
Role and Perspective of Molecular Simulation-Based Investigation of RNA–Ligand Interaction: From Small Molecules and Peptides to Photoswitchable RNA Binding


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Targeting RNA Trinucleotide Repeat Expansions: HTT RNA CAG
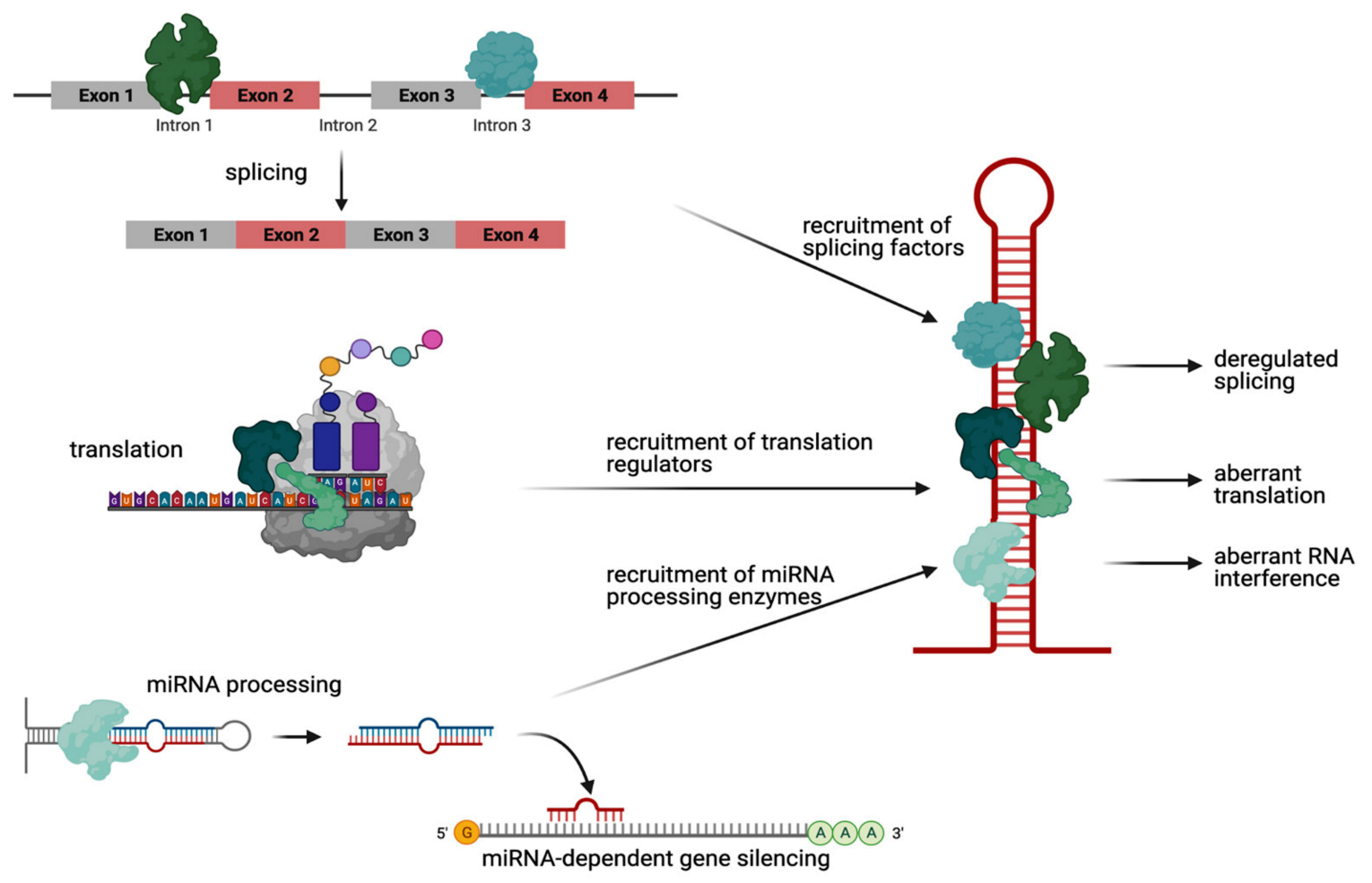
2.1. RNA-Mediated Toxicity: Aberrant RNA Hairpin–Protein Interactions in HD
2.2. MD Simulations of HTT RNA CAG with Small Molecules
3. Targeting HIV-1 TAR RNA Hairpin
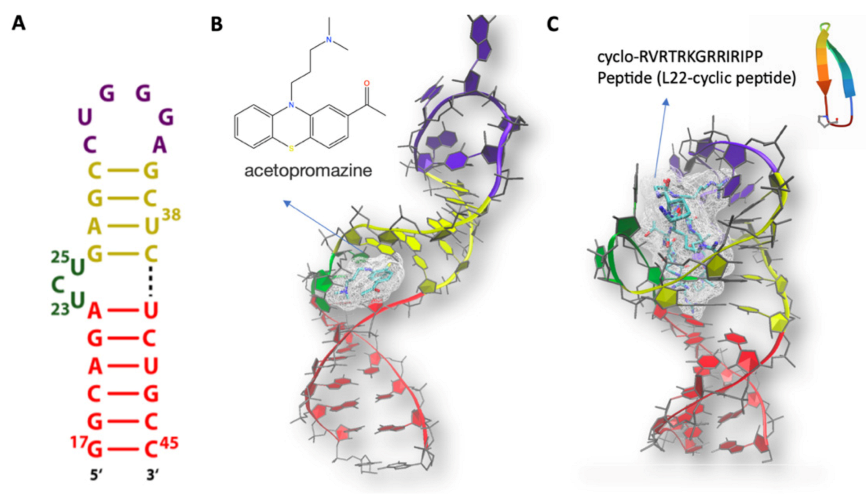
MD Simulations of HIV TAR with Ligands
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Petrov, A.S.; Gulen, B.; Norris, A.M.; Kovacs, N.A.; Bernier, C.R.; Lanier, K.A.; Fox, G.E.; Harvey, S.C.; Wartell, R.M.; Hud, N.V.; et al. History of the ribosome and the origin of translation. Proc. Natl. Acad. Sci. USA 2015, 112, 15396–15401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luhrmann, R.; Kastner, B.; Bach, M. Structure of spliceosomal snRNPs and their role in pre-mRNA splicing. Biochim. Biophys. Acta 1990, 1087, 265–292. [Google Scholar] [CrossRef]
- Scott, W.G. RNA catalysis. Curr. Opin. Struct. Biol. 1998, 8, 720–726. [Google Scholar] [CrossRef]
- Choi, S.I.; Ryu, K.; Seong, B.L. RNA-mediated chaperone type for de novo protein folding. RNA Biol. 2009, 6, 21–24. [Google Scholar] [CrossRef]
- Balcerak, A.; Trebinska-Stryjewska, A.; Konopinski, R.; Wakula, M.; Grzybowska, E.A. RNA-protein interactions: Disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol. 2019, 9, 190096. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Daley, D.T.; Luscombe, N.M.; Berman, H.M.; Thornton, J.M. Protein-RNA interactions: A structural analysis. Nucleic Acids Res. 2001, 29, 943–954. [Google Scholar] [CrossRef]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [Green Version]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Marelli, C.; Maschat, F. The P42 peptide and Peptide-based therapies for Huntington’s disease. Orphanet J. Rare Dis. 2016, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.R.; Hergenrother, P.J. Targeting RNA with small molecules. Chem. Rev. 2008, 108, 1171–1224. [Google Scholar] [CrossRef]
- Connelly, C.M.; Moon, M.H.; Schneekloth, J.S., Jr. The Emerging Role of RNA as a Therapeutic Target for Small Molecules. Cell Chem. Biol. 2016, 23, 1077–1090. [Google Scholar] [CrossRef]
- Falese, J.P.; Donlic, A.; Hargrove, A.E. Targeting RNA with small molecules: From fundamental principles towards the clinic. Chem. Soc. Rev. 2021, 50, 2224–2243. [Google Scholar] [CrossRef]
- Bernat, V.; Disney, M.D. RNA Structures as Mediators of Neurological Diseases and as Drug Targets. Neuron 2015, 87, 28–46. [Google Scholar] [CrossRef] [Green Version]
- Kruger, D.M.; Neubacher, S.; Grossmann, T.N. Protein-RNA interactions: Structural characteristics and hotspot amino acids. RNA 2018, 24, 1457–1465. [Google Scholar] [CrossRef]
