy storage. Mater. Chem. Phys. 2010, 122, 533–536. [Google Scholar] [CrossRef]
- Tahan Latibari, S.; Mehrali, M.; Mehrali, M.; Indra Mahlia, T.M.; Cornelis Metselaar, H.S. Synthesis, characterization and thermal properties of nanoencapsulated phase change materials via sol–gel method. Energy 2013, 61, 664–672. [Google Scholar] [CrossRef]
- Meng, D.; Zhao, K.; Zhao, W.; Jiang, G. Preparation and characterization of CA-MA eutectic/silicon dioxide nanoscale composite phase change material from water glass via sol-gel method. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2017, 32, 1048–1056. [Google Scholar] [CrossRef]
- Fu, Z.; Dai, L.; Yi, Y.; Luo, J.; Li, B. Structure and thermal properties of stearic acid/silica composites as form-stable phase change materials. J. Sol-Gel Sci. Technol. 2018, 87, 419–426. [Google Scholar] [CrossRef]
- Wang, C.; Cai, Z.; Wang, T.; Chen, K. Preparation and thermal properties of shape-stabilized 1, 8-octanediol /SiO2 composites via sol gel methods. Mater. Chem. Phys. 2020, 250, 123041. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Li, X.; Wu, X. Preparation of hydrophobic lauric acid/SiO2 shape-stabilized phase change materials for thermal energy storage. J. Energy Storage 2019, 21, 611–617. [Google Scholar] [CrossRef]
- Marske, F.; E Silva, J.M.D.S.; Wehrspohn, R.B.; Hahn, T.; Enke, D. Synthesis of monolithic shape-stabilized phase change materials with high mechanical stability: Via a porogen-assisted in situ sol-gel process. RSC Adv. 2020, 10, 3072–3083. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Sun, G.; Liu, M.; Zhang, J.; Wang, Q.; Wei, Q. Fabrication and characterization of capric-lauric-palmitic acid/electrospun SiO2 nanofibers composite as form-stable phase change material for thermal energy storage/retrieval. Sol. Energy 2015, 118, 87–95. [Google Scholar] [CrossRef]
- Zhang, J.; Narh, C.; Lv, P.; Cai, Y.; Zhou, H.; Hou, X.; Wei, Q. Preparation of novel form–stable composite phase change materials with porous silica nanofibrous mats for thermal storage/retrieval. Colloids Surf. A 2019, 570, 1–10. [Google Scholar] [CrossRef]
- Nemati, S.; Pircheraghi, G. Fabrication of a form-stable phase change material with green fatty acid and recycled silica nanoparticles from spent lead-acid battery separators with enhanced thermal conductivity. Acta 2020, 693, 178781. [Google Scholar] [CrossRef]
- Luo, Z.; Zhang, H.; Gao, X.; Xu, T.; Fang, Y.; Zhang, Z. Fabrication and characterization of form-stable capric-palmitic-stearic acid ternary eutectic mixture/nano-SiO2 composite phase change material. Energy Build. 2017, 147, 41–46. [Google Scholar] [CrossRef]
- Reddy, V.J.; Yadav, J.S.; Chattopadhyay, S. Phase change material loaded form-stable composites for low temperature thermal buffering application. Mater. Chem. Phys. 2020, 247, 122859. [Google Scholar] [CrossRef]
- Vennapusa, J.R.; Konala, A.; Dixit, P.; Chattopadhyay, S. Caprylic acid based PCM composite with potential for thermal buffering and packaging applications. Mater. Chem. Phys. 2020, 253, 123453. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, T.D.; Zheng, H.; Feng, H.X. Stearic acid/silica fume composite as form-stable phase change material for thermal energy storage. Energy Build. 2011, 43, 2365–2370. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Wang, B.; Lv, M.; Zhu, Y.; Gao, J. Fabrication and characterization of novel shape-stabilized stearic acid composite phase change materials with tannic-acid-templated mesoporous silica nanoparticles for thermal energy storage. RSC Adv. 2017, 7, 15625–15631. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Gao, H.; Dong, W.; Tang, J.; Wang, J.; Yang, M.; Wang, G. Shape-Stabilized Phase Change Materials Based on Stearic Acid and Mesoporous Hollow SiO2 Microspheres (SA/SiO2) for Thermal Energy Storage. Eur. J. Inorg. Chem. 2017, 2017, 2138–2143. [Google Scholar] [CrossRef]