- Mirza, S.B.; Lee, R.C.H.; Chu, J.J.H.; Salmas, R.E.; Mavromoustakos, T.; Durdagi, S. Discovery of selective dengue virus inhibitors using combination of molecular fingerprint-based virtual screening protocols, structure-based pharmacophore model development, molecular dynamics simulations and in vitro studies. J. Mol. Graph. Model. 2018, 79, 88–102. [Google Scholar] [CrossRef]
- Bochicchio, A.; Rossetti, G.; Tabarrini, O.; Krauß, S.; Carloni, P. Molecular view of ligands specificity for CAG repeats in anti-Huntington therapy. J. Chem. Theory Comput. 2015, 11, 4911–4922. [Google Scholar] [CrossRef]
- Matthes, F.; Massari, S.; Bochicchio, A.; Schorpp, K.; Schilling, J.; Weber, S.; Offermann, N.; Desantis, J.; Wanker, E.E.; Carloni, P.; et al. Reducing mutant Huntingtin protein expression in living cells by a newly identified RNA CAG binder. ACS Chem. Neurosci. 2018, 9, 1399–1408. [Google Scholar] [CrossRef] [Green Version]
- Bottaro, S.; Bussi, G.; Kennedy, S.D.; Turner, D.H.; Lindorff-Larsen, K. Conformational ensembles of RNA oligonucleotides from integrating NMR and molecular simulations. Sci. Adv. 2018, 4, eaar8521. [Google Scholar] [CrossRef] [Green Version]
- Musiani, F.; Rossetti, G.; Capece, L.; Gerger, T.M.; Micheletti, C.; Varani, G.; Carloni, P. Molecular dynamics simulations identify time scale of conformational changes responsible for conformational selection in molecular recognition of HIV-1 transactivation responsive RNA. J. Am. Chem. Soc. 2014, 136, 15631–15637. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.G.; Tan, Z.; Spasic, A.; Dutta, D.; Salas-Estrada, L.A.; Grossfield, A.; Mathews, D.H. Chemically Accurate Relative Folding Stability of RNA Hairpins from Molecular Simulations. J. Chem. Theory Comput. 2018, 14, 6598–6612. [Google Scholar] [CrossRef]
- Šponer, J.; Bussi, G.; Krepl, M.; Banáš, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurečka, P.; et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denesyuk, N.A.; Hori, N.; Thirumalai, D. Molecular Simulations of Ion Effects on the Thermodynamics of RNA Folding. J. Phys. Chem. B 2018, 122, 11860–11867. [Google Scholar] [CrossRef] [PubMed]
- Draper, D.E. A guide to ions and RNA structure. RNA 2004, 10, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlýnský, V.; Bussi, G. Exploring RNA structure and dynamics through enhanced sampling simulations. Curr. Opin. Struct. Biol. 2018, 49, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Leach, A.R.; Ar, L. Molecular Modelling; Pearson Education: London, UK, 2001; p. 744. [Google Scholar]
- Tuckerman, M.E.; Martyna, G.J. Understanding Modern Molecular Dynamics: Techniques and Applications. J. Phys. Chem. B 2000, 104, 159–178. [Google Scholar] [CrossRef]
- Yang, M. Discoveries of Tat-TAR interaction inhibitors for HIV-1. Curr. Drug Targets Infect. Disord. 2005, 5, 433–444. [Google Scholar] [CrossRef]
- La Spada, A.R.; Taylor, J.P. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 2010, 11, 247–258. [Google Scholar] [CrossRef]
- Verma, A.K.; Khan, E.; Bhagwat, S.R.; Kumar, A. Exploring the Potential of Small Molecule-Based Therapeutic Approaches for Targeting Trinucleotide Repeat Disorders. Mol. Neurobiol. 2020, 57, 566–584. [Google Scholar] [CrossRef]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef]
- Ursu, A.; Childs-Disney, J.L.; Andrews, R.J.; O’Leary, C.A.; Meyer, S.M.; Angelbello, A.J.; Moss, W.N.; Disney, M.D. Design of small molecules targeting RNA structure from sequence. Chem. Soc. Rev. 2020, 49, 7252–7270. [Google Scholar] [CrossRef]
- Kiliszek, A.; Kierzek, R.; Krzyzosiak, W.J.; Rypniewski, W. Atomic resolution structure of CAG RNA repeats: Structural insights and implications for the trinucleotide repeat expansion diseases. Nucleic Acids Res. 2010, 38, 8370–8376. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, K.; Krzyzosiak, W.J. Imperfect CAG repeats form diverse structures in SCA1 transcripts. J. Biol. Chem. 2004, 279, 41563–41572. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, K.; Krzyzosiak, W.J. CAG repeats containing CAA interruptions form branched hairpin structures in spinocerebellar ataxia type 2 transcripts. J. Biol. Chem. 2005, 280, 3898–3910. [Google Scholar] [CrossRef] [Green Version]
- Nalavade, R.; Griesche, N.; Ryan, D.P.; Hildebrand, S.; Krauss, S. Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 2013, 4, e752. [Google Scholar] [CrossRef] [Green Version]
- Schilling, J.; Griesche, N.; Krauß, S. Mechanisms of RNA-Induced Toxicity in Diseases Characterised by CAG Repeat Expansions; John Wiley & Sons, Ltd.: Chichester, UK, 2016. [Google Scholar]
- Schilling, J.; Broemer, M.; Atanassov, I.; Duernberger, Y.; Vorberg, I.; Dieterich, C.; Dagane, A.; Dittmar, G.; Wanker, E.; van Roon-Mom, W.; et al. Deregulated Splicing Is a Major Mechanism of RNA-Induced Toxicity in Huntington’s Disease. J. Mol. Biol. 2019, 431, 1869–1877. [Google Scholar] [CrossRef]
- Mykowska, A.; Sobczak, K.; Wojciechowska, M.; Kozlowski, P.; Krzyzosiak, W.J. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011, 39, 8938–8951. [Google Scholar] [CrossRef] [Green Version]
- Gipson, T.A.; Neueder, A.; Wexler, N.S.; Bates, G.P.; Housman, D. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. 2013, 10, 1647–1652. [Google Scholar] [CrossRef] [Green Version]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.; Roos, R.A.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef] [Green Version]
- Neueder, A.; Dumas, A.A.; Benjamin, A.C.; Bates, G.P. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat. Commun. 2018, 9, 3955. [Google Scholar] [CrossRef] [Green Version]
- Franich, N.R.; Hickey, M.A.; Zhu, C.; Osborne, G.F.; Ali, N.; Chu, T.; Bove, N.H.; Lemesre, V.; Lerner, R.P.; Zeitlin, S.O.; et al. Phenotype onset in Huntington’s disease knock-in mice is correlated with the incomplete splicing of the mutant huntingtin gene. J. Neurosci. Res. 2019, 97, 1590–1605. [Google Scholar] [CrossRef]