- Gao, J.; Lv, M.; Lu, J.; Chen, Y.; Zhang, Z.; Zhang, X.; Zhu, Y. Enhanced Thermal Properties of Novel Latent Heat Thermal Storage Material Through Confinement of Stearic Acid in Meso-Structured Onion-Like Silica. JOM 2017, 69, 2785–2790. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Tang, X.; Liu, Y.; Han, Z.; Chen, Y. Comparison study between mesoporous silica nanoscale microsphere and active carbon used as the matrix of shape-stabilized phase change material. Sci. Rep. 2019, 9, 16056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Chen, Y.; Guo, X.; Tao, W.; Wang, J.; Gao, S.; Gao, J. Mesoporous silica nanoparticles with wrinkled structure as the matrix of myristic acid for the preparation of a promising new shape-stabilized phase change material via simple method. RSC Adv. 2018, 8, 34224–34231. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Ma, G.; Xie, S.; Sun, J.; Jia, Y.; Jing, Y. Preparation and characterization of the shape-stabilized phase change material based on sebacic acid and mesoporous MCM-41. J. Anal. Calorim. 2017, 130, 935–941. [Google Scholar] [CrossRef]
- Huang, X.; Liu, Z.; Xia, W.; Zou, R.; Han, R.P.S. Alkylated phase change composites for thermal energy storage based on surface-modified silica aerogels. J. Mater. Chem. A 2015, 3, 1935–1940. [Google Scholar] [CrossRef]
- Wan, X.; Chen, C.; Tian, S.; Guo, B. Thermal characterization of net-like and form-stable ML/SiO2 composite as novel PCM for cold energy storage. J. Energy Storage 2020, 28, 101276. [Google Scholar] [CrossRef]
- Mitran, R.-A.; Berger, D.; Matei, C. Improving thermal properties of shape-stabilized phase change materials containing lauric acid and mesocellular foam silica by assessing thermodynamic properties of the non-melting layer. Acta 2018, 660, 70–76. [Google Scholar] [CrossRef]
- Sarı, A.; Bicer, A.; Al-Ahmed, A.; Al-Sulaiman, F.A.; Zahir, M.H.; Mohamed, S.A. Silica fume/capric acid-palmitic acid composite phase change material doped with CNTs for thermal energy storage. Sol. Energy Mater. Sol. Cells 2018, 179, 353–361. [Google Scholar] [CrossRef]
- Lin, Y.; Cong, R.; Chen, Y.; Fang, G. Thermal properties and characterization of palmitic acid/nano silicon dioxide/graphene nanoplatelet for thermal energy storage. Int. J. Energy Res. 2020, 44, 5621–5633. [Google Scholar] [CrossRef]
- Zhang, M.; Xiao, Q.; Chen, C.; Li, L.; Yuan, W. Developing a heat-insulating composite phase change material with light-to-thermal conversion performance from graphene oxide/silica hybrid aerogel. Appl. Eng. 2020, 174, 115303. [Google Scholar]
- Ding, J.; Wu, X.; Shen, X.; Cui, S.; Chen, X. A promising form-stable phase change material composed of C/SiO2 aerogel and palmitic acid with large latent heat as short-term thermal insulation. Energy 2020, 210, 118478. [Google Scholar] [CrossRef]
- Liu, P.; Gao, H.; Chen, X.; Chen, D.; Lv, J.; Han, M.; Cheng, P.; Wang, G. In situ one-step construction of monolithic silica aerogel-based composite phase change materials for thermal protection. Compos. Part B 2020, 195, 108072. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, S.; Zhai, M.; Sui, J.; Lan, X.; Gao, J. Thermal Properties of Nano-Sized Polyethylene Glycol Confined in Silica Gels for Latent Heat Storage. Acta 2017, 655, 211–218. [Google Scholar] [CrossRef]
- Yang, H.; Feng, L.; Wang, C.; Zhao, W.; Li, X. Confinement Effect of SiO2 Framework on Phase Change of PEG in Shape-Stabilized PEG/SiO2 Composites. Eur. Polym. J. 2012, 48, 803–810. [Google Scholar] [CrossRef]