- Mason, M.A.; Gomez-Paredes, C.; Sathasivam, K.; Neueder, A.; Papadopoulou, A.S.; Bates, G.P. Silencing Srsf6 does not modulate incomplete splicing of the huntingtin gene in Huntington’s disease models. Sci. Rep. 2020, 10, 14057. [Google Scholar] [CrossRef]
- Krauss, S.; Griesche, N.; Jastrzebska, E.; Chen, C.; Rutschow, D.; Achmuller, C.; Dorn, S.; Boesch, S.M.; Lalowski, M.; Wanker, E.; et al. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat. Commun. 2013, 4, 1511. [Google Scholar] [CrossRef] [Green Version]
- Griesche, N.; Schilling, J.; Weber, S.; Rohm, M.; Pesch, V.; Matthes, F.; Auburger, G.; Krauss, S. Regulation of mRNA translation by MID1: A common mechanism of expanded CAG repeat RNAs. Front. Cell. Neurosci. 2016, 10, 226. [Google Scholar] [CrossRef] [Green Version]
- Trockenbacher, A.; Suckow, V.; Foerster, J.; Winter, J.; Krauss, S.; Ropers, H.H.; Schneider, R.; Schweiger, S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 2001, 29, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Knutzen, C.A.; Krauss, S.; Schweiger, S.; Chiang, G.G. Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc. Natl. Acad. Sci. USA 2011, 108, 8680–8685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Fiszer, A.; Mykowska, A.; Sobczak, K.; de Mezer, M.; Krzyzosiak, W.J. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell 2007, 25, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.W.; Rudnicki, D.D.; Yu, L.; Margolis, R.L. A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum. Mol. Genet. 2011, 20, 3467–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, K.T.; O’Keefe, L.V.; Samaraweera, S.E.; van Eyk, C.L.; McLeod, C.J.; Maloney, C.A.; Dang, T.H.; Suter, C.M.; Richards, R.I. Double-stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum. Mol. Genet. 2011, 20, 3757–3768. [Google Scholar] [CrossRef] [Green Version]
- Banez-Coronel, M.; Porta, S.; Kagerbauer, B.; Mateu-Huertas, E.; Pantano, L.; Ferrer, I.; Guzman, M.; Estivill, X.; Marti, E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012, 8, e1002481. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Guo, P.; Lin, X.; An, Y.; Sze, K.-H.; Lau, M.H.Y.; Chen, Z.S.; Wang, Q.; Li, W.; Sun, J.K.-L.; et al. CAG RNAs induce DNA damage and apoptosis by silencing NUDT16expression in polyglutamine degeneration. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar]
- Disney, M.D. Targeting RNA with Small Molecules to Capture Opportunities at the Intersection of Chemistry, Biology, and Medicine. J. Am. Chem. Soc. 2019, 141, 6776–6790. [Google Scholar] [CrossRef]
- Juru, A.U.; Patwardhan, N.N.; Hargrove, A.E. Understanding the Contributions of Conformational Changes, Thermodynamics, and Kinetics of RNA–Small Molecule Interactions. ACS Chem. Biol. 2019, 14, 824–838. [Google Scholar] [CrossRef]
- Sledz, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Park, H.-J. Virtual Screening for RNA-Interacting Small Molecules. Biophys. Approaches Transl. Control Gene Expr. 2013, 1, 235. [Google Scholar]
- Tessaro, F.; Scapozza, L. How ‘Protein-Docking’ Translates into the New Emerging Field of Docking Small Molecules to Nucleic Acids? Molecules 2020, 25, 2749. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Filikov, A.V.; Mohan, V.; Vickers, T.A.; Griffey, R.H.; Cook, P.D.; Abagyan, R.A.; James, T.L. Identification of ligands for RNA targets via structure-based virtual screening: HIV-1 TAR. J. Comput. Aided Mol. Des. 2000, 14, 593–610. [Google Scholar] [CrossRef]
- Moitessier, N.; Westhof, E.; Hanessian, S. Docking of aminoglycosides to hydrated and flexible RNA. J. Med. Chem. 2006, 49, 1023–1033. [Google Scholar] [CrossRef]
- Guilbert, C.; James, T.L. Docking to RNA via root-mean-square-deviation-driven energy minimization with flexible ligands and flexible targets. J. Chem. Inf. Model. 2008, 48, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Morley, S.D.; Afshar, M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using Ribodock. J. Comput. Aided Mol. Des. 2004, 18, 189–208. [Google Scholar] [CrossRef]
- Pfeffer, P.; Gohlke, H. DrugScoreRNA--knowledge-based scoring function to predict RNA-ligand interactions. J. Chem. Inf. Model. 2007, 47, 1868–1876. [Google Scholar] [CrossRef]
- Salsbury, A.M.; Lemkul, J.A. Recent developments in empirical atomistic force fields for nucleic acids and applications to studies of folding and dynamics. Curr. Opin. Struct. Biol. 2021, 67, 9–17. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules J. Am. Chem. Soc. 1995, 117, 5179− 5197. J. Am. Chem. Soc. 1996, 118, 2309. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, J.D.; Condon, D.E.; Kennedy, S.D.; Hauser, M.; Bevilacqua, P.C.; Turner, D.H. The nuclear magnetic resonance of CCCC RNA reveals a right-handed helix, and revised parameters for AMBER force field torsions improve structural predictions from molecular dynamics. Biochemistry 2013, 52, 996–1010. [Google Scholar] [CrossRef]
- Sponer, J.; Banas, P.; Jurecka, P.; Zgarbova, M.; Kuhrova, P.; Havrila, M.; Krepl, M.; Stadlbauer, P.; Otyepka, M. Molecular Dynamics Simulations of Nucleic Acids. From Tetranucleotides to the Ribosome. J. Phys. Chem. Lett. 2014, 5, 1771–1782. [Google Scholar] [CrossRef]
- Perez, A.; Marchan, I.; Svozil, D.; Sponer, J.; Cheatham, T.E., 3rd; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [Green Version]
- Zgarbova, M.; Otyepka, M.; Sponer, J.; Mladek, A.; Banas, P.; Cheatham, T.E., 3rd; Jurecka, P. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Perez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goni, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, I.; Stern, H.A.; Kennedy, S.D.; Tubbs, J.D.; Turner, D.H. Reparameterization of RNA chi Torsion Parameters for the AMBER Force Field and Comparison to NMR Spectra for Cytidine and Uridine. J. Chem. Theory Comput. 2010, 6, 1520–1531. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.A.; Garcia, A.E. High-resolution reversible folding of hyperstable RNA tetraloops using molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 16820–16825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, I. Improvement of RNA Simulations with Torsional Revisions of the AMBER Force Field. Methods Mol. Biol. 2019, 2022, 55–74. [Google Scholar] [PubMed]