- Qian, Y.; Wei, P.; Jiang, P.; Li, Z.; Yan, Y.; Ji, K.; Deng, W. Preparation of Shape-Stabilized Co-Crystallized Poly (Ethylene Glycol) Composites as Thermal Energy Storage Materials. Energy Convers. Manag. 2013, 76, 101–108. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, T. Influence of SiO2 Pore Structure on Phase Change Enthalpy of Shape-Stabilized Polyethylene Glycol/Silica Composites. J. Mater. Sci. 2013, 48, 3716–3721. [Google Scholar] [CrossRef]
- Liu, Z.; Wei, H.; Tang, B.; Xu, S.; Shufen, Z. Novel Light–Driven CF/PEG/SiO2 Composite Phase Change Materials with High Thermal Conductivity. Sol. Energy Mater. Sol. Cells 2018, 174, 538–544. [Google Scholar] [CrossRef]
- Weng, Z.; Wu, K.; Luo, F.; Xiao, F.; Zhang, Q.; Wang, S.; Lu, M. Fabrication of High Thermal Conductive Shape-Stabilized Polyethylene Glycol/Silica Phase Change Composite by Two-Step Sol Gel Method. Compos. Part A Appl. Sci. Manuf. 2018, 110, 106–112. [Google Scholar] [CrossRef]
- Sun, K.; Kou, Y.; Zheng, H.; Liu, X.; Tan, Z.; Shi, Q. Using Silicagel Industrial Wastes to Synthesize Polyethylene Glycol/Silica-Hydroxyl Form-Stable Phase Change Materials for Thermal Energy Storage Applications. Sol. Energy Mater. Sol. Cells 2018, 178, 139–145. [Google Scholar] [CrossRef]
- Serrano, A.; Martín del Campo, J.; Peco, N.; Rodriguez, J.F.; Carmona, M. Influence of Gelation Step for Preparing PEG–SiO2 Shape-Stabilized Phase Change Materials by Sol–Gel Method. J. Sol-Gel Sci. Technol. 2019, 89, 731–742. [Google Scholar] [CrossRef]
- Wan, X.; Su, L.; Guo, B. Design and Preparation of Novel Shapeable PEG/SiO2/AA Shape-Stabilized Phase Change Materials Based on Double-Locked Network with Enhanced Heat Storage Capacity for Thermal Energy Regulation and Storage. Powder Technol. 2019, 353, 98–109. [Google Scholar] [CrossRef]
- Liu, Z.; Tang, B.; Zhang, S. Novel Network Structural PEG/PAA/SiO2 Composite Phase Change Materials with Strong Shape Stability for Storing Thermal Energy. Sol. Energy Mater. Sol. Cells 2020, 216, 110678. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, W.; Shi, X.; Yao, C.; Kuai, C. Use of PEG/SiO2 Phase Change Composite to Control Porous Asphalt Concrete Temperature. Constr. Build. Mater. 2020, 245, 118459. [Google Scholar] [CrossRef]
- Yan, D.; Tang, B.; Zhang, S. Preparation and Performances of Sunlight-Induced Phase Change PEG/SiO2-Dye Composite for Solar Energy Conversion and Storage. Sol. Energy Mater. Sol. Cells 2020, 215, 110657. [Google Scholar] [CrossRef]
- Xu, J.; Yang, T.; Xu, X.; Guo, X.; Cao, J. Processing Solid Wood into a Composite Phase Change Material for Thermal Energy Storage by Introducing Silica-Stabilized Polyethylene Glycol. Compos. Part A Appl. Sci. Manuf. 2020, 139, 106098. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X.; Fang, Y.; Ding, J. Preparation and Performance of Form-Stable Polyethylene Glycol/Silicon Dioxide Composites as Solid-Liquid Phase Change Materials. Appl. Energy 2009, 86, 170–174. [Google Scholar] [CrossRef]
- Qian, T.; Li, J.; Min, X.; Deng, Y.; Guan, W.; Ning, L. Radial-like Mesoporous Silica Sphere: A Promising New Candidate of Supporting Material for Storage of Low-, Middle-, and High-Temperature Heat. Energy 2016, 112, 1074–1083. [Google Scholar] [CrossRef]
- Gao, J.; Tao, W.; Chen, D.; Guo, X.; Chen, Y.; Jiang, Y. High Performance Shape-Stabilized Phase Change Material with Nanoflower-like Wrinkled Mesoporous Silica Encapsulating Polyethylene Glycol: Preparation and Thermal Properties. Nanomaterials 2018, 8, 385. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ding, H.; Wang, B.; Shi, Q.; Gao, J.; Cui, Z.; Wan, Y. Dopamine Functionalization for Improving Crystallization Behaviour of Polyethylene Glycol in Shape-Stable Phase Change Material with Silica Fume as the Matrix. J. Clean. Prod. 2019, 208, 951–959. [Google Scholar] [CrossRef]
- Gao, J.; Zhou, J.; Zhang, X.; Shi, Q.; Han, Z.; Chen, Y. Facile Functionalized Mesoporous Silica Using Biomimetic Method as New Matrix for Preparation of Shape-Stabilized Phase-Change Material with Improved Enthalpy. Int. J. Energy Res. 2019, 43, 8649–8659. [Google Scholar] [CrossRef]
- Kumar, A.; Jain, H.; Tripathi, B.P. Synthesis and Nanoencapsulation of Poly(Ethylene Glycol)-Distearates Phase Change Materials for Latent Heat Storage and Release. ACS Appl. Energy Mater. 2020, 3, 5965–5976. [Google Scholar] [CrossRef]
- Abbasi Hattan, H.; Madhkhan, M.; Marani, A. Thermal and Mechanical Properties of Building External Walls Plastered with Cement Mortar Incorporating Shape-Stabilized Phase Change Materials (SSPCMs). Constr. Build. Mater. 2020, 121385. [Google Scholar] [CrossRef]
- Li, B.; Shu, D.; Wang, R.; Zhai, L.; Chai, Y.; Lan, Y.; Cao, H.; Zou, C. Polyethylene Glycol/Silica (PEG@SiO2) Composite Inspired by the Synthesis of Mesoporous Materials as Shape-Stabilized Phase Change Material for Energy Storage. Renew. Energy 2020, 145, 84–92. [Google Scholar] [CrossRef]
- Feng, D.; Feng, Y.; Li, P.; Zang, Y.; Wang, C.; Zhang, X. Modified Mesoporous Silica Filled with PEG as a Shape-Stabilized Phase Change Materials for Improved Thermal Energy Storage Performance. Microporous Mesoporous Mater. 2020, 292, 109756. [Google Scholar] [CrossRef]
- Wang, A.; Chen, C.; Xu, G. Silica/acetamide composite as form-stable phase change material for latent heat thermal energy storage. J. Adv. Microsc. Res. 2012, 7, 286–291. [Google Scholar] [CrossRef]
- Pethurajan, V.; Sivan, S.; Konatt, A.J.; Reddy, A.S. Facile approach to improve solar thermal energy storage efficiency using encapsulated sugar alcohol based phase change material. Sol. Energy Mater. Sol. Cells 2018, 185, 524–535. [Google Scholar] [CrossRef]
- Zhou, X.; Xiao, H.; Feng, J.; Zhang, C.; Jiang, Y. Preparation, properties and thermal control applications of silica aerogel infiltrated with solid-liquid phase change materials. J. Exp. Nanosci. 2012, 7, 17–26. [Google Scholar]
- Toyoda, T.; Narisada, R.; Suzuki, H.; Hidema, R.; Komoda, Y. Fabrication process of silica hard-shell microcapsule (HSMC) containing phase-change materials. Chem. Lett. 2014, 43, 820–821. [Google Scholar] [CrossRef]
- Zhai, M.; Zhang, S.; Sui, J.; Tian, F.; Lan, X.Z. Solid–solid phase transition of tris(hydroxymethyl)aminomethane in nanopores of silica gel and porous glass for thermal energy storage. J. Anal. Calorim. 2017, 129, 957–964. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Y.; Sun, R.; Lai, M.; Du, R.; Zhang, Z. The anti-supercooling effect of surface-modified nano-scaled SiO2 in hydrated salts phase transition system. J. Phys. Conf. Ser. 2009, 188, 012046. [Google Scholar] [CrossRef] [Green Version]
- Ling, Z.; Liu, J.; Wang, Q.; Lin, W.; Fang, X.; Zhang, Z. MgCl2·6H2O-Mg(NO3)2·6H2O eutectic/SiO2 composite phase change material with improved thermal reliability and enhanced thermal conductivity. Sol. Energy Mater. Sol. Cells 2017, 172, 195–201. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Q.; Liu, Y.; Wang, D.; Song, W.; Chen, Y.; Liu, J. Calcium chloride hexahydrate/nano-SiO2 composites as form-stable phase change materials for building energy conversation: The influence of pore size of nano-SiO2. Energy Build. 2020, 208, 109672. [Google Scholar] [CrossRef]
- Gao, C.-F.; Wang, L.-P.; Li, Q.-F.; Wang, C.; Nan, Z.-D.; Lan, X.-Z. Tuning thermal properties of latent heat storage material through confinement in porous media: The case of (1-CnH2n+1NH3)2ZnCl4 (n=10 and 12). Sol. Energy Mater. Sol. Cells 2014, 128, 221–230. [Google Scholar] [CrossRef]