- Wales, D.J.; Yildirim, I. Improving Computational Predictions of Single-Stranded RNA Tetramers with Revised α/γ Torsional Parameters for the Amber Force Field. J. Phys. Chem. B 2017, 121, 2989–2999. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Jurečka, P.; Banáš, P.; Havrila, M.; Šponer, J.; Otyepka, M. Noncanonical α/γ Backbone Conformations in RNA and the Accuracy of Their Description by the AMBER Force Field. J. Phys. Chem. B 2017, 121, 2420–2433. [Google Scholar] [CrossRef]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef]
- Bergonzo, C.; Grishaev, A. Accuracy of MD solvent models in RNA structure refinement assessed via liquid-crystal NMR and spin relaxation data. J. Struct. Biol. 2019, 207, 250–259. [Google Scholar] [CrossRef]
- Kührová, P.; Mlýnský, V.; Zgarbová, M.; Krepl, M.; Bussi, G.; Best, R.B.; Otyepka, M.; Šponer, J.; Banáš, P. Improving the Performance of the Amber RNA Force Field by Tuning the Hydrogen-Bonding Interactions. J. Chem. Theory Comput. 2019, 15, 3288–3305. [Google Scholar] [CrossRef]
- Mlynsky, V.; Kuhrova, P.; Kuhr, T.; Otyepka, M.; Bussi, G.; Banas, P.; Sponer, J. Fine-Tuning of the AMBER RNA Force Field with a New Term Adjusting Interactions of Terminal Nucleotides. J. Chem. Theory Comput. 2020, 16, 3936–3946. [Google Scholar] [CrossRef]
- Tan, D.; Piana, S.; Dirks, R.M.; Shaw, D.E. RNA force field with accuracy comparable to state-of-the-art protein force fields. Proc. Natl. Acad. Sci. USA 2018, 115, E1346–E1355. [Google Scholar] [CrossRef] [Green Version]
- Foloppe, N.; MacKerell, A.D., Jr. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Banavali, N.K. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000, 21, 105–120. [Google Scholar] [CrossRef]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; MacKerell, A.D. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lu, C.; Jing, Z.; Wu, C.; Piquemal, J.-P.; Ponder, J.W.; Ren, P. AMOEBA Polarizable Atomic Multipole Force Field for Nucleic Acids. J. Chem. Theory Comput. 2018, 14, 2084–2108. [Google Scholar] [CrossRef]
- Lemkul, J.A.; MacKerell, A.D. Polarizable force field for RNA based on the classical drude oscillator. J. Comput. Chem. 2018, 39, 2624–2646. [Google Scholar] [CrossRef]
- Ebenezer, O.; Jordaan, M.A.; Damoyi, N.; Shapi, M. Discovery of Potential Inhibitors for RNA-Dependent RNA Polymerase of Norovirus: Virtual Screening, and Molecular Dynamics. Int. J. Mol. Sci. 2020, 22, 171. [Google Scholar] [CrossRef]
- Roy, S.; Ali, A.; Bhattacharya, S. Theoretical Insight into the Library Screening Approach for Binding of Intermolecular G-Quadruplex RNA and Small Molecules through Docking and Molecular Dynamics Simulation Studies. J. Phys. Chem. B 2021. [Google Scholar] [CrossRef]