- Li, Q.F.; Wang, C.; Lan, X.Z. Solid-solid phase transition of (1-C14H29NH3)2ZnCl4 in nanopores of silica gel for thermal energy storage. Chin. Chem. Lett. 2017, 28, 49–54. [Google Scholar] [CrossRef]
- Garay Ramirez, B.M.L.; Glorieux, C.; San Martin Martinez, E.; Flores Cuautle, J.J.A. Tuning of thermal properties of sodium acetate trihydrate by blending with polymer and silver nanoparticles. Appl. Eng. 2014, 62, 838–844. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, T. Preparation and characterization of hydrated salts/silica composite as shape-stabilized phase change material via sol-gel process. Acta 2014, 591, 10–15. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, T. The dependence of phase change enthalpy on the pore structure and interfacial groups in hydrated salts/silica composites via sol–gel. J. Colloid Interface Sci. 2015, 448, 100–105. [Google Scholar] [CrossRef]
- Peng, S.; Huang, J.; Wang, T.; Zhu, P. Effect of fumed silica additive on supercooling, thermal reliability and thermal stability of Na2HPO4·12H2O as inorganic PCM. Acta 2019, 675, 1–8. [Google Scholar] [CrossRef]
- Zou, T.; Fu, W.; Liang, X.; Wang, S.; Gao, X.; Zhang, Z.; Fang, Y. Preparation and performance of form-stable TBAB hydrate/SiO2 composite PCM for cold energy storage. Int. J. Refrig. 2019, 101, 117–124. [Google Scholar] [CrossRef]
- Fang, Y.; Su, J.; Tang, Y.; Liang, X.; Wang, S.; Gao, X.; Zhang, Z. Form-stable Na2SO4·10H2O-Na2HPO4·12H2O eutectic/hydrophilic fumed silica composite phase change material with low supercooling and low thermal conductivity for indoor thermal comfort improvement. Int. J. Energy Res. 2020, 44, 3171–3182. [Google Scholar] [CrossRef]
- Fu, W.; Zou, T.; Liang, X.; Wang, S.; Gao, X.; Zhang, Z.; Fang, Y. Preparation and properties of phase change temperature-tuned composite phase change material based on sodium acetate trihydrate–urea/fumed silica for radiant floor heating system. Appl. Eng. 2019, 162, 114253. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, T. Enthalpy of solid−liquid phase change confined in porous materials. Ind. Eng. Chem. Res. 2016, 55, 11536–11541. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, T. Preparation and Characterization of Sodium Sulfate/Silica Composite as a Shape-stabilized Phase Change Material by Sol-gel Method. Chin. J. Chem. Eng. 2014, 22, 360–364. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, T. Study on preparation and thermal properties of sodium nitrate/silica composite as shape-stabilized phase change material. Acta 2015, 613, 66–70. [Google Scholar] [CrossRef]
- Wu, X.; Ding, J.; Kong, Y.; Sun, Z.; Shao, G.; Li, B.; Wu, J.; Zhong, Y.; Shen, X.; Cui, S. Synthesis of a novel three-dimensional Na2SO4@SiO2@Al2O3-SiO2 phase change material doped aerogel composite with high thermal resistance and latent heat. Ceram. Int. 2018, 44, 21855–21865. [Google Scholar] [CrossRef]
- Milián, Y.E.; Reinaga, N.; Grágeda, M.; Ushak, S. Development of new inorganic shape stabilized phase change materials with LiNO3 and LiCl salts by sol-gel method. J. Sol-Gel Sci. Technol. 2020, 94, 22–33. [Google Scholar]
- Deng, Z.F.; Tan, H.; Zhang, G.Q.; Xu, G.Z.; Yang, C.Y.; Chang, L.; Du, Z.L. Effects of SiO2/SiC particles on the thermal stability of carbonate eutectic salt/ceramic composite. IOP Conf. Ser. Mater. Sci. Eng. 2019, 504, 012034. [Google Scholar] [CrossRef]
- Mitran, R.-A.; Petrescu, S.; Şomǎcescu, S.; Mocioiu, O.C.; Buhǎlţeanu, L.; Berger, D.; Matei, C. Nanocomposite phase change materials based on NaCl–CaCl2 and mesoporous silica. J. Anal. Calorim. 2019, 138, 2555–2563. [Google Scholar] [CrossRef]