- Kumar, A.; Parkesh, R.; Sznajder, L.J.; Childs-Disney, J.L.; Sobczak, K.; Disney, M.D. Chemical correction of pre-mRNA splicing defects associated with sequestration of muscleblind-like 1 protein by expanded r(CAG)-containing transcripts. ACS Chem. Biol. 2012, 7, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Poltronieri, P.; Sun, B.; Mallardo, M. RNA Viruses: RNA Roles in Pathogenesis, Coreplication and Viral Load. Curr. Genom. 2015, 16, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Salmon, L.; Bascom, G.; Andricioaei, I.; Al-Hashimi, H.M. A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings: The basis for interhelical motions revealed. J. Am. Chem. Soc. 2013, 135, 5457–5466. [Google Scholar] [CrossRef] [Green Version]
- Bardaro, M.F., Jr.; Shajani, Z.; Patora-Komisarska, K.; Robinson, J.A.; Varani, G. How binding of small molecule and peptide ligands to HIV-1 TAR alters the RNA motional landscape. Nucleic Acids Res. 2009, 37, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Lind, K.E.; Du, Z.; Fujinaga, K.; Peterlin, B.M.; James, T.L. Structure-Based Computational Database Screening, In Vitro Assay, and NMR Assessment of Compounds that Target TAR RNA. Chem. Biol. 2002, 9, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Lind, K.E.; James, T.L. Structure of TAR RNA complexed with a Tat-TAR interaction nanomolar inhibitor that was identified by computational screening. Chem. Biol. 2002, 9, 707–712. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Davidson, A.; Leeper, T.C.; Athanassiou, Z.; Patora-Komisarska, K.; Karn, J.; Robinson, J.A.; Varani, G. Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. USA 2009, 106, 11931–11936. [Google Scholar] [CrossRef] [Green Version]
- Aboul-ela, F.; Karn, J.; Varani, G. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 1996, 24, 3974–3981. [Google Scholar] [CrossRef] [Green Version]
- Aboul-ela, F.; Karn, J.; Varani, G. The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J. Mol. Biol. 1995, 253, 313–332. [Google Scholar] [CrossRef]
- Faber, C.; Sticht, H.; Schweimer, K.; Rosch, P. Structural rearrangements of HIV-1 Tat-responsive RNA upon binding of neomycin B. J. Biol. Chem. 2000, 275, 20660–20666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murchie, A.I.; Davis, B.; Isel, C.; Afshar, M.; Drysdale, M.J.; Bower, J.; Potter, A.J.; Starkey, I.D.; Swarbrick, T.M.; Mirza, S.; et al. Structure-based drug design targeting an inactive RNA conformation: Exploiting the flexibility of HIV-1 TAR RNA. J. Mol. Biol. 2004, 336, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Afshar, M.; Varani, G.; Murchie, A.I.; Karn, J.; Lentzen, G.; Drysdale, M.; Bower, J.; Potter, A.J.; Starkey, I.D.; et al. Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic “hot spots”. J. Mol. Biol. 2004, 336, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.; Patora-Komisarska, K.; Robinson, J.A.; Varani, G. Essential structural requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat protein. Nucleic Acids Res. 2011, 39, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Davidson, A.; Begley, D.W.; Lau, C.; Varani, G. A small-molecule probe induces a conformation in HIV TAR RNA capable of binding drug-like fragments. J. Mol. Biol. 2011, 410, 984–996. [Google Scholar] [CrossRef] [Green Version]
- Do, T.N.; Ippoliti, E.; Carloni, P.; Varani, G.; Parrinello, M. Counterion Redistribution upon Binding of a Tat-Protein Mimic to HIV-1 TAR RNA. J. Chem. Theory Comput. 2012, 8, 688–694. [Google Scholar] [CrossRef]
- Levintov, L.; Vashisth, H. Role of conformational heterogeneity in ligand recognition by viral RNA molecules. Phys. Chem. Chem. Phys. 2021, 23, 11211–11223. [Google Scholar] [CrossRef]
- Do, T.N.; Carloni, P.; Varani, G.; Bussi, G. RNA/Peptide Binding Driven by Electrostatics-Insight from Bidirectional Pulling Simulations. J. Chem. Theory Comput. 2013, 9, 1720–1730. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Chen, Y.-L.; Pollack, L.; Kirmizialtin, S. The structural plasticity of nucleic acid duplexes revealed by WAXS and MD. Sci. Adv. 2021, 7, eabf6106. [Google Scholar] [CrossRef]
- Fröhlking, T.; Bernetti, M.; Calonaci, N.; Bussi, G. Toward empirical force fields that match experimental observables. J. Chem. Phys. 2020, 152, 230902. [Google Scholar] [CrossRef]
- Howe, J.A.; Wang, H.; Fischmann, T.O.; Balibar, C.J.; Xiao, L.; Galgoci, A.M.; Malinverni, J.C.; Mayhood, T.; Villafania, A.; Nahvi, A.; et al. Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526, 672–677. [Google Scholar] [CrossRef]
- Berdnikova, D.V. Visible-range hemi-indigo photoswitch: ON-OFF fluorescent binder for HIV-1 RNA. Chem. Commun. 2019, 55, 8402–8405. [Google Scholar] [CrossRef] [Green Version]
- Berdnikova, D.V. Design, synthesis and investigation of water-soluble hemi-indigo photoswitches for bioapplications. Beilstein J. Org. Chem. 2019, 15, 2822–2829. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berdnikova, D.V.; Carloni, P.; Krauß, S.; Rossetti, G. Role and Perspective of Molecular Simulation-Based Investigation of RNA–Ligand Interaction: From Small Molecules and Peptides to Photoswitchable RNA Binding. Molecules 2021, 26, 3384. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113384
Berdnikova DV, Carloni P, Krauß S, Rossetti G. Role and Perspective of Molecular Simulation-Based Investigation of RNA–Ligand Interaction: From Small Molecules and Peptides to Photoswitchable RNA Binding. Molecules. 2021; 26(11):3384. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113384
Chicago/Turabian StyleBerdnikova, Daria V., Paolo Carloni, Sybille Krauß, and Giulia Rossetti. 2021. "Role and Perspective of Molecular Simulation-Based Investigation of RNA–Ligand Interaction: From Small Molecules and Peptides to Photoswitchable RNA Binding" Molecules 26, no. 11: 3384. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113384