- Mitran, R.A.; Lincu, D.; Ioniţǎ, S.; Deaconu, M.; Jerca, V.V.; Mocioiu, O.C.; Berger, D.; Matei, C. High temperature shape–Stabilized phase change materials obtained using mesoporous silica and NaCl–NaBr–Na2MoO4 salt eutectic. Sol. Energy Mater. Sol. Cells 2020, 218, 110760. [Google Scholar] [CrossRef]
- Milian, Y.E.; Ushak, S. Design of synthesis route for inorganic shape-stabilized phase change materials. Direct sol–gel process versus vacuum impregnation method. J. Sol-Gel Sci. Technol. 2020, 94, 67–79. [Google Scholar] [CrossRef]
- Shin, S.J.; Guzman, J.; Yuan, C.W.; Liao, C.Y.; Boswell-Koller, C.N.; Stone, P.R.; Dubon, O.D.; Minor, A.M.; Watanabe, M.; Beeman, J.W.; et al. Embedded binary eutectic alloy nanostructures: A new class of phase change materials. Nano Lett. 2010, 10, 2794–2798. [Google Scholar] [CrossRef]
- Wei, H.; Wang, C.; Yang, S.; Han, J.; Yang, M.; Zhang, J.; Lu, Y.; Liu, X. A strategy for designing microencapsulated composite phase change thermal storage materials with tunable melting temperature. Sol. Energy Mater. Sol. Cells 2019, 203, 110166. [Google Scholar] [CrossRef]
















| Heat Storage | Sensible Heat | Latent Heat | Chemical Heat | |
|---|---|---|---|---|
| Properties | ||||
| Energy density | <600–800 kJ kg−1 0.8–1.7 J g−1 K−1 [4] | ~100–1800 kJ kg−1 * | 300–3000 kJ kg−1 [4] | |
| Temperature difference needed for storage, ΔT | 25–1200 °C [4] | 0–50 °C * | 100–500 °C [4] | |
| Volume change | ~1% [4] | 10–40% [5] | >1000% (at 1 atm) [4] | |
| Complexity | Very simple | Simple | Complex | |
| Maturity | Industrial scale | Pilot scale | Laboratory scale | |
| Type | Property | Value | Benefits |
|---|---|---|---|
| Thermal properties | Heat of fusion | High | Increased energy storage density |
| Specific heat | High | ||
| Thermal conductivity | High | Increased power density, lower temperature gradients | |
| Melting point | Determines operating temperature | ||
| Physical properties | Volume change on transition | Low | Increases stability, minimizes leakage |
| Vapor pressure | Low | Decreases evaporative loss of material | |
| Crystallization rate | High | Decreases the hysteresis between charging and discharging | |
| Supercooling degree | Low | ||
| Chemical properties | Thermal & chemical stability | High | Increases life cycle |
| Reactivity/corrosiveness | Low | ||
| Non-toxic, non-flammable, non-explosive | High | Increases safety and decreases system complexity | |
| Wettability & surface tension | High | Formation of shape stabilized materials with the silica matrix and higher PCM loading | |
| Economic properties | Cost | Low | Improved economic efficiency and decreased risk |
| Abundance | High | ||
| Environmental impact | Low |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitran, R.-A.; Ioniţǎ, S.; Lincu, D.; Berger, D.; Matei, C. A Review of Composite Phase Change Materials Based on Porous Silica Nanomaterials for Latent Heat Storage Applications. Molecules 2021, 26, 241. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010241
Mitran R-A, Ioniţǎ S, Lincu D, Berger D, Matei C. A Review of Composite Phase Change Materials Based on Porous Silica Nanomaterials for Latent Heat Storage Applications. Molecules. 2021; 26(1):241. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010241
Chicago/Turabian StyleMitran, Raul-Augustin, Simona Ioniţǎ, Daniel Lincu, Daniela Berger, and Cristian Matei. 2021. "A Review of Composite Phase Change Materials Based on Porous Silica Nanomaterials for Latent Heat Storage Applications" Molecules 26, no. 1: 241. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26010